
Abstract
The technology to convert adult human non-neural cells into neural lineages, through induced pluripotent stem cells (iPSCs), somatic cell nuclear transfer, and direct lineage reprogramming or transdifferentiation has progressed tremendously in recent years. Reprogramming-based approaches aimed at manipulating cellular identity have enormous potential for disease modeling, high-throughput drug screening, cell therapy, and personalized medicine. Human iPSC (hiPSC)-based cellular disease models have provided proof of principle evidence of the validity of this system. However, several challenges remain before patient-specific neurons produced by reprogramming can provide reliable insights into disease mechanisms or be efficiently applied to drug discovery and transplantation therapy. This review will first discuss limitations of currently available reprogramming-based methods in faithfully and reproducibly recapitulating disease pathology. Specifically, we will address issues such as culture heterogeneity, interline and inter-individual variability, and limitations of two-dimensional differentiation paradigms. Second, we will assess recent progress and the future prospects of reprogramming-based neurologic disease modeling. This includes three-dimensional disease modeling, advances in reprogramming technology, prescreening of hiPSCs and creating isogenic disease models using gene editing.
Introduction
T
The Need for Human Neurologic Disease Models
Until recently, the genetic basis for many neurologic diseases was largely unknown. Thanks to the increasing scope and declining cost of genome sequencing, candidate genes that underlie or predispose individuals to disorders of the nervous system ranging from autism to Alzheimer's disease are now being discovered at an accelerated pace [9 –12]. Yet, even for well-understood monogenic disorders such as Friedreich's ataxia or Huntington's disease, the cellular and molecular links between causative mutations and the symptoms exhibited by affected patients are incompletely understood [13 –16]. One barrier to studying biological mechanisms and discovering drugs for rare human disorders is the lack of availability or access to large enough patient cohorts. In addition, even for more common diseases, the high cost of clinical trials restricts the number of potential therapeutics that can be tested in humans.
For these reasons, animal models have been extensively used to study disease mechanisms and identify candidate therapeutics. However, the relevance of these studies is ambiguous due to inherent differences between the rodent and human nervous system [17 –19]. For example, differences in lifespan may explain why animal models often fail to recapitulate key aspects of the pathology of late onset diseases like Alzheimer's disease [20]. Similarly, aspects of cognitive function and social behavior that are unique to humans are challenging to evaluate in animal models of neurodevelopmental disorders such as autism and schizophrenia [21 –23]. Finally, the human nervous system significantly differs from rodents in its overall structure and cell type composition. For example, the human brain is gyrencephalic, has a proportionately larger upper cortical layer [19], and a better developed prefrontal and temporal cortex implicated in higher cognition [17,18]. An important example of a molecular difference between the developing human and mouse brain was recently reported by Lui et al. Here, the authors show that the growth factor PDGFD and its downstream signaling pathway contribute to neurogenesis in human, but not mouse cortex [24]. Other examples include the presence of a layer of neural progenitors called the outer subventricular zone in the developing human cortex, which does not exist in rodents [25,26]. The origin and subtype identity of cortical interneurons might also differ between humans and rodents [27]. Accordingly, many drugs that display efficacy in animal models have not successfully translated to humans [28 –30]. Therefore, creating disease models using human neurons generated through reprogramming may offer improved insights into the molecular and cellular bases of neurologic disorders.
One method to produce human neurons suitable for disease modeling is by differentiating human iPSCs (hiPSCs) or human embryonic stem cells (hESCs) into desired neural lineages, such as cortical pyramidal neurons, striatal interneurons, motor neurons, or dopaminergic neurons [31 –42]. Importantly, hiPSC-derived neurons are functionally active, and can respond to synaptic stimulation and specific sensory response-evoking ligands [43 –49]. In addition, Livesey and colleagues showed that hiPSCs subjected to directed neural differentiation follow the same temporal sequence as in vivo corticogenesis [38]. Similar findings have been reported for forebrain interneurons [50]. Despite limitations, these methods have been used to model and study several neurodevelopmental and neurodegenerative disorders [30,51,52]. Encouragingly, iPSC-based neurologic disease models have identified previously unknown aspects of disease biology [47,48,53 –58]. There is proof of principle evidence that they can recapitulate the pathology observed in animal disease models [55,58 –64], including diseases associated with abnormal imprinting [65,66]. More recently, researchers succeeded in reproducing the pathophysiology of neuronal activity found in patients diagnosed with amyotrophic lateral sclerosis (ALS), in motor neurons derived from patient-specific iPSCs [49]. In addition, there are alternative strategies for generating disease phenotypes in a dish. These include, direct differentiation of iPSCs into neurons by bypassing normal developmental sequence [67], and direct lineage reprogramming (or transdifferentiation) of non-neural cells such as fibroblasts into neurons or neural stem cells (NSCs) (Box 1) [68 –71]. However, despite considerable progress, current technology has several limitations that, if overcome, could significantly improve the utility of reprogramming-based human in vitro models of neurologic diseases.
