
Abstract
Human embryonic stem cells (hESCs) have long been considered as a promising source for cell replacement therapy. However, one major obstacle for the use of these cells is immune compatibility. Histocompatible human parthenogenetic ESCs have been reported as a new method for generating human leukocyte antigen (HLA)-matched hESCs. To further investigate the possibility of obtaining histocompatible stem cells from uniparental embryos, we tried to produce androgenetic haploid human embryos by injecting a single spermatozoon into enucleated human oocyte, and establish human androgenetic embryonic stem (hAGES) cell lines from androgenetic embryos. In the present study, a diploid hAGES cell line has been established, which exhibits typical features of human ESCs, including the expression of pluripotency markers, having differentiation potential in vitro and in vivo, and stable propagation in an undifferentiated state (>P40). Bisulfite sequencing of the H19, Snrpn, Meg3, and Kv imprinting control regions suggested that hAGES cells maintained to a certain extent a sperm methylation pattern. Genome-wide single nucleotide polymorphism, short tandem repeat, and HLA analyses revealed that the hAGES cell genome was highly homozygous. These results suggest that hAGES cells from spermatozoon could serve as a useful tool for studying the mechanisms underlying genomic imprinting in humans. It might also be used as a potential resource for cell replacement therapy as parthenogenetic stem cells.
Introduction
N
Human ESCs (hESCs) are considered to be the most promising source of differentiated cell types for regenerative medicine [14]. However, one major obstacle for the application of hESCs in regenerative medicine is immune compatibility [15]. Immunocompatible pluripotent stem cells can be obtained by somatic cell nuclear transfer [16], the induction of pluripotent stem cells [17], or the establishment of a hESC bank that contains diverse human leukocyte antigen (HLA) genotypes [18]. Histocompatible human parthenogenetic ESCs (pESCs) have been obtained in 2007 [19], which represent a new method for generating HLA-matched hESCs. Homozygous parthenogenetic hESCs from uniparental embryos could benefit a larger population [20]. For example, 55 randomly selected homozygous pESC lines could completely match 80% of the Japanese patients at three important HLA loci (A, B, and DR) [21]. Moreover, human pESCs could provide β cells, fibroblasts, cardiomyocytes, and neurons in vitro [20,22], suggesting that it might be possible to use these cells as an optimal source of genetically matched hESCs for cell therapy in female patients.
However, it is unclear whether human androgenetic ESCs can also be isolated from androgenetic embryos and utilized for cell therapy. In this study, we established diploid human homozygous androgenetic ESCs from human androgenetic haploid embryos. To the best of our knowledge, this is the first article reporting the derivation of homozygous human androgenetic ESCs.
Materials and Methods
Informed consent for egg and sperm donation
This study was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University (Approval Reference No. 2013-49, Issue date: February 28, 2013). Written informed consent was obtained from each infertile couple before donating oocytes for research. Oocytes were donated from couples with no sperm available for intracytoplasmic sperm injection (ICSI) from September 2009 to June 2013 at the Reproductive Medical Center of the First Affiliated Hospital of Sun Yat-sen University. Healthy sperm donors were also recruited. Written informed consent was obtained from each donor before donating oocytes or sperm for researches.
Superovulation and oocyte collection
Ovulation stimulation protocols followed established clinical IVF guidelines, and all patients received a long protocol using a GnRH agonist and GONAL-f (Merck-Serono) for stimulation. Oocyte retrieval was performed 34–36 h after the administration of 10,000 IU human chorionic gonadotrophin. Cumulus-oocyte complexes (COCs) were collected from aspirates and placed in G-MOPS medium (Vitrolife) supplemented with 5% human serum albumin (HSA) (Vitrolife) for 4–6 h at 37°C in a humidified atmosphere of 6% CO2, 5% O2, and 89% N2. COCs were treated with hyaluronidase to disaggregate cumulus and granulosa cells. Mature oocytes [metaphase II (MII)] were selected for further studies.
Derivation and growth of androgenetic embryos
Mature oocytes were placed into separate 10 μL manipulation droplets of G-MOPS with 5% HSA and covered with tissue culture oil. After the first polar body of the oocytes reached 12 o'clock, partial zona pellucida dissection was performed before enucleation (Supplementary Fig. S1; Supplementary Data are available online at

Derivation of human androgenetic embryos.
