
Abstract
Hepatoblasts are somatic progenitor cells in the fetal liver, which retain a high proliferative capacity and differentiate into both hepatocytes and cholangiocytes in vivo. Although efficient expansion of hepatoblasts in vitro has been difficult without genetic modification, we have previously demonstrated that the interaction with mesenchymal cells is important for expansion of hepatoblasts in vitro. In this study, we show cell signaling pathways regulating the long-term proliferative ability of hepatoblasts. Individual primary hepatoblasts derived from mouse fetal livers formed large colonies when cocultured with mesenchymal feeder cells; however, secondary colony formation was unsuccessful, indicating that in vitro culture could induce short-term, but not long-term, proliferation. When the MEK inhibitor, PD0325901, was added to these cultures, hepatoblasts formed large colonies containing many Ki-67-positive cells. Expression of p16/19cdkn2a, a cyclin-dependent kinase inhibitor, was induced after 3–6 days culture of hepatoblasts, whereas PD0325901 significantly suppressed this expression. Consistent with these observations, fetal hepatoblasts derived from p16/19cdkn2a knockout mice showed long-term proliferation without PD0325901, suggesting that MEK activity induced cell cycle arrest through accumulation of p16/19cdkn2a. In transplantation assays, we could demonstrate that in vitro expanded hepatoblasts could proliferate and differentiate into hepatocytic and cholangiocytic cells in injured livers. It should also be noted that ERK in primary hepatoblasts was not highly activated during fetal liver development. Collectively, all these findings suggest that the MEK/ERK-independent pathway in the fetal liver is involved in hepatoblast proliferation to avoid accumulation of cyclin-dependent kinase inhibitor.
Introduction
T
In the early stage of liver development, the foregut endoderm migrates into the septum transversum and differentiates into the liver bud [2,3]. This liver bud contains many hepatoblasts, fetal hepatic progenitor cells, which have a high proliferative activity and capacity for bipotent differentiation [4]. In the mid-to-late fetal livers, most hepatoblasts differentiate into immature and mature hepatocytes that comprise the parenchymal cells in the adult liver. Oncostatin M, an interleukin-6 family cytokine, is produced by hematopoietic cells in the fetal liver and induces maturation of hepatoblasts and immature hepatocytes [5]. Cholangiocytic differentiation occurs in the periportal region during mid-to-late fetal liver development [6]. Identification and purification of hepatoblasts have been achieved using specific cell surface antigens and fluorescence-activated cell sorting. CD29+CD49flow cells derived from mid-fetal livers show a colony-forming activity, which have been named hepatic colony-forming unit cells (H-CFU-Cs) [7]. In addition, Dlk, Liv2, CD13, and CD133 are expressed on hepatoblasts during fetal liver development [8 –10]. Hepatic progenitor cells also exist in normal and injured adult livers [11 –15]. For example, CD13 and CD133 are specific markers of adult liver progenitor cells, and CD13+CD133+ cells in adult livers exhibit a high proliferative ability and can differentiate into hepatocytic and cholangiocytic cells.
Hepatoblasts expand and differentiate into a large number of hepatocytes in vivo during development [8]. Transplanted hepatoblasts also proliferate and repopulate injured livers in vivo [10,16]. In contrast, few hepatoblasts can proliferate in long-term culture in vitro without genetic modification [17]. These observations suggest that the high proliferative potential of hepatic cells is suppressed during in vitro culture, but not in vivo. The interactions between hepatoblasts and other nonparenchymal cells are important for the differentiation and proliferation of hepatoblasts. Thy1+ mesenchymal cells derived from fetal livers induce maturation of hepatoblasts and embryonic stem cell-derived hepatic cells in coculture systems [18]. Paracrine signals from mesenchymal cell populations also regulate the expansion and differentiation of human hepatic stem/progenitor cells [19]. However, long-term expansion remains difficult for hepatic stem and progenitor cells in vitro.
In this study, we established a new expansion system for long-term proliferation of hepatoblasts derived from fetal mouse livers. Hepatoblasts cocultured with mouse embryonic fibroblasts (MEFs) formed more colonies than those cultured without MEFs. However, these colonies could not undergo long-term proliferation because of the induction of p16/19cdkn2a. We found that the addition of an MEK inhibitor, PD0325901, suppressed the expression of p16/19cdkn2a and induced hepatoblast proliferation. Limited numbers of hepatoblasts could expand in coculture with MEFs in the presence of PD0325901 and compensate for injured liver hepatocytes in vivo. Proliferation and survival of many types of cells are known to be regulated by the MEK-ERK pathway. For example, the MEK-ERK pathway is important for the progression of G0–G1 cell cycle progression [20,21]. However, our results suggested that a high level of activation of the MEK-ERK pathway in fetal hepatoblasts induced the accumulation of cyclin-dependent kinase (cdk) inhibitors and cell cycle arrest.