Reprogramming: Conversion of cell fate from one lineage to another. Example- conversion of somatic cells (such as fibroblasts) into iPSCs.
Direct lineage reprogramming or transdifferentiation: Conversion of one mature cell fate into another, without an intermediate pluripotent stage. Example- conversion of fibroblasts into neurons.
Direct neural differentiation: Quick and almost direct conversion of iPSCs into neurons, usually by overexpressing lineage-specifying genes that function during in vivo neurogenesis; involves a brief transient neural progenitor stage.
Directed neural differentiation: Step by step differentiation of iPSCs into neurons using protocols based on in vivo neural development; involves long neural progenitor and immature neuronal stages.
iPSCs, induced pluripotent stem cells.
Challenges for Reprogramming-Based Studies of Neurologic Diseases
Numerous studies have shown that current technology is more amenable to studying disease onset and processes leading to it [72,73] than progression. This is also true for diseases with known mutations and a cell intrinsic phenotype where the affected cell or tissue type is known from prior clinical and animal studies [48,55,56,58,74]. Here, we outline some of the challenges in adapting reprogramming-based manipulation of cellular identity to model more complex disorders of the nervous system.
Limitations of current neural differentiation protocols
Heterogeneity within long-term cultures
Directed neural differentiation can require many months of culture to generate mature neurons and glia (Box 1) [33,34,36 –38]. This is a barrier to certain aspects of personalized medicine, such as the generation of disease-relevant neural cells from individual patients for drug testing. In addition, the long time course of in vitro differentiation can contribute to heterogeneity within the culture [75]. For example, subtle differences in the proportions of distinct NSC subtypes that arise early during differentiation may be amplified as they expand and differentiate into fate-restricted precursors and postmitotic neurons (Fig. 1A′). In addition, due to the asynchronous nature of these cultures, neural progenitors of diverse ages and lineage-commitments coexist with neurons at various stages of differentiation and maturation. This type of heterogeneity is a problem because it can increase variability between technical replicates and potentially mask a phenotype specific to only a subset of cells in culture [76]. Another potential source of bias can arise from artificial selection of NSC subtypes that survive and grow better in vitro than other possibly more disease-relevant subtypes.

Challenges of current reprogramming- based methods for studying neurologic diseases.
Most hiPSC-derived NSCs, neurons and glial cells have been characterized using generic markers such as Sox2 and Nestin (pan-NSC), and β-Tubulin and Map2 (pan-neuronal), which label cells with diverse subtype identities. For many cell types in the nervous system, region-, subtype-, and developmental stage-specific markers have not been identified. Consequently, during in vitro differentiation, expression of one or two markers may not be sufficient to establish the true identity of the neuron, since these markers may be expressed by neurons in other regions of the brain or during other developmental stages. In many human disease models for which an in vitro phenotype has been identified, information on the specific subtype of neuron that was affected or assayed is lacking. Moreover, many protocols designed to generate a certain neuronal subtype also produce other types of neurons with varying frequency, which further complicates phenotypic analyses [19].
Limited subtype-, region-, and stage-specific differentiation protocols
The number and type of neurologic diseases that can be accurately modeled is limited by the availability of differentiation protocols that produce the neural cell type relevant to the disease. The selectivity of the protocol, with regard to age and function of derived neurons, is critical for the following reasons. First, in certain neurodegenerative conditions, the disease preferentially impacts specific neuronal subtypes while sparing other neurons. For example, because ALS preferentially impacts motor neurons, one would not wish to model this disorder using cortical neurons that are typically spared in the patients [76]. Second, the late onset of neurodegenerative diseases, such as Alzheimer's or Parkinson's disease, suggests that aging neurons may have different disease susceptibility than younger neurons of the same subtype. Most iPSC-derived neurons seem to more closely resemble embryonic neurons than mature and aged neurons [77]. Therefore, to best model these disorders, it may be necessary to devise protocols that generate neurons with aging or degeneration- associated features (Fig. 1A′′).