Fluorescence in situ hybridization analysis of haploid androgenetic embryos
Fluorescence in situ hybridization (FISH) analysis of chromosomes 18, X and Y was performed in haploid androgenetic embryos at day 6, including both morula and blastocyst stage embryos. Haploid androgenetic embryos were dissolved in a lysis buffer (0.01 N HCl, 0.1% Tween 20) to remove the zona pellucida and the cytoplasm. Nuclei were dispersed and fixed in methanol:acetic acid (3:1). Fixed nuclei were air dried and stored at −20°C before FISH analysis. FISH analysis was performed by sequential hybridization using centromere probes for chromosomes 18 (05J09-018; Vysis), X and Y (05J10-051; Vysis). Slides were examined and recorded using an Olympus fluorescence-equipped inverted microscope and LUCIA FISH software (LUCIA Cytogenetics; Laboratory Imaging, S.R.O.).
Human androgenetic ESC isolation and culture
Human stem cells were isolated and cultured as described previously [10], with slight modification. After removal of the zona pellucida using 0.5% protease (Sigma), the inner cell mass (ICM) was mechanically isolated with a needle. The isolated ICMs were plated on mitomycin-C-irradiated mouse embryonic fibroblast (MEF) cells for 5–9 days. Then, primary ESC colonies were dissociated mechanically and cultured in the human ES cell culture medium consisting of 80% knockout Dulbecco's modified Eagle's medium (DMEM; Gibco), 20% serum replacement (Gibco), 1 mM glutamine (Sigma), 1% nonessential amino acid (Gibco), 0.1 mM 2-mercaptoethanol (Sigma), 50 UI/mL penicillin (Sigma), and 50 UI/mL streptomycin (Sigma). Primary colonies were cultured for 5–9 days, and the ESCs were selected and replated on new feeder cells. When the ESCs appeared to proliferate stably, ES colonies were dissociated every 4–5 days by mechanical or digestive methods. All ESCs were cultured at 37°C with 5% CO2 in a humidified atmosphere.
Characterization of human androgenetic ES cell lines
Immunofluorescence staining was carried out as previously described [10]. Human AGES cells were plated on glass coverslips coated with MEF cells and cultured for 3–4 days. ES cell clones were fixed with 4% paraformaldehyde (PFA) at room temperature (RT) and permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 20 min, blocked with 10% goat serum in PBS (Sigma) for 1 h, and incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: OCT4 (octamer-binding transcription factor 4; Abcam), SOX2 (sex determining region Y-box 2; Abcam), SSEA-3 and SSEA-4 (stage-specific embryonic antigen 3, 4) (Developmental Studies Hybridoma Bank at the University of Iowa), TRA-1-60 (tumor resistance antigen 1-60; Chemicon), and TRA-1-81 (tumor resistance antigen 1-81; Chemicon). Next, the cells were incubated for 1 h at RT with a fluorescent-conjugated secondary antibody, and ESCs nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma). Specimens were observed using a Zeiss LSM 510 Meta Confocal Microscope (Carl Zeiss Microscopy GmbH).
Alkaline phosphatase (AP) staining was performed using the BCIP/NBT (5-Bromo-4-Chloro-3-Indolyl Phosphate/nitro blue tetrazolium) Alkaline Phosphatase Color Development Kit (Beyotime) according to the manufacturer's instructions.
RNA extraction and quantitative real-time polymerase chain reaction
RNA was isolated using the TRIzol Reagent (Invitrogen), and 2 μg of total RNA was used in the reverse transcription reaction using the Moloney murine leukemia virus reverse transcriptase (M-MLV; Promega) according to the manufacturer's protocol. The expression of imprinting genes (H19, Ube3a, Snrpn, and Igf2) and pluripotent genes (Oct4, Nanog, Sox2, and Rex1) was detected by real-time polymerase chain reaction (RT-PCR) using the SYBR-Green PCR Master Mix (Applied Biosystems) and normalized to Gapdh. The RT-PCR primers are listed in Supplementary Table S1.
Karyotype analysis and flow analysis
Karyotyping analysis was carried out as previously described [10]. Human AGES colonies were incubated with 0.2 μg/mL colchicine (Invitrogen) for 2 h at 37°C. Cells were collected and trypsinized, washed with PBS, and incubated with 0.075 M potassium chloride for 10 min at 37°C. Cells were fixed with methanol:glacial acetic acid (1:3) three times and dropped onto glass slides. Chromosome spreads were Giemsa banded and photographed. Karyotypes were assessed by the normal G-banding procedure, and 50 MII spreads were examined for each sample; a normal karyotype displayed normal chromosome numbers and G-banding patterns.
Human AGES cells were fixed in cold ethanol overnight (−20°C). Fixed cells were centrifuged, and the pellet was resuspended in PBS. Human AGES cells were treated with RNase (37°C, 30 min) and stained with propidium iodide (50 μg/mL) at RT for 10 min. The samples were analyzed on a Cyan analyzer (Beckman Coulter), and H9 human ESCs served as a control.