Materials and Methods
Materials
C57BL/6NCrSlc mice, ICR mouse, and green fluorescent protein (GFP)-transgenic (Tg) mice were purchased from Nihon SLC (Shizuoka, Japan). p16/19cdkn2a knockout (KO) mice were obtained from the Mouse Models of Human Cancers Consortium in the National Cancer Institute (Frederick, MD). Animal experiments were performed with approval of the Institutional Animal Care and Use Committee of the Institute of Medical Science, the University of Tokyo, and the Institutional Animal Care and Use Committee of Tokai University. Dulbecco's modified Eagle's medium (DMEM), DMEM/Ham's F12 medium, penicillin/streptomycin/
Preparation of MEFs
Embryonic day (E) 13.5 ICR mice embryos were dissected, and the head and internal organs were completely removed. The torso was minced and dissociated in 0.05% trypsin-EDTA (Sigma) for 30 min. After washing, cells were expanded in DMEM supplemented with 10% FBS and penicillin/streptomycin/
In vitro culture of fetal hepatoblasts
Minced embryonic liver tissues from E13.5 mice were dissociated with a 0.05% collagenase solution. Dissociated liver cells were washed with phosphate-buffered saline (PBS) containing 3% FBS, and then incubated with antibodies against cell surface markers for 60 min at 4°C. After washing with PBS containing 3% FBS and staining the dead cells with propidium iodide, the cells were analyzed and sorted using the MoFlo™ fluorescence-activated cell sorter (DAKO, Glostrup, Denmark and a FACS Aria [BD Biosciences, San Jose, CA).
The standard culture medium was a 1:1 mix of H-CFU-C medium (DMEM/Ham's F12 medium supplemented with 10% FBS, 1 × insulin–transferrin–selenium X, 10 mM nicotinamide, 10−7 M dexamethasone, 2.5 mM HEPES, 1 × penicillin/streptomycin/
After first 6 days of culture (the primary culture in Supplementary Fig. S1A; Supplementary Data are available online at
Overexpression of cyclin kinase inhibitors using retroviruses
The retroviral vector pGCDNsam, with a long terminal repeat derived from murine stem cell virus (MSCV), has intact splice donor and splice acceptor sequences for generation of subgenomic mRNA [23]. The cDNAs of p16ink4a and p19ARF were subcloned into an upstream sequence of an internal ribosomal entry site in a pGCDNsam vector. This vector has the sequence of GFP from an internal ribosomal entry site. Retroviruses were generated as previously described [24]. Virus titers were determined by infection of NIH3T3 cells. Purified hepatoblasts were sorted onto feeder cells at a low density and infected with retrovirus in the presence of 1 μg/mL protamine sulfate (Sigma). After 24 h of incubation, the cells were washed and cultured in the standard culture medium with HGF and EGF.
Messenger RNA detection by RT-PCR
Dlk+CD133+ cells in the nonhematopoietic cell fraction from GFP-Tg mice were purified using the cell sorter. After 3 or 6 days of coculture with MEFs, GFP-positive cells were purified using the cell sorter. Total RNA was extracted from primary Dlk+CD133+ cells and cultured cells using an RNeasy Micro Kit (Qiagen, Venlo, The Netherlands). First-strand cDNA synthesized using a High-Capacity cDNA reverse transcription Kit (Life Technologies) was used as a template for quantitative RT-PCR. The expression of cyclins and cyclin inhibitors was analyzed using the TaqMan Low-Density Array real-time PCR system (Life Technologies). cDNA samples were normalized by the expression of glyceraldehyde-3-phosphate dehydrogenase. In some quantitative RT-PCR assays, we used the Universal ProbeLibrary system (Roche Diagnosis, Basel, Switzerland), and cDNA samples were normalized by the expression of hypoxanthine phosphoribosyltransferase. The probe number for each gene is shown in Supplementary Table S1.
In-droplet cell staining
To quantify phospho-ERK expression in individual cells, we used an in-droplet staining method [25]. Briefly, Dlk+CD133+ cells derived from fetal livers were sorted onto poly-
Transplantation of expanded hepatoblasts into livers with hepatocellular injury
Recipient KSN/Slc nude mice were treated weekly with retrorsine (60 mg/kg i.p.) for 3 weeks to inhibit replication of endogenous hepatocytes [14]. At 3 days after the final retrorsine injection, the mice underwent a 70% hepatectomy under anesthesia. Donor cells were isolated from E13.5 fetuses of heterozygous GFP-Tg mice. A total of 2 × 103 Dlk+CD133+ cells in the CD45−Ter119−c-Kit− fraction were purified and cocultured with MEFs for 5 days in the presence of PD0325901. Expanded cells were suspended in 0.1 mL DMEM containing 5% KSR, and then injected into the splenic pulp using a 30G needle. All mice were sacrificed at 7–20 weeks after transplantation. Transplanted liver tissues were fixed with 4% paraformaldehyde. Cryostat sections were cut from frozen tissues embedded in optimal cutting temperature compound (Sakura Finetechnical, Tokyo, Japan). Donor cell expansion was analyzed using immunostaining.
Immunostaining
Cultured cells were washed with PBS, and then fixed with 4% paraformaldehyde/PBS. After three washes with PBS, the cells were permeabilized with 0.25% Triton/PBS for 10 min. After washing with PBS, the cells were incubated with 5% donkey serum/PBS for 1 h at room temperature, and then diluted primary antibodies overnight at 4°C. The primary antibodies were goat anti-albumin (Bethyl Laboratories, Montgomery, TX), rabbit anti-cytokeratine 19 (CK19, a gift from Prof. A. Miyajima, The University of Tokyo) [9], rabbit anti-GFP (Life Technologies), and rabbit anti-Ki67 (Abcam, Cambridge, MA) antibodies. The cells were then washed with PBS and incubated for 1 h at room temperature with anti-rabbit IgG-Alexa 488, anti-rabbit IgG-Alexa 546, or anti-goat IgG-Alexa 546 antibodies (Life Technologies). The cells were washed with PBS and their nuclei were counterstained with 40,6-diamidine-20-phenylindole dihydrochloride (DAPI; Sigma).