Low efficiency and variable specificity of direct conversion methods
One goal of disease modeling is high-throughput screening (HTS) for potentially therapeutic compounds. Disease-relevant neural cells could be a powerful tool to identify drugs with high efficacy and low toxicity to individual patients. Proof of principle studies for diseases such as ALS and Rett Syndrome have been performed using mouse iPSCs [35,78]. HTS can also serve as an effective strategy to identify novel drug targets [30]. While most reported HTS assays utilized mouse ESCs [79,80], few labs have successfully demonstrated the use of hiPSC-derived neural progenitors for drug screening [81].
One reason for the limited success of hiPSCs in HTS is the difficulty in generating large numbers of a pure population of disease-relevant neurons, highlighting the need for improved derivation protocols or selection methods (Fig. 1A′). One attractive, alternative strategy to produce purer populations of desired neuronal subtypes is direct lineage reprogramming or transdifferentiation of somatic cells into neurons [68,69]. However, this technology is still rather new and its relative utility compared to iPSC- derived neurons has not been clearly established. In addition, the efficiency of most direct conversion protocols may not be sufficient to generate a pure or nearly pure population for reproducible screening of thousands of compounds (Fig. 1A′′′), or even high enough to make enrichment by fluorescence- activated cell sorting (FACS) logistically and economically feasible [68,69]. Despite recent advances in generating subtype-specific neurons [70,71], most direct conversion protocols are not selective. Often, the same combination of transcription factors gives rise to neurons belonging to functionally and developmentally independent lineages. For example, Wernig and his colleagues noted the induction of both central nervous system and peripheral nervous system neurons from human fibroblasts upon overexpression of Brn2, Ascl1, Myt1l, and NeuroD1 [82]. Recent developments and future prospects in the field are discussed in the section Direct Neuronal Reprogramming and Transdifferentiation.
Limitations of two-dimensional neural cultures and cellular models
Numerous studies have shown that it is possible to model neurologic diseases that have a strong and obvious in vitro phenotype, such as, neuronal degeneration or cell death, and the inability to differentiate, proliferate, or grow dendritic processes in culture [57,83 –86]. Yet, certain aspects of neuronal differentiation and function are likely to require cues that may not exist in vitro. This could complicate the analyses of cellular features, in normal and disease conditions, that are dependent on the dynamic processes occurring within a three-dimensional microenvironment.
Present methods that largely employ two-dimensional cultures of heterogeneous neural subtypes are quite distinct from the more precisely specified three-dimensional arrangements of cell types and microenvironments that exist in the developing brain. Neurologic disorders that involve a defect in one or more of the following parameters might be challenging to model using two-dimensional neural cultures: (1) Complex neural connectivity between different brain regions such as the cortex, basal ganglia, thalamus, and hippocampus (Fig. 1B′, B′′) [20,48,75,87]. Neuropsychiatric disorders like Alzheimer's disease are likely to involve defects in such neural circuitry; (2) Relative proportions of various neuronal subtypes; (3) Cellular migration within a defined milieu of extracellular matrix (ECM) and axon guidance molecules [88]; (4) Temporal and developmental stage-specific neurogenesis [89,90]; (5) Noncell autonomous effects on neuronal properties and function (Fig. 1B′′′, B′′′′) [89,91,92]. Despite these challenges, however, cell extrinsic effects of diseased glial cells on unaffected neurons has been modeled for some diseases [54,93,94]. Promising solutions to these issues, including three-dimensional disease models, are discussed in the section Three-Dimensional Cellular Models and Improved Microenvironments.