Assessment of the differentiation capacity in vitro and in vivo
The embryoid body (EB) and teratoma method were used to assess the differentiation ability of hAGES cells in vitro and in vivo, as previously described [10].
Human AGES cells were removed from culture dishes using 1 mg/mL collagenase IV and cultured in suspension. Spontaneous EBs were grown in a medium consisting of 80% knockout DMEM, supplemented with 20% serum replacement, 0.1 mM 2-mercaptoethanol, and 1% nonessential amino acids. After 7–14 days, the differentiated EB samples were collected, and RT-PCR was performed to analyze the expression of marker genes for the three embryonic germ layers: alpha-fetoglobulin (Afp, mesoderm), Gata4 and Acta2 (endoderm), and Nestin and Tubb3 (ectoderm). Alternatively, EBs were transferred to a gelatin-coated plate and cultured for 7–14 days. Then, differentiated EBs were immunostained with alpha-fetoglobulin (AFP; Millipore), fibronectin (FN; Millipore), and glial fibrillary acidic protein (GFAP; Millipore).
Teratoma production was used to assess the differentiation ability of hAGES cells in vivo. Approximately 5–6 million undifferentiated hAGES cells from passages 17–19 were harvested and injected into the groin of 4-week-old male SCID-mice. After 2 months, the mice were sacrificed. Various tissues from teratoma were dissected, fixed in Bouin's solution overnight, processed, sectioned according to standard procedures, and counterstained with Hematoxylin and eosin. Sections were examined using bright field light microscopy and photographed.
Bisulfite sequencing for DNA methylation analysis
Bisulfite sequencing was performed to confirm the DNA methylation state of the hAGES cells. Sodium bisulfite treatment of genomic DNA was performed using the EZ DNA Methylation-Direct Kit (Zymo Research). PCR amplification was performed using the PCR amplification with TAKARA HS DNA polymerase (TAKARA, JAN) and specific primers for H19, Snrpn, Meg3, and Kv (KCNQ1OT1) differentially methylated region (DMR). The PCR products were gel extracted, subcloned into pMD18T vector (TAKARA, JAN), and sequenced. Methylation was analyzed using the web-based tool “QUMA” (
Short tandem repeat typing
Genomic DNA samples were extracted from hAGES cells as well as the peripheral blood of sperm and oocyte donors using the phenol–chloroform method, as described previously [10]. Sixteen short tandem repeat (STR) loci were coamplified in one PCR reaction. Alleles of STR loci were distinguished by fluorescence detection after electrophoretic separation. The STR typing process was performed according to the manufacturer's recommendations using the PowerPlex 16 system (Promega). An ABI PRISM 310 Genetic Analyzer was used for fluorescence detection of amplified fragments.
Array comparative genomic hybridization analysis of genomic integrity
Human AGES cells (P14–P20) (105 cells) were subjected to whole genome amplification (WGA) using the SurePlex Kit (BlueGnome Ltd.). The amplified products were labeled with different fluorescent dyes (CY3/CY5) and hybridized to 24 Sure V3/24 Sure+ chips (BlueGnome Ltd.) (average 2–5 Mb effective resolution of all 24 chromosomes). After washing, the chips were scanned using the InnoScan 710 scanner (Innopsys), and the data were analyzed using the BlueFuse Multi software (BlueGnome Ltd.). H9 human ESCs were used as a control.
Single nucleotide polymorphism analysis of genomic homozygosity
Human AGES cells (P14–P20) (105 cells) were subjected to WGA using the REPLI-g Midi Kit (Qiagen). WGA products from human AGES cells were used as the input for a single genomic single nucleotide polymorphism (SNP) array (HumanCytoSNP-12; Illumina) consisting of 299,140 SNP markers with a median marker spacing of 10 kb. DNA amplification, tagging, and hybridization were performed according to the manufacturer's protocol. The array slides were scanned on an iScan Reader (Illumina, Inc.), and data analysis was performed using GenomeStudio version 2010.1 and KaryoStudio version 1.2 (standard settings; Illumina). The HapMap control set provided by the manufacturer and H9 human ESCs were used as controls.
The B allele frequency (BAF) of HumanCytoSNP-12 helps to recognize and exclude false positives. The BAF is a value between 0 and 1, and it represents the proportion contributed by one SNP allele (B) to the total copy number. A BAF value of 0.5 indicates a heterozygous genotype (AB), whereas 0 and 1 indicate homozygous genotypes.