For the transplantation samples, after cryostat sections were washed with PBS, the cells were incubated with 5% donkey serum/PBS for 1 h at room temperature, and then diluted primary antibodies overnight at 4°C. For hepatocytic cell staining, the cryostat sections were stained with both goat anti-albumin and rabbit anti-GFP antibodies. For cholangiocytic cell staining, the cryostat serial sections were stained with either rabbit anti-CK19 or rabbit anti-GFP antibodies. After incubation, sections were washed and stained with anti-rabbit IgG-Alexa 488, anti-rabbit IgG-Alexa 546, or anti-goat IgG-Alexa 546 antibodies. Samples were washed with PBS and their nuclei were counterstained with DAPI.
Fluorescence images were obtained with a Carl Zeiss Axio Observer Z1 using AxioVision version 4.8 software (Carl Zeiss, Jena, Germany).
Statistics
Microsoft Excel 2007 for Windows (Microsoft, Redmond, WA) was used to calculate standard deviations and statistically significant differences between samples with Student's two-tailed t-test.
Other methods are shown in the Supplementary Materials and Methods section.
Results
The MEK-ERK pathway in hepatoblast colony formation assay
During liver development, fetal hepatoblasts are highly proliferative and have the ability to differentiate into both mature hepatocytes and cholangiocytes. Purified hepatoblasts form colonies in low-density culture on extracellular matrix-coated dishes. However, when hepatoblasts are cultured without nonparenchymal cells, the efficiency of colony formation is low. Mesenchymal cells in fetal livers are important for the proliferation and differentiation of fetal hepatoblasts. We recently found that coculture with MEFs supports the proliferation of hepatic cells derived from early fetal livers [26]. In this study, the colony formation of mid-fetal (E13.5) hepatoblasts was analyzed in coculture with MEFs (Supplementary Fig. S1A). Fetal hepatoblasts, CD45−Ter119−c-Kit−Dlk+CD133+ cells, cultured on collagen-coated dishes mainly differentiated into CK19-positive cholangiocytic cells (Fig. 1A). In contrast, when cocultured with MEFs, most hepatoblasts differentiated into albumin-positive hepatocytic cells and some cells in the marginal region of colonies differentiated into cholangiocytic cells. In addition, the efficiency of colony formation increased by coculture with MEFs (Supplementary Fig. S1B, C). When hepatoblasts (150 cells/well) were cocultured with MEFs, we observed more than 80 colonies. These results suggest that the coculture system with mesenchymal cells is suitable for expansion of fetal hepatoblasts. Next, we attempted long-term expansion of fetal hepatoblasts. E13.5 fetal liver-derived hepatoblasts were cocultured with MEFs. After 6–7 days of coculture (the primary culture), the cells were dissociated with trypsin and plated onto new MEF feeder cells (Supplementary Fig. S1A). Almost no colony formation was observed in the secondary culture (Fig. 1B, Mock), indicating that hepatoblasts expanded in the primary culture could not undergo long-term proliferation in this culture system. Using several signal molecule-specific inhibitors (a ROCK inhibitor [Y-27632], GSK-3β inhibitor [CHIR99021], MEK inhibitor [PD0325901], and ALK5 kinase inhibitor [A83-01]), we analyzed the molecular mechanism regulating the proliferation of hepatoblasts. We added these inhibitors in the primary culture and analyzed colony formation. When the hepatoblasts were cultured on collagen-coated dishes, the MEK-ERK pathway was strictly required for cell expansion (Supplementary Fig. S1B, PD). In contrast, the MEK-ERK signaling inhibitors did not significantly inhibit the colony formation of hepatoblasts in the coculture system with MEFs (Fig. 1C and Supplementary Fig. S1C). Hepatoblasts expanded in the presence of Y-27632, CHIR99021, or A83-01, similar to control cells, barely formed colonies in the secondary culture. In contrast, after hepatoblasts were expanded with PD0325901 in the primary culture, these cells could proliferate in the secondary culture (Fig. 1B and Supplementary Fig. S2A). This result suggests that inhibition of the MEK-ERK pathway during expansion is important to maintain the proliferative activity of hepatoblasts. We analyzed phosphorylation of ERK in hepatoblast cultures in the presence or absence of PD0325901. Treatment of hepatoblasts with 1 μM PD0325901 completely suppressed phosphorylation of ERK1/2 stimulated by the addition of HGF and EGF. In contrast, moderate phosphorylation of ERK1/2 was detected in hepatoblasts treated with 0.25 μM PD0325901 (Supplementary Fig. S2B).

Inhibition of the MEK-ERK pathway is required to maintain hepatic progenitor activity in vitro.
HGF and EGF are known to be important factors for expansion of hepatoblasts. In addition, we recently reported that insulin-like growth factor (IGF) 1, 2, and TWEAK are involved in proliferation of hepatoblasts derived from pluripotent stem cells [27]. To evaluate the effect of MEK inhibitor on the feeder MEFs, we analyzed whether expression of these soluble factors was changed by the addition of MEK inhibitor. Expression of Egf was not detected in MEF [27]. Expression of Hgf, Igf1, Igf2, and Tweak in MEF was not changed in the presence of PD0325901 (Supplementary Fig. S3). It is suggested that these cytokines had activated MEF-ERK signaling in fetal hepatoblasts in our culture system and MEK inhibitor barely changed expression of soluble factors related with hepatoblast proliferation derived from feeder cells.