Interline and inter-individual variability among hiPSC lines
Differences in the developmental potential of hiPSC lines
Three kinds of variability-related issues have been reported in reprogramming studies: intraline variability, interline variability, and inter-individual variability. Intraline variability are inconsistencies between independent experiments and between different passage numbers of the same hiPSC lines subjected to identical differentiation protocols and phenotypic assays [95]. Such variability has also been reported among hESC lines [96]. Intraline variability may arise from several sources. First, there is evidence that the properties of iPSCs change with increasing passage number, possibly due to increasing accumulation of genomic mutations and epigenetic changes during in vitro culture and passaging [97,98]. Second, passaging can introduce a selection bias, where one might inadvertently select and expand cells carrying a specific genetic composition from a population of iPSCs with preexisting genetic variability. Third, variability could also arise from the use of growth factors, cytokines, and small molecule inhibitors that can differ in activity between experiments depending on storage conditions or batch number.
Different hiPSC lines derived by the same reprogramming method and from the same donor population seem to be heterogeneous, for example, in their propensity to commit to specific lineages (Fig. 1C′) (personal observations, [18,95,99 –101]). Such interline variability has been noted by independent groups. This is of particular concern in studies where only one or two patients and/or one or two lines per patient are available [76,85,86]. The source of interline variability could range from differences in the cohort of mutations accumulated by donor cells [95], to the level of epigenetic resetting, and status of X-chromosome inactivation (and therefore dosage compensation) during reprogramming, all of which could affect lineage specification and the properties of differentiated neurons [102 –104].
Reliable disease modeling is further complicated by inter-individual variability, the high level of diversity between iPSC lines derived from different patients diagnosed with the same disease. These lines often differ in the onset and severity of the phenotype following in vitro neural differentiation (Fig. 1C′′) [20,30]. Possible causes include, differences in the genetic background and the cohort of mutations accumulated by different patients, and the contribution of individual-specific genetic modifiers to disease phenotype. While such modifiers might be useful in understanding disease pathology, they are of concern when they influence the generation of specific neuronal subtypes in a way that is not related to the disease being modeled.
Genomic instability and epigenetic aberrations in iPSC lines contribute to interline variability
Despite considerable progress in reprogramming methods, including nonintegrating methods and stoichiometry-controlled protocols, one concern about the present generation of iPSCs is that they may fail to accurately phenocopy the disease [95,105]. This could be due to several reasons. First, iPSCs harbor genomic mutations derived from at least three sources: somatic mutations specific to the donor cells, reprogramming-associated mutations, and mutations that arise during culture or expansion. Each of these could impact the developmental potential of these lines. For example, the population of donor cells (such as fibroblasts) could be genetically heterogeneous, with different cells carrying different somatic mutations. When reprogrammed, independent iPSC lines would inherit the mutations unique to the donor cell they were reprogrammed from, resulting in lines with different cohorts of genomic mutations. In addition, the reprogramming process itself could introduce unknown and random mutations into the reprogrammed cells [106,107]. To this end, substituting viral vectors with integration-free approaches have helped alleviate this issue [108 –111]. Nevertheless, several reports have shown an increase in the number of acquired genomic mutations such as copy number variants (CNVs) and aneuploidies during reprogramming, and with increasing passage number and days in culture [95,98,105,106,112,113]. This would cause independent lines that are the result of independent reprogramming events, to be genetically different. Moreover, in vitro culture conditions, passaging, and handling might inconsistently introduce unique and unknown mutations into iPSC lines derived from the same donor population, rendering them heterogeneous.
In addition, cellular reprogramming involves genome-wide epigenetic changes that are not typical of normal embryonic development, which may result in imperfect, aberrant, or incomplete resetting of the epigenetic landscape of the donor cell [114,115]. Recent improvements in reprogramming protocols have repeatedly shown that chemical manipulations that increase the accessibility of the chromatin to the cell's transcriptional machinery, can contribute to improved reprogramming efficiency [116 –119]. Additional studies also indicate that more complete erasure of the donor cell's epigenetic profile might be needed for complete restoration of pluripotency (reviewed in Cantone and Fisher [114]). Incomplete epigenetic resetting causes independent iPSC lines to uniquely inherit the epigenetic features of the specific donor cells they were reprogrammed from. This retention of “epigenetic memory” can contribute to developmental diversity between lines [120 –122]. Aberrant methylation patterns can also result from the reprogramming process itself [105,113,123], and from culturing of iPSCs [105,124], all of which could contribute to interline variability. Furthermore, the variable epigenetic status of iPSC lines might confer them distinct responses to the same initial differentiation conditions, leading to differences in cell fate specification. Finally, the epigenetic status of iPSCs may not be stable over time, introducing more variability into this system. Interestingly, Rouhani et al. recently showed that the influence of epigenetic memory on the transcription profile of an iPSC line is minimal; in comparison, the genetic background of the individuals these lines are derived from contribute to greater heterogeneity in neural differentiation assays [125].