HLA typing analysis
To carry out HLA typing analysis, hAGES cells were transferred onto gelatin-plated dishes for further culture. Approximately 1×105 cells were collected for HLA typing. WGA products from human AGES cells as well as lymphocytes from sperm and oocyte donors were purified and amplified using the REPLI-g Midi Kit (Qiagen). HLA typing was performed using the Sanger sequencing-based typing by BGI Tech Solutions Co., Ltd. (BGI Tech).
Results
Generation of human androgenetic haploid embryos
Four patients with male factor infertility (aged 21–32 years) donated their oocytes due to no sperm available for ICSI. Totally 40 mature MII oocytes were retrieved (range of 7–28; mean of 13 oocytes per cycle) to generate haploid human androgenetic embryos. After enucleation using the SpindleView system (Table 1), 37 MII oocytes survived and were microinjected with single spermatozoon. Twenty-eight reconstructed embryos formed single, visible pronuclei (PN) (76.7%±8.8%). Most of the haploid androgenetic embryos developed to the 8-cell stage (22/28, 62.8%±25.6%), and seven developed to the blastocyst stage (19.4%±5.8%) (Fig. 1a). In addition, four morulas (stop further development) were observed 6 days postmanipulation. In total, the efficiency of blastocyst formation for human androgenetic embryos was lower than that of normal ICSI embryos (52.2%±17.1%, Table 1).
The embryos of 33 patients in the ICSI cycle were included as controls.
Values with different superscripts lowercase letters in the same column represent a significant difference (p<0.05).
“Fertilized” refers to pronuclear stage for androgenetic embryos.
ICSI, intracytoplasmic sperm injection.
Next, we performed FISH analysis to explore the ploidy of the human androgenetic embryos, and we observed diploidy (two 18 chromosomes and sex chromosomes in one blastomere), haploidy (single 18 chromosome and single sex chromosome in one blastomere), and aneuploidy (the blastomere contained different numbers of 18 and sex chromosomes) blastomeres. In total, four human androgenetic morulas and four human androgenetic blastocysts were examined. Human androgenetic morulas contained 27.0±13.3 blastomeres, including haploid (21.0±13.3, 77.8%±29.4%), diploid (2.3±3.3, 8.7%±14.6%), and aneuploid (3.8±3.5, 13.5%±14.9%) blastomeres (Supplementary Table S2). A Y chromosome was observed in three human androgenetic morulas, and an X chromosome was found in another human androgenetic morula. Human AG blastocysts contained 87.5±51.7 blastomeres, including haploid (52.5±41.0, 52.0%±34.1%), diploid (26.8±19.9, 35.5%±30.3%), and aneuploid (8.3±4.1, 12.4%±8.4%) blastomeres. (The former figure represented the number of cells with different ploidies, the latter one was the percentage of these cells). Human androgenetic blastocysts only contained the X chromosome (Supplementary Table S3 and Fig. 1b). The percentage of diploid cells in blastocysts was greater than in the morula stage (35.5%±30.3% vs. 8.7%±14.6%). These data indicated that the X chromosome is essential for androgenetic blastocyst development, which was also reported previously in mice [11,23,24].
Derivation of hAGES cell line from human androgenetic blastocysts
Three human androgenetic blastocysts were used to derive ES cell line, and they were plated into four-well plates precoated in feeder cells. One outgrowth was observed 8 days later. After repeated mechanical dissection every 6 or 7 days, one stable cell line was derived (hAGES) (Fig. 2a) and was maintained for more than 40 passages. Karyotype analysis and chromosome G-banding revealed that the hAGES ES cell line exhibited normal diploid karyotypes of 46 XX (Fig. 2b). Array CGH analysis indicated that the hAGES cells contained an intact genome, without observable chromosome deletion or duplication (Fig. 2c). DNA content analysis confirmed that the hAG-1 ESCs were diploid (Fig. 2d).