When the cells were cultured for 6 days, the total number of hepatoblasts in the primary culture did not change in the presence or absence of 0.25 μM PD0325901. In contrast, the cell number decreased in the presence of 1 μM PD0325901 (Fig. 1D). In the secondary culture, hepatoblast proliferation required inhibition of the MEK-ERK signaling pathway in the primary expansion step. When the cells were cultured in the absence of PD0325901, the number of hepatoblasts did not increase in the secondary culture. In contrast, when hepatoblasts were expanded with 0.25 or 1 μM PD0325901 in the primary culture, these cells could proliferate in the secondary culture. Interestingly, the number of hepatoblasts in cultures treated with 0.25 μM PD0325901 was higher than that in cultures treated with 1 μM PD0325901. Because moderate activation of MEK was detected in hepatoblasts treated with 0.25 μM PD0325901, a low level of MEK-ERK activity might be important for an unknown proliferative pathway in fetal hepatoblasts. In addition, we analyzed whether conditioned medium is required for expansion of fetal hepatoblasts in this culture. When hepatoblasts were cultured in conventional H-CFU-C culture, we added conditioned medium derived from E14.5 fetal liver culture to our standard culture medium [22]. However, the conditioned medium was not required for short-term and long-term expansion of hepatoblasts in the coculture system (Fig. 1D). Therefore, we did not use the condition medium in the following experiments.
Next, we examined expression of cell surface markers after in vitro expansion. Dlk and CD133 were hepatoblast cell surface markers derived from fetal livers [14]. However, it is reported that expression of Dlk decreased during in vitro culture [9]. Similar to the previous study, expression of Dlk decreased after expansion in the primary culture (Supplementary Fig. S4A). CD133 was also downregulated after expansion in the primary culture without MEK inhibitor. In contrast, expression of CD133 was maintained in expanded cells cultured with MEK inhibitor, suggesting that the addition of MEK inhibitor contributed to maintain expression of hepatic progenitor marker, CD133. In addition, these CD133-positive expanded cells could form albumin+ and CK19+ colonies in the secondary culture (Supplementary Fig. S4B).
Proliferation of hepatoblasts treated with MEK inhibitor in vitro
The proliferative ability of hepatoblasts was analyzed by immunostaining for Ki67. Hepatoblasts were cultured on MEFs in the presence or absence of 0.25 μM PD0325901 for 10 days. As shown in Fig. 2, colonies in the control culture had few Ki67-positive cells. In contrast, colonies treated with PD0325901 had many Ki67-positive cells in the marginal zone. In addition, we analyzed expression of proliferation-related genes in this culture system (Supplementary Fig. S5). The well-known oncogene, c-Myc, was not changed by the addition of MEK inhibitor. However, expression of Pcna and Ki67 was significantly upregulated by the addition of MEK inhibitor. These data show that inhibition of the MEK-ERK pathway is important for long-term proliferation of hepatoblasts in vitro.