The Future of Reprogramming-Based Study of Neurologic Disorders and Related Clinical Applications
Improvements in directed and direct neural differentiation protocols
To faithfully model polygenic, multifactorial, and sporadic diseases within the context of an individual's unique genetic background, neural differentiation paradigms should be robust, reproducible, and reliable. Principles of developmental biology have been used to optimize such protocols by adding growth factors or small molecules that mimic the endogenous environment or selectively block or activate known developmental pathways [45,126]. However, for many neuronal subtypes, little is known about their endogenous developmental sequence. Two recent reports outline methods that may broaden the number of appropriate in vitro developmental paradigms. In one study, Tarunina et al. developed an HTS approach to devise methods for stepwise differentiation of iPSCs into specific neural cells at specific stages of development [127]. In the second study, Maury et al. performed combinatorial screening of small molecules to identify methods to rapidly and efficiently generate specific neural subtypes from hPSCs [128]. Other developments and future prospects are discussed below.
Generating homogenous cultures of NSCs from iPSCs
Derivation of intermediate neural progenitors: Neurologic disease modeling could benefit from differentiation protocols that produce a homogeneous and synchronized starting population of neural progenitors. Generating intermediate neural progenitors that are multipotent and can be expanded for a prolonged period of time is a step in this direction [129 –134]. This will be particularly useful when large numbers of a specific neuronal subtype is required either for HTS or disease modeling. Factors regulating neural cell fate specification and neuronal diversity in the developing brain are still being investigated [135,136]. The outcome of these studies will be critical in establishing protocols to generate specific NSC subtypes either by differentiating iPSCs or by transdifferentiating non-neural cells [called induced NSCs (iNSCs)]. Alternatively, one could identify combinations of transcription factors that could reprogram donor cells into iPSCs with strong neurogenic potential. These could then be differentiated rapidly and efficiently into a uniform population of NSCs that can self-renew without changing their developmental potential. How the differentiation properties of these progenitors will vary over time and with increasing number of passages needs to be determined. A promising finding was made recently when iNSCs were found to differentiate and integrate efficiently into the host neural circuitry upon transplantation into adult mouse brain [137].
Enrichment of NSC subtypes by FACS: Most directed differentiation protocols generate mixed populations of NSCs and neurons at various stages of differentiation and maturation and a few glial precursors. Reducing culture heterogeneity might make it easier to detect subtle subtype-specific phenotypic differences, especially in cell numbers and proportions. To resolve this heterogeneity and avoid assay artifacts, some groups have used FACS to enrich their desired cell types (Fig. 2A) [138 –141]. For example, to analyze iPSC models of Timothy Syndrome and identify differences in the generation of subtype-specific neurons, the Dolmetsch lab performed single cell gene expression analysis on reporter-labeled neurons isolated by FACS [57]. In the future, differentiating iPSCs carrying fluorescent reporters tagged to novel NSC subtype-specific markers [24] could be sorted at the start of differentiation into homogenous NSC cultures.