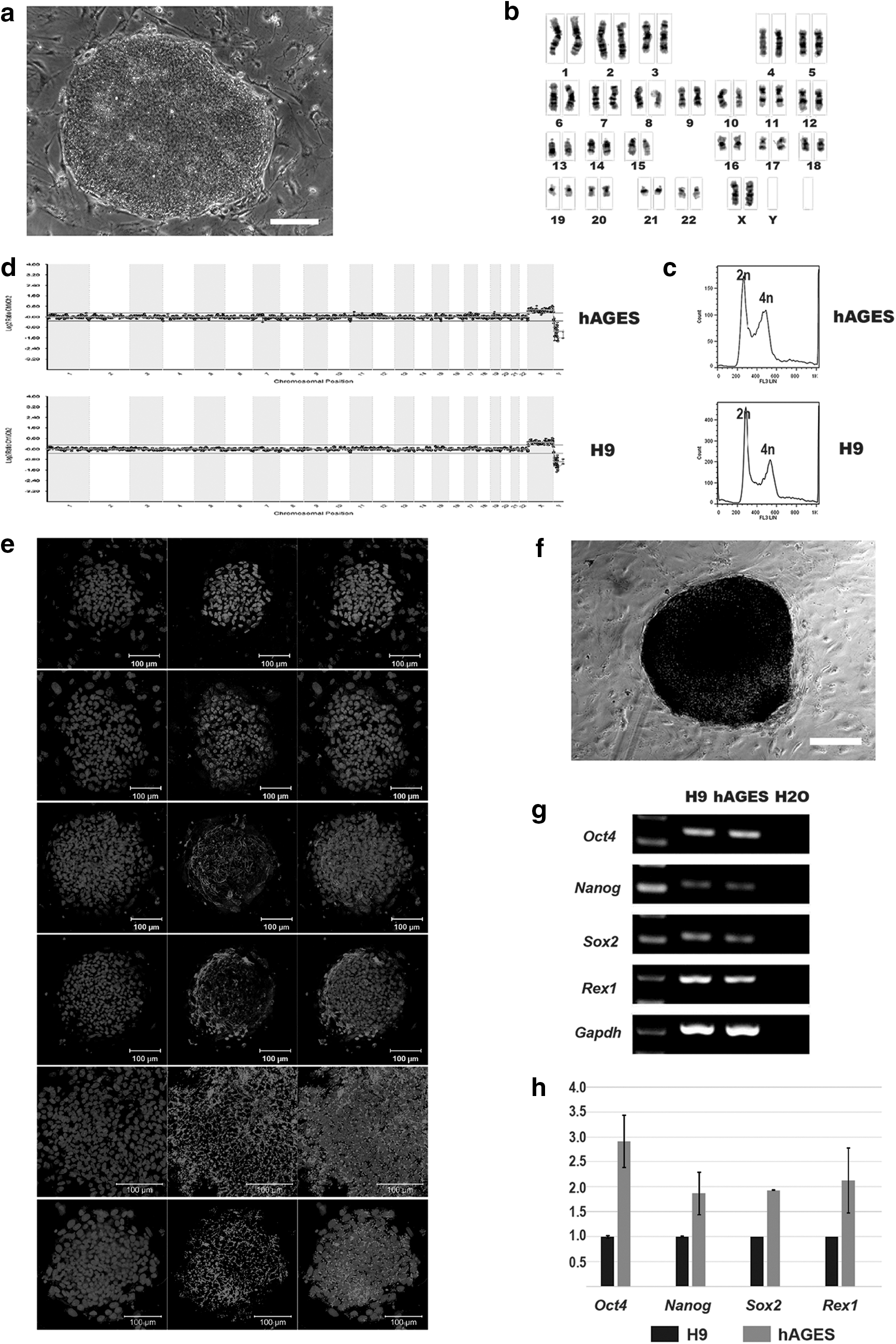
Derivation of human androgenetic embryonic stem cells (ESCs).
The hAGES colonies exhibited typical human pluripotent stem cell morphology (Fig. 2a) and expressed key pluripotent markers, including OCT4, NANOG, SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 (Fig. 2e), and they displayed high levels of AP activity (Fig. 2f). Real-time PCR analysis demonstrated that the hAGES cells expressed key pluripotency genes compared with H9 ESCs, including Oct4, Nanog, Sox2, and Rex1 (Fig. 2g, h).
Pluripotency of hAGES cells
Human AGES cells were capable of differentiating into EBs in vitro when cultured in basic medium (Fig. 3a). RT-PCR analysis revealed that spontaneous differentiated EBs expressed markers of all three germ layers (Fig. 3b). Similarly, immunostaining indicated that differentiated EB cells expressed markers of all three embryonic layers, including AFP (endoderm), FN (mesoderm), and GFAP (ectoderm) (Fig. 3c).