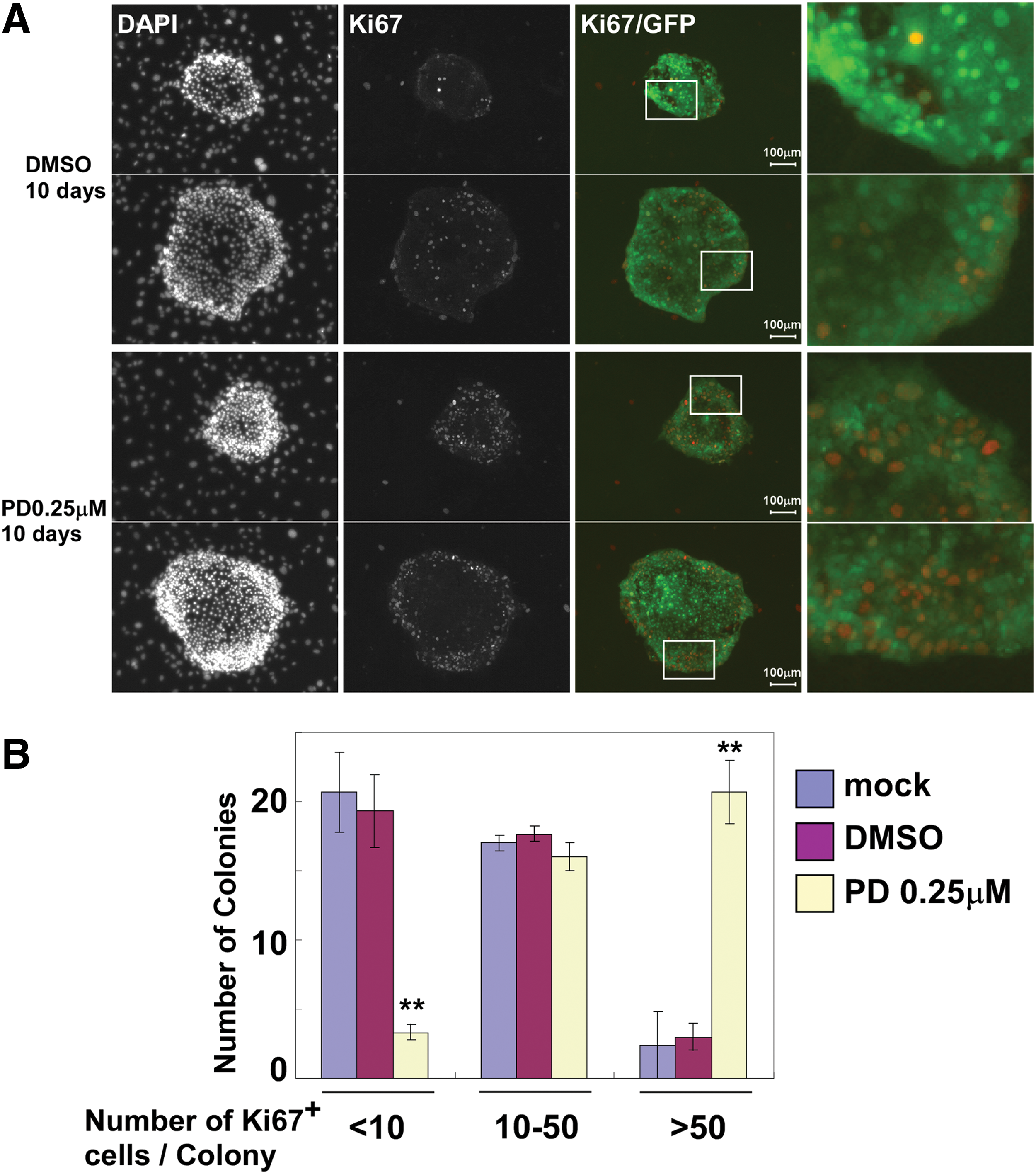
Long-term proliferation of fetal hepatoblasts is suppressed by activation of MEK. E13.5 CD45−Ter119−c-Kit−Dlk+CD133+ cells (derived from GFP-Tg mice) were cocultured with MEFs in the presence of 0.25 μM PD032501. After 10 days of coculture, the cells were fixed with 4% paraformaldehyde, and then immunostained with an anti-Ki67 antibody.
Regulation of cell cycle molecules in hepatoblasts by the MEK-ERK signaling pathway
The proliferation of somatic stem/progenitor cells is regulated by cell cycle molecules, cyclins, and cdk inhibitors. Bmi1 and Rign1B, polycomb component genes, regulate the proliferation of several types of somatic stem cells (eg, hematopoietic stem cells, neural stem cells, and hepatic stem cells) through inhibition of cdk inhibitors [17,28,29]. E13.5 hepatoblasts were purified and cocultured with MEFs for 3 or 6 days, and then the expression of cell cycle molecules was analyzed by real-time quantitative PCR (Fig. 3). Primary adult hepatocytes express few cyclins because mature hepatocytes in normal adult livers barely proliferate in vivo. In contrast, primary hepatoblasts purified from fetal livers express several cyclins (cyclin B1, B2, D1, and E1). Hepatoblasts cocultured with MEFs also expressed these cyclins. Expression of cyclin D2 in hepatoblasts cultured without PD0325901 was much higher than that in primary hepatoblasts (Fig. 3A). Induction of cyclin D2 expression was suppressed by the addition of PD0325901, suggesting that abnormal induction of cyclin D2 expression might be involved in the inhibition of hepatoblast proliferation in vitro.
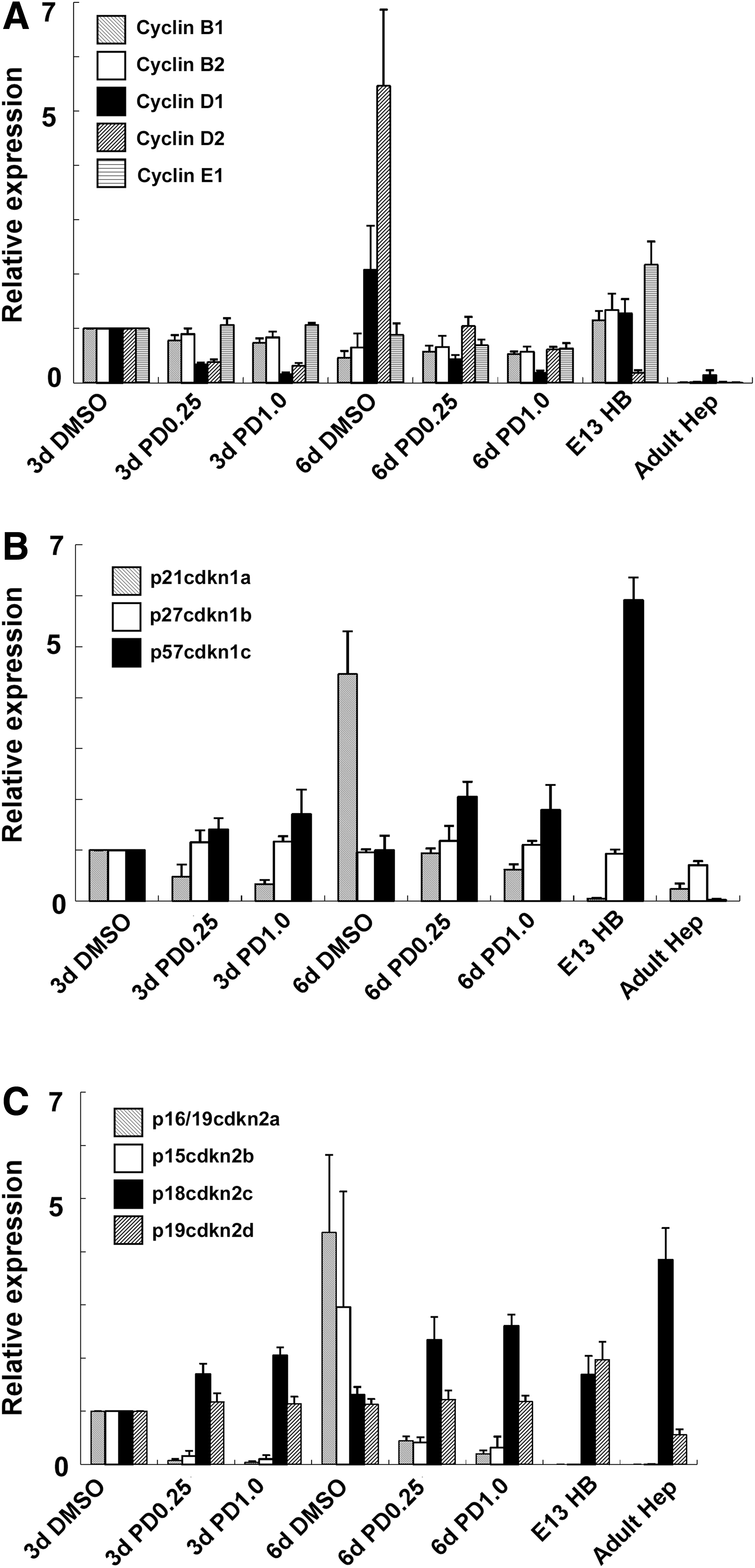
Expression of cell cycle-regulating molecules in expanding fetal hepatoblasts. E13.5 CD45−Ter119−c-Kit−Dlk+CD133+ cells (derived from GFP-Tg mice) were cocultured with MEFs in the presence of 0.25 or 1 μM PD032501. After 3 or 6 days of coculture, the cells were dissociated with trypsin and GFP+ cells were isolated by fluorescence-activated cell sorting. Expression of cell cycle-regulating molecules was analyzed by reverse transcription–polymerase chain reaction. Expression of genes in 3 days culture of hepatoblasts with DMSO (3d DMSO) was set to 1.0. Results are represented as the mean expression ± SD of samples from three independent experiments. DMSO, 0.1% DMSO added as a negative control; E13HB, fresh primary fetal hepatoblasts (E13.5 CD45−Ter119−c-Kit−Dlk+CD133+ cells); Adult Hep, fresh primary adult hepatocytes.