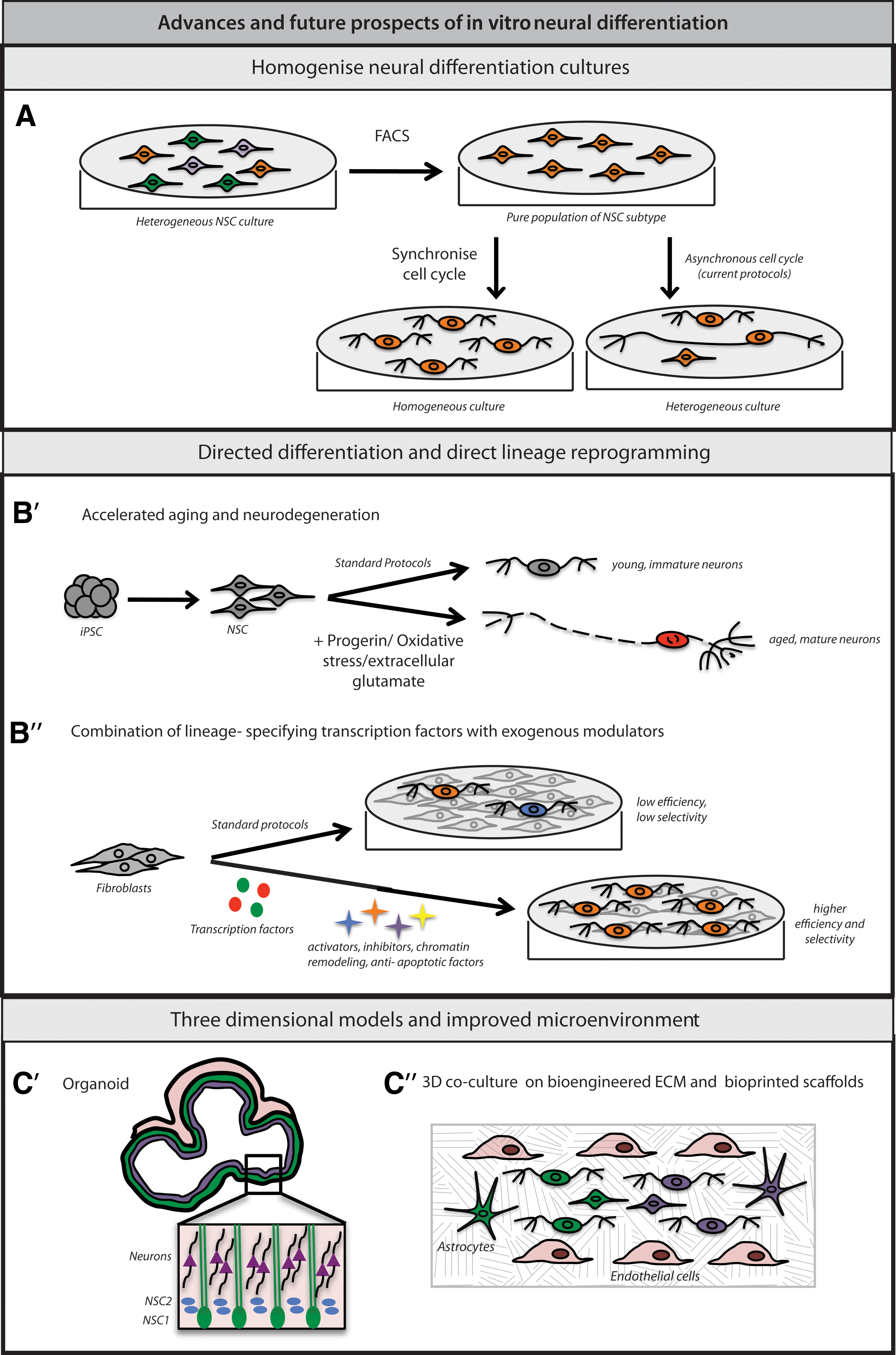
Recent advances and future prospects in in vitro neural differentiation.
Synchronization of cell cycle during directed neural differentiation: Asynchronous cell cycle in hiPSC-derived neural cultures is responsible not only for the heterogeneity within the culture, but could also contribute to variable differentiation potential of these lines [142]. Any means to modulate and synchronize cell cycle progression, including the use of dimethyl sulfoxide (DMSO) [143], could prove valuable in regulating their development and in isolating a disease-specific phenotype (Fig. 2A). Currently known methods of synchronizing mammalian cells have several disadvantages (reviewed in Rosner et al. [144]), calling for novel or improved strategies.
Characterization of hiPSC-derived neural differentiation cultures
Molecular techniques such as single cell expression profiling [57] and microarray analyses [37] have been used by researchers to characterize individual hiPSC- derived neurons. Such high-throughput characterization methods might become a necessity in future disease modeling studies.