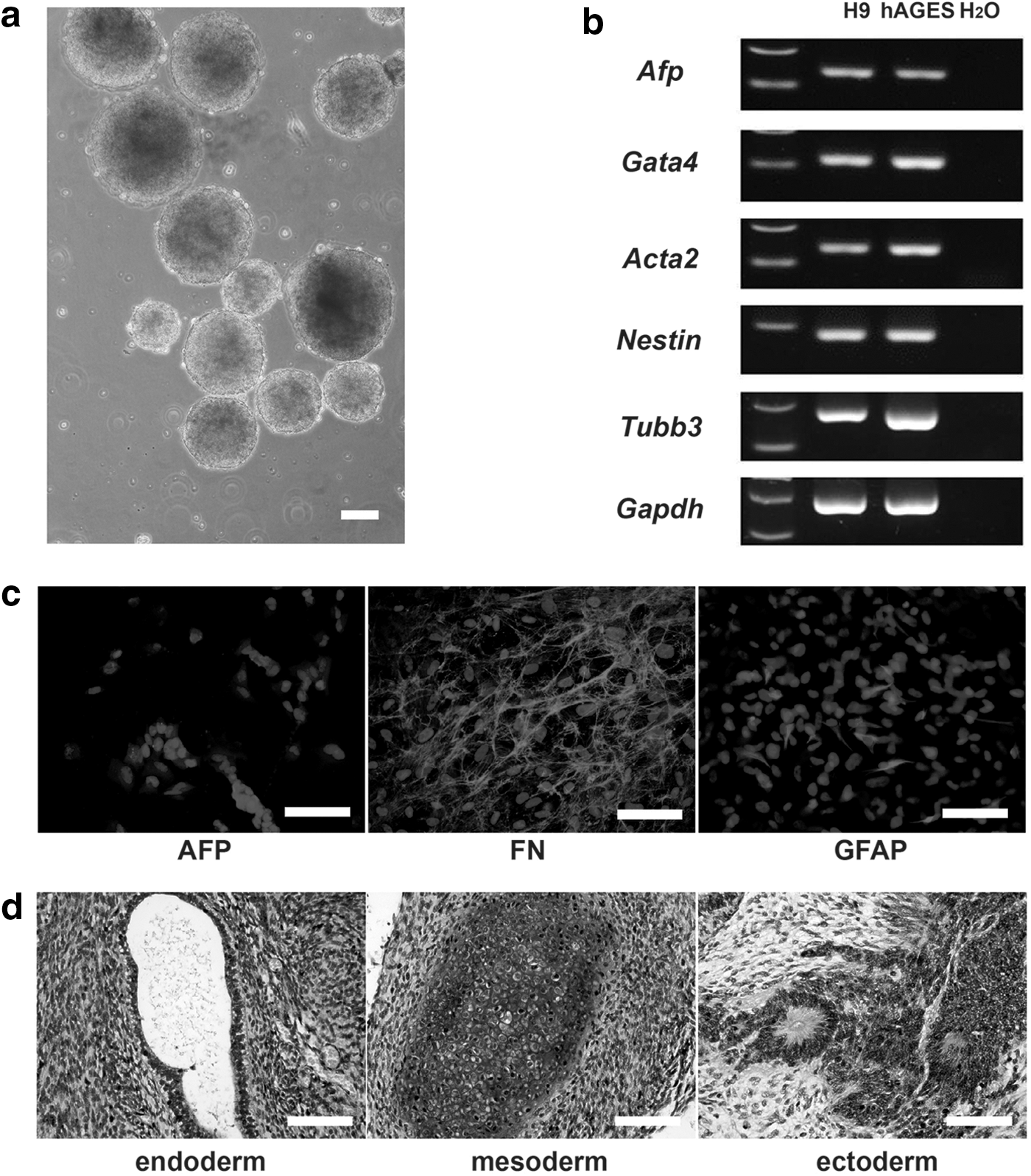
In vitro and in vivo differentiation potential of the hAGES cell line.
Next, we assessed the differentiation ability of hAGES cells in vivo by teratoma formation. One or two million hAGES cells were subcutaneously injected into four SCID mice, and two formed teratomas 2 months later. The H9 ESCs were used as control, and the teratoma formation rate was 83.3% (10/12).
Histological analysis revealed the presence of various tissues, including glandular epithelia (endoderm), cartilage cells (mesoderm), and neural rosettes (ectoderm) in the teratomas (Fig. 3d and Supplementary Fig. S2). These results demonstrated that hAGES cells are pluripotent and capable of differentiating into all three embryonic germ layers in vivo.
The homozygosity of hAGES cells
To confirm the autologous origin, an STR study was performed using hAGES cell line as well as lymphocytes from the sperm and oocyte donors. STR sequencing results from 16 DNA loci indicated that hAGES cells were genetically identical and comparable (more than 99.85%) to the lymphocytes from the sperm donor, but not the oocyte donors (Supplementary Fig. S3 and Supplementary Table S4). In addition, all 15 STR loci analyzed and the Amelogenin genes were homologous in hAGES cells.
HLA typing of the HLA-A, -B, -C, and HLA-DRB1 and -DQB1 loci of hAGES cells and lymphocytes of the sperm and oocyte donors revealed that all hAGES cell loci were homozygous and that they were all identical to one allele of the sperm donor, but did not match the allele of the oocyte donor (Table 2).
hAGES, human androgenetic embryonic stem; HLA, human leukocyte antigen.