Primary hepatoblasts expressed p27cdkn1b, p57cdkn1c, p18cdkn2c, and p19cdkn2d. In particular, expression of p21cdkn1a, p16/19cdkn2a, and p15cdkn2b was hardly detected in both primary hepatoblasts and adult hepatocytes. In contrast, expression of p21cdkn1a, p16/19cdkn2a, and p15cdkn2b increased in hepatoblasts cultured without PD0325901 (Fig. 3B, C). Expression of these cyclin inhibitors was suppressed by the addition of PD0325901. Thus, we assumed that activation of the MEK-ERK pathway inhibited long-term proliferation of hepatoblasts in vitro through upregulation of p21cdkn1a, p16/19cdkn2a, and p15cdkn2b. We could not detect activation of senescence-activated β-galactosidase or apoptotic signals of TUNEL assays in hepatoblast colonies cultured in the presence or absence of PD0325901 (Supplementary Fig. S6).
Proliferation of fetal hepatoblasts in colony formation assays can be affected by several factors such as cell attachment and cell–cell communication. Therefore, we analyzed the long-term proliferative ability of these cells in normal cell density culture. E13.5 fetal hepatoblasts were sorted and seeded in collagen type I-coated 96-well culture plates at a normal cell density (4,000 cells/well). After 6 days of the primary culture, the hepatoblasts formed an epithelial-like monolayer (Supplementary Fig. S7A). In contrast, after passaging for the secondary culture, the cells could not expand efficiently (Supplementary Fig. S7B). In addition, we also found induction of cdkn2a (p16ink4a and p19ARF) expression in the secondary culture (Supplementary Fig. S7C).
Long-term proliferation of hepatoblasts is induced by gene inactivation of p16/19cdkn2a without the addition of an MEK inhibitor
We analyzed the effect of gene inactivation of p16/19cdkn2a on fetal hepatoblast culture. E13.5 CD45−Ter119−c-Kit−Dlk+CD133+ cells were isolated from p16/19cdkn2a+/+, +/−, and −/− mice, and then cultured on MEF feeder cells. We found that the proportion of hepatoblasts, CD45−Ter119−c-Kit−Dlk+CD133+ cells, in total liver cells was similar in E13.5 wild-type and p16/19cdkn2a−/− mouse livers (Supplementary Fig. S8). In the primary culture, hepatoblasts from both p16/19cdkn2a+/− and −/− mice formed many colonies expressing albumin and CK19 (Fig. 4A). In particular, p16/19cdkn2a−/− hepatoblasts cultured without PD0325901 formed many large colonies (Fig. 4C). After p16/19cdkn2a+/+ and +/− hepatoblasts were expanded without PD0325901, these cells formed few small colonies in subcultures (Fig. 4B, D). In contrast, the defect of p16/19cdkn2a significantly induced secondary colony formation of hepatoblasts expanded without PD0325901 (Fig. 4D). These results suggest that p16/19cdkn2a expression induced by the MEK-ERK signaling pathway leads to cell cycle arrest of hepatoblasts in long-term culture. Interestingly, suppression of MEK-ERK signaling was required for long-term proliferation of p16/p19cdkn2a WT hepatoblasts, whereas the proliferation rate in cultures of p16/p19cdkn2a KO hepatoblasts treated with PD0325901 was significantly lower than that in cultures without PD0325901 (Fig. 4C, D). These findings indicate that MEK-ERK signaling is involved in both proliferation and cell cycle arrest of fetal hepatoblasts.