Accelerated aging of hiPSC-derived neurons to model late-onset neurologic diseases
Because directed neural differentiation appears to partly recapitulate in vivo development, the neurons generated in these protocols often resemble immature or embryonic neurons [18,77]. However, many of the most devastating neurologic diseases seem to affect more mature neurons in young adults or aged individuals [30,76]. Whether this is due to changes in neuronal gene expression over time or mutations and/or epigenetic changes that accompany aging is an important question. Both oxidative stress [61,74,145] and overexpression of progerin, a protein associated with premature aging [146], have been used to artificially induce the aging process in hiPSC-derived neurons (Fig. 2B′). In addition, exposure to extracellular glutamate, thought to mimic in vivo excitotoxicity, can induce degeneration in susceptible neurons (Fig. 2B′) [147]. Although promising, the utility and physiological relevance of these perturbations in modeling neurodegenerative diseases will require additional study.
Direct neuronal reprogramming and transdifferentiation
Scientists have applied the knowledge gained from developmental studies in animal models to identify transcription factor combinations that can convert non-neural cells such as fibroblasts into specific neural lineages [148]. These promising approaches have been reviewed extensively [68,69]. Here, we will outline areas where they may be adapted or improved to generate large numbers of desired neuronal subtypes. For example, in one recent report, Ladewig et al. converted fibroblasts into neurons at more than 80% purity, by combining ectopic expression of Ascl1 and Ngn2 with small molecule-mediated inhibition of GSK3β and SMAD signaling [149]. In another study, Victor at al. used histone deacetylase inhibitor, valproic acid, and overexpressed antiapoptotic factor Bcl-xl, in addition to lineage-specifying transcription factors and microRNAs to convert fibroblasts into striatal interneurons at high efficiency [150]. This suggests that lineage-specific transdifferentiation could be improved by simultaneously suppressing antagonistic developmental pathways, improving chromatin accessibility to the transcriptional machinery, or blocking cell death (Fig. 2B′′). It should be noted that direct neuronal reprogramming appears to bypass certain phases of early lineage specification and other developmental processes that might be necessary for accurate manifestation of disease phenotype. Therefore, in some cases, directly reprogrammed neurons might be better suited for HTS rather than disease modeling.
Three-dimensional cellular models and improved microenvironments
Animal studies have shown that abnormal development of specific brain regions such as the neocortex and basal ganglia may underlie neurodevelopmental conditions such as schizophrenia or autism spectrum disorders [151]. Therefore, disease modeling could be improved by aiming to recapitulate key aspects of in vivo tissue and organ development [148]. Neural development is tightly coordinated, both temporally and spatially [152], and is influenced both by cell intrinsic properties and by extracellular cues such as morphogen gradients, paracrine and feedback signaling, ECM and cell–cell contact [89,136,153 –155]. Some evidence suggests neuronal differentiation may be improved when the structural and molecular properties of the microenvironment closely mimics the developing nervous system [156 –159]. For example, Warmflash et al. recently showed how geometric confinement alone could influence the differentiation of pluripotent stem cells [160], by ensuring a more accurate reproduction of the signaling events and cell–cell interactions occurring during neurogenesis in vivo. In another study, Choi et al. differentiated a human NSC line carrying mutations associated with familial Alzheimer's disease, within a three-dimensional system. They were able to recapitulate both extracellular deposition of amyloid plaques and intracellular aggregation of phosphorylated tau, hallmarks of Alzheimer's disease pathology [156]. Other developments are discussed below.
Organoids mimic the architecture of developing brain
Miniature three-dimensional tissue assemblies called organoids have been generated for the liver [161], kidney [162], intestine [157,163], pancreas [164], the optic cup [159], cerebellum [165], and the cortex [158,166]. In a recent study, young human liver organoids, when transplanted into mice, were shown to integrate with the host vasculature and grow and mature in response to this nutritional support [161]. Cortical organoids mimic in vivo tissue architecture (Fig. 2C′), which may make them particularly useful to model diseases with defects in subtype-specific cell number and proportion, noncell autonomous signaling, neural circuitry, and anatomical structures [20,75]. For example, synaptic communication within a cortical organoid could mimic the neural connectivity between different areas of the brain, such as neocortex and basal ganglia. Isogenic three-dimensional disease models can be constructed by introducing disease-specific mutations into hiPSCs before generating organoids. Furthermore, inducible or cell type- specific promoters will be useful in carrying out such genetic manipulations at precise time points during in vitro differentiation. These or similar approaches could help to identify molecular mechanisms underlying neurodevelopmental disorders involving patterning that depends on the interactions of multiple neuronal subtypes or on particular circuit architectures [88].