The homozygosity of hAGES cells was also determined by genome-wide SNP analysis using the HumanCytoSNP-12 array (Fig. 4a and Supplementary Fig. S4). Among 298,563 SNP sites, 287,738 SNP sites were hybridized and 99.39% SNP sites (285,985) from the hAGES cells were homozygous sites composed of AA, GG, CC, and TT (Fig. 4a). Fertilized human ESCs (H9) displayed only 72.85% homozygous SNP sites (Fig. 4a). These data confirmed that the hAGES cells were homogeneous.
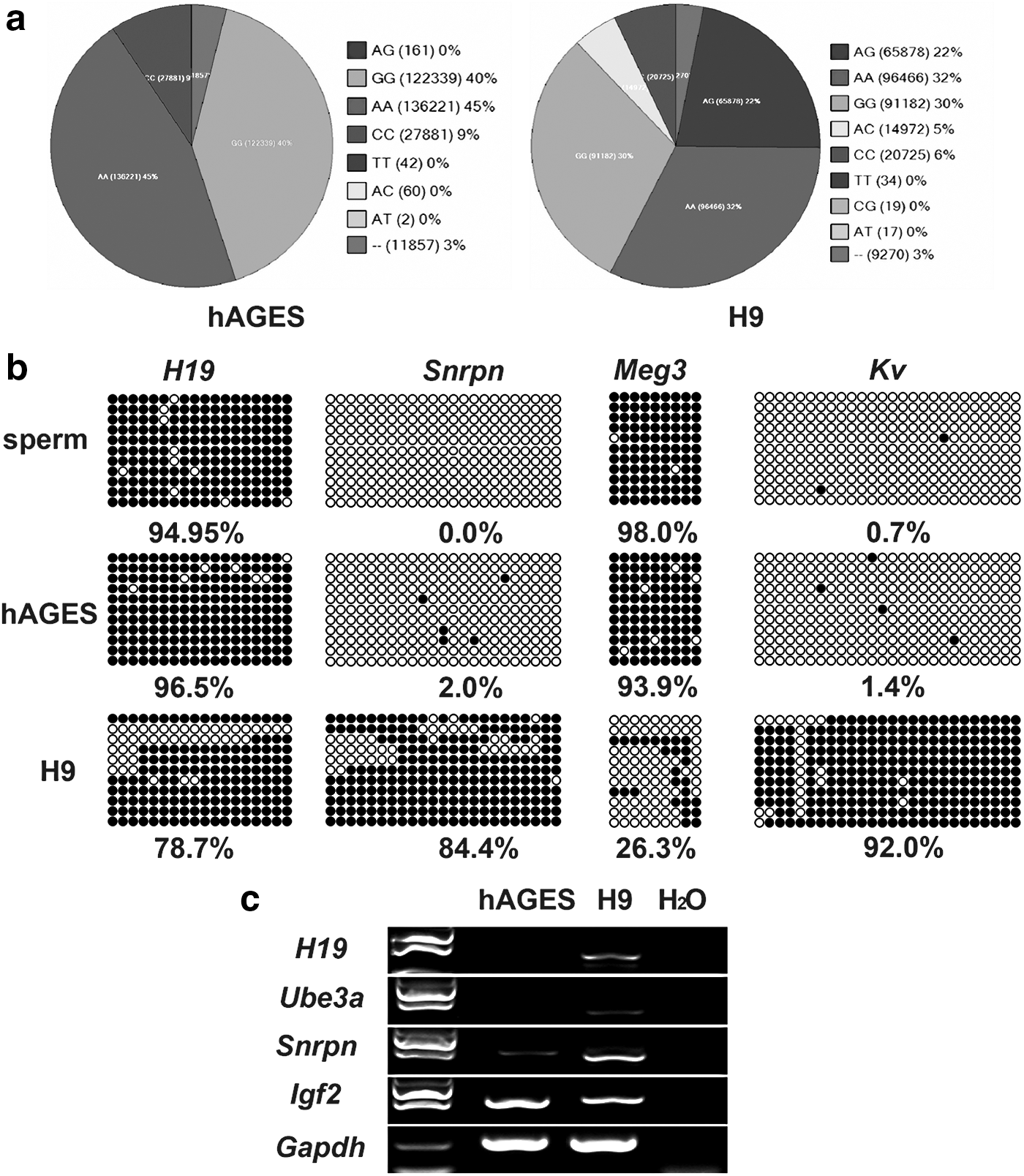
Characteristics of hAGES cells.