Induction of the proliferation of fetal hepatoblasts derived from p16/19cdkn2a knockout (KO) mice.
The cdkn2a gene encodes two cell cycle regulators, p16ink4a and p19ARF. Therefore, we analyzed which regulator induced the cell cycle arrest of hepatoblasts in culture. E13.5 CD45−Ter119−c-Kit−Dlk+CD133+ cells were isolated from p16/19cdkn2a+/− and −/− mice, and then seeded on MEF feeder cells. After infection with mock, p16ink4a, or p19ARF expression retroviruses, the cells were cultured for 6 days. As shown in Fig. 4E, overexpression of either p16ink4a or p19ARF suppressed colony formation of p16/19cdkn2a−/− hepatoblasts, suggesting that expression of either p16ink4a or p19ARF is sufficient for cell cycle arrest of fetal hepatoblasts.
Next, we analyzed the expression of cell cycle regulators in cultured hepatoblasts derived from p16/19cdkn2a KO mice (Supplementary Fig. S9). Cyclin D1 and D2 expression in hepatoblasts was highly suppressed by PD0325901 treatment, whereas the expression of these genes was not completely suppressed by inactivation of p16/19cdkn2a. Expression of p21cdkn1a, the downstream target gene of p16/19cdkn2a, was suppressed in p16/19cdkn2a−/− hepatoblasts, even without PD0325901. Furthermore, PD0325901 highly suppressed the expression of p15cdkn2b that was expressed even in p16/19cdkn2a KO hepatoblasts. These results suggest that suppression of p16/19cdkn2a and p21cdkn1a, but not p15cdkn2b, is important for long-term expansion of fetal hepatoblasts.
Activation of the MEK-ERK signaling pathway is inhibited in primary fetal hepatoblasts
Activation of the MEK-ERK pathway induced expression of p16/19cdkn2a and cell cycle arrest of hepatoblasts in vitro. Therefore, we analyzed whether hepatoblasts express phospho-ERK during in vivo liver development. Primary hepatoblasts derived from E13.5 livers were sorted onto glass slides for analysis. ERK phosphorylation in individual cells was detected by the in-droplet assay [25]. Positive control cells stimulated with HGF and EGF for 5 min expressed highly phosphorylated ERKs (Fig. 5). In contrast, the phosphorylation level of ERK in primary hepatoblasts was similar to that in negative control (cells incubated with an MEK inhibitor), suggesting that ERK in hepatoblasts is not highly activated in the liver during fetal development.

Phosphorylation of ERK in primary hepatoblasts. E13.5 CD45−Ter119−c-Kit−Dlk+CD133+ cells were directly sorted onto glass slides and fixed. Some cells were incubated for 10 min with hepatocyte growth factor (HGF) and epidermal growth factor (EGF) or with PD0325901 (+HGF+EGF or +PD). Cells were immunostained with an anti-phospho ERK antibody. Data of three samples are shown.
In vivo transplantation of hepatoblasts expanded with the MEK inhibitor
Primary hepatoblasts can expand and differentiate into functional hepatic cells in injured livers upon transplantation into preconditioned recipient mice. In this study, we checked whether in vitro expanded hepatoblasts could engraft and proliferate in the injured livers. After a limited number of hepatoblasts (3–6 × 103 cells per recipient mouse) were expanded in vitro, we transplanted these cells into injured livers. E13.5 CD45−Ter119−c-Kit−Dlk+CD133+ cells were purified and cultured on MEF feeder cells with or without PD0325901 for 6 days. A total of 3–6 × 103 hepatoblasts were expanded to almost 1–2 × 105 cells after in vitro culture. These expanded cells were transplanted into retrorsine-treated and partially hepatectomized livers. When CD45−Ter119−c-Kit−Dlk+CD133+ cells were expanded in vitro with PD0325901, proliferation of GFP-positive donor cells was detected in the recipient livers after transplantation (Supplementary Fig. S10A). We transplanted into eight mouse livers and detected in vivo expansion of hepatoblasts in six mouse livers. In addition, macroscopic observation revealed that no tumor nodule-like structures were seen in the recipient KSN nude mouse livers. GFP-positive donor cells differentiated into albumin-positive hepatocytic cells in recipient livers (Supplementary Fig. S10B). In addition, some donor cells differentiated into CK19-positive cholangiocytic cells (Supplementary Fig. S10C). In contrast, when CD45−Ter119−c-Kit−Dlk+CD133+ cells were expanded in vitro without PD0325901, we detected a limited proliferation of donor cells in the recipient livers after transplantation (Supplementary Fig. S10A). These results suggest that hepatoblasts maintain their progenitor potency (the ability of high proliferation and bipotent differentiation) after in vitro expansion with MEK inhibitor.