Coculturing different hiPSC-derived cell types to recreate complete tissues
Since hiPSCs can be differentiated into multiple lineages, those cell types that are known to coexist in vivo can be combined and cocultured. For example, growing iPSC-derived neural progenitors and neurons with astrocytes, fibroblasts, and endothelial precursors within precisely constructed scaffolds could provide structural, functional, and vascular support to differentiating neural tissue (Fig. 2C′′). Endothelial cells also secrete vascular endothelial growth factor that can stimulate proliferation and migration of neuronal precursors, and neuronal maturation [167]. Recently, hiPSC-derived hepatocytes and endothelial cells were cocultured to create functional liver tissue [168]. In addition, iPSC-derived neurons are routinely cocultured with astrocytes to promote synapse formation [53,169]. Similar approaches could also be applied to organoids since lack of vascularization (and hence an efficient means of nutrient exchange) will limit the extent to which an organoid can grow and mature in vitro.
Modeling within bioengineered matrices and bioprinted scaffolds
With the advent of three-dimensional bioprinting of biomaterials and live cells, it is now possible to construct intricate scaffolds of bioengineered substrates and ECM proteins within which iPSCs could be allowed to differentiate [170,171]. In addition, different regions within a compartmentalized scaffold can be bioprinted with different types of live iPSC-derived neural progenitors and support cells (astrocytes, fibroblasts, and endothelial cells) (Fig. 2C′′). Two factors are critical for the success of this approach: the mechanical and chemical properties of the matrix and the composition of cells seeded into it. In neurogenesis, the ECM is required for structural support, providing cues for cell migration, and presenting extracellular growth factors to target cell surface receptors [172]. Murphy et al. recently reviewed the significance of the mechanical properties of ECM in determining stem cell fate [173]. The significance of ECM composition in hiPSC-derived neural cultures has also been shown [174]. Altogether, one could recreate the necessary mechanical properties (elasticity and stiffness) [175,176], relative cellular positioning, vascular innervations, and morphogen gradients characteristic of in vivo neural development [177]. Three-dimensional bioprinting could also streamline drug toxicity testing, and eventually have significant cell therapy applications (
Improvements in reprogramming methods
Recent efforts to improve hiPSC quality and safety have focused on reducing aberrant genomic and epigenetic alterations during the process of reprogramming. A few promising strategies to establish better quality and physiologically relevant hiPSC lines are discussed below.
Extracellular manipulation of prodifferentiation and antiproliferative signals in donor cells (Fig. 3A)
In a recent reprogramming study by Ichida et al., the authors inhibited Notch signaling extracellularly to suppress the differentiation of transforming keratinocytes. This led to improved reprogramming efficiency and potentially safer hiPSCs [178]. In another study, Bar-Nur et al. added ascorbic acid and GSK3β inhibitor to mouse donor cells, leading to more rapid and uniform reprogramming [179].
Resetting hiPSCs to the ground state of pluirpotentcy
While mouse iPSCs resemble the primitive and uncommitted cells of the inner cell mass (ICM), most currently available hiPSCs resemble the primed and developmentally restricted mouse epiblast stem cells (Fig. 3B) [180,181]. Recently, several groups have generated naïve hiPSCs with reduced global DNA methylation and molecular properties comparable to mouse ESCs and human ICM [180,182]. Others have shown that a small cohort of naïve cells exist within traditionally derived hiPSC populations, which can potentially be purified, cultured, and expanded further [183]. In addition, silencing mechanisms that contribute to genomic instability and epigenetic aberrations could reduce the variability between hiPSC lines. This was highlighted in a recent study by Chen et al. where Tet1, an enzyme involved in DNA demethylation and transcriptional reactivation [184], was shown to improve the quality of mouse iPSC lines [185].

Recent advances and future prospects in reprogramming methods.
Alternative strategies for streamlining the reprogramming process
Considerable efforts have been directed toward identifying and designing novel transcription factors that can achieve better reprogramming. For example, the Jaenisch lab recently reported a novel combination of transcription factors that generated mouse iPSC lines of better quality and genomic stability [186]. Others have attempted to generate synthetic factors using gene editing tools (discussed later) (Fig. 3A), that could