Bisulfite sequencing of the DMR of Snrpn, H19, Meg3, and Kv indicated that the hAGES cells maintained hypermethylation on the DMR of H19 and Meg3 (Fig. 4b) and hypomethylation in the DMR of Snrpn and Kv, which was consistent with the sperm and different from normal ESCs (H9). Moreover, the hAGES cells expressed imprinted genes (Snrpn and Igf2), but not other imprinted genes (H19 and Ube3a) (Fig. 4c).
Discussion
In this study, we successfully derived a diploid human androgenetic embryonic stem cell line (hAGES) from reconstructed human haploid androgenetic embryos. The hAGES cells showed typical human ESCs characteristics, normal morphology, strong self-renewal potential, and could differentiate into various cells both in vitro and in vivo. Moreover, hAGES cells were homogeneous in HLA typing, SNPs and STR loci among all of the genome theoretically, which is a valuable trait for cell transplantation-based therapies. In addition, the uniparental origin of hAGES cells maintains to a certain extent paternal imprinting, making it a useful tool for studying gene imprinting in humans.
Human haploid androgenetic embryos can be generated by removing female PN from abnormally fertilized 3PN embryos, or by injecting single spermatozoon into enucleated oocytes. In a previous study, 2PN were removed from abnormally fertilized 3PN embryos to produce haploid human embryos [25]. The developmental ability of those haploid embryos was seriously impaired, as a few haploid embryos could reach the blastocyst stage (2.7%) and no ES cell line was successfully derived from these embryos. Moreover, researchers could not confirm the genetic origin of the haploid embryos because the female or male PNs of human 3PN embryos cannot be distinguished morphologically.
Alternatively, a single spermatozoon can be injected into enucleated oocyte to ensure the paternal origin of haploid embryos. Previous reports showed that only 11%–12% of mouse haploid androgenetic embryos produced by this method could reach the blastocyst stage [26,27], indicating that haploid embryos have impaired developmental potential [26,28]. In our study, similar results were observed in human androgenetic embryos, as the blastocyst formation rate was lower than that of normal diploid fertilized embryos (Table 1).
However, the developmental ability of human haploid androgenetic embryos in the present study was higher than that reported previously. Only the X chromosome could be found in the human androgenetic blastocysts, consistent with previous studies that Y androgenetic embryos were arrested before the blastocyst stage [26,28]. At the same time, there were complete moles reported in the literature that were karyotypically normal, but contained no fetal tissue. Ninety percent of them are 46, XX and 10% of them are 46, XY. No 46, YY moles have been observed or reported at the present time [29,30].
In the present study, diploidization was also found in our human haploid androgenetic embryos, although few diploid cells exist in the morula of human haploid androgenetic embryos. The rate of diploid cells was significantly increased in blastocyst embryos. It was possible due to duplication of genome in haploid blastomeres or proliferation of diploid blastomeres. Currently, we did not know whether the hAGES cell line came from diploid ICM cells or duplication of haploid androgenetic ESCs. Further investigation is needed to clarify the origin of hAGES.
The autologous origin of hAGES cells makes them a potential cell source for regenerative medicine, as pESCs. Additionally, the homozygosity of HLA loci makes hAGES cells a potential source of cell replacement that would avoid immune rejection in allotransplantation. In previous studies, mouse androgenetic ESCs could form neural and hematopoietic progenitor cells in vivo and in vitro [31,32]. The potential of hAGES cells should be partly speculated in the future.
The HLA homozygosity of hAGES cells means that they can be matched to other patients with less risk of immunological rejection [33], and the number of ES cell lines needed in the stem cell bank for HLA matching could be remarkably reduced. In other words, the human androgenetic ESCs in the present study would be as valuable as human pESCs as in the previous reports. Further efforts were needed to optimize the generation of homozygous hAGES cells and investigate the safety of these cells to facilitate their possible application in cell replacement therapy in the future.
In addition, hAGES cells exhibited to a certain extent the paternal DNA methylation profile originated from the sperm in the DMRs of some imprinting genes. In our study, human AGES cells also differentiated into various cell types, making them a complementary tool to study the mechanism of human genomic imprinting during the developmental process and to facilitate drug screening for diseases caused by aberrant genomic imprinting.
Footnotes
Acknowledgments
The authors thank all of the members of Drs. Q.Z.'s and X.Z.'s laboratory for contributing to the discussion. This study was supported by grants from the Key Laboratory of Reproductive Medicine of Guangdong Province, the Priming Scientific Research Foundation for the National Natural Science Foundation of China (81100472), National Basic Research Program of China (973 Program) (grant no. 2012CB947600), the junior teachers of medicine in Sun Yat-sen University (12ykpy23), the China National Basic Research Program Grants (no. 2012CBA01300), and the National Key Basic Research Program of China (no. 2011CB965301).
Author Disclosure Statement
No competing financial interest exists.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.