Discussion
In this study, we found that activation of the MEK-ERK signaling pathway induced cell cycle arrest of hepatoblasts derived from fetal livers by accumulation of p16/19cdkn2a. Inhibition of MEK activity by addition of PD0325901 during expansion maintained the proliferative ability of hepatoblasts cocultured with MEFs. This result suggests that a high level of MEK-ERK signal activation is not required for hepatoblast proliferation under the coculture condition. In contrast, when hepatoblasts were cultured alone, these cells barely proliferated in the presence of PD0325901. These results show that the mechanism regulating cell survival and proliferation is different under feeder cell-dependent and -independent culture conditions. The interaction with MEFs might upregulate MEK-ERK-independent cell survival and proliferation of hepatoblasts. We found that inhibition of the PI3K pathway by LY294002 significantly suppressed colony formation of hepatoblasts cocultured with MEFs (data not shown). The PI3K signaling pathway is known to be involved in the survival and proliferation of several cell types. Therefore, PI3K is one of the candidate signaling molecules regulating hepatoblast survival in our coculture system. In addition, MEFs expressed several soluble factors related with hepatoblast proliferation [27]. In this study, HGF, IGF2, and TWEAK were expressed in MEFs and these expressions were not changed by the stimulation of MEK inhibitor. However, another effect of MEK inhibitor on MEFs in this culture system might be involved in hepatoblast proliferation and it would be a fascinating area of future research.
When hepatoblasts were expanded in vitro, we found significant upregulation of several cdk inhibitors, such as p21cdkn1a, p15cdkn2b, and p16/p19cdkn2a, which were involved in cell cycle arrest of hepatoblasts in long-term culture. When a small amount of PD0325901 (0.25 μM) was added to cocultures, we detected slight phosphorylation of ERK. However, we found significant suppression of the induction of cdk inhibitors with 0.25 μM PD0325901, suggesting that a high level of MEK-ERK signal activation was required for induction of these cdk inhibitors. Using p16/19cdkn2a KO mice, we found that induction of p16/p19cdkn2a, but not p15cdkn2b, regulated the cell cycle of hepatoblasts in vitro. Interestingly, p21cdkn1a expression in p16/p19cdkn2a KO hepatoblasts was significantly decreased, suggesting that induction of p21cdkn1a in cultured hepatoblasts is directly regulated by p16/p19cdkn2a. PD0325901 was required for long-term proliferation of p16/p19cdkn2a+/+ hepatoblasts. In contrast, the proliferative capacity of p16/p19cdkn2a KO hepatoblasts was significantly suppressed by PD0325901. These results suggest that the MEK/ERK signaling pathway has two distinct functions: (1) proliferation induction through an unknown mechanism and (2) an antiproliferative function through induction of p16/p19cdkn2a. It is previously described that the MEK-ERK pathway is important for the progression of G0–G1 cell cycle progression of hepatocytes [20,21]. In contrast, we found that the MEK-ERK pathway inhibited the cell cycle progression by the increase of cdk inhibitors in this study. Primary colonies derived from p16/p19cdkn2a+/− and p16/p19cdkn2a−/− hepatoblasts expressed both albumin and CK19, indicating that the p16/19cdkn2a KO hepatoblasts also have a bipotent differentiation ability. However, albumin production was downregulated and the number of CK19+ cells was increased in some colonies derived from p16/19cdkn2a KO hepatoblasts cultured without PD0325901 (Fig. 4B, p16/19−/−, dimethyl sulfoxide [DMSO]) in the secondary culture. p16/19 KO hepatoblasts showed a high proliferation rate in colony culture without the MEK inhibitor. Thus, an excessive proliferation rate might affect hepatocytic differentiation of hepatoblasts in long-term culture.
Activation of the MEK/ERK pathway in vitro induced cell cycle arrest of hepatoblasts by upregulation of cdk inhibitors. An increase of either p16ink4a or p19ARF was sufficient to inhibit hepatoblast proliferation. Therefore, the expressional control of cdk inhibitors, in particular p16ink4a or p19ARF, may be very important for fetal liver expansion during development. Hepatoblasts derived from mid-fetal livers hardly expressed p16/p19cdkn2a. A high level of MEK/ERK signal activation was not detected in hepatoblasts derived from mid-fetal livers. In addition, transplanted fetal hepatoblasts expanded efficiently for a long period in the recipient liver. These data suggest the existence of a molecular mechanism that suppresses MEK/ERK activation and cdk inhibitor expression in the in vivo environment.
The liver is the largest organ in mammals. Therefore, regenerative medicine for the treatment of liver injury and several diseases requires a large number of functional hepatic cells. Hepatic stem and progenitor cells show a high proliferative ability and differentiation potential for functional hepatocytes in vivo. Upon liver injury, normal mature hepatocytes also exhibit a proliferative ability in vivo. However, suitable culture conditions for expansion of these hepatic progenitor cells and mature hepatocytes in vitro remained unknown. In this study, we found that coculture with MEF feeder cells in the presence of an MEK inhibitor can expand a small number of hepatoblasts in vitro. We succeeded that 100 hepatoblasts proliferated into almost 1.8 × 104 cells in the primary culture and 3 × 105 cells in the secondary culture. This technique might allow a small number of hepatic progenitor cells to be used for cell transplantation through in vitro expansion. Our findings might be useful for future regenerative therapies of severe liver diseases.
Footnotes
Acknowledgments
Some of analyses were assisted by the Education and Research Support Center of Tokai University. The authors thank Mr. Yuji Yamazaki (Japan Science and Technology Agency, NAKAUCHI Stem Cell and Organ Regeneration Project) for the flow cytometric analyses and Dr. Masataka Kasai (The Institute of Medical Science, The University of Tokyo) for critical reading of the manuscript. The authors also thank Ms. Kinuyo Ida for the experimental assistance.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (23689040, 24249056, 25670373, and 26293178) and Grants-in-Aid for Scientific Research from the Ministry of Health, Labour and Welfare of Japan.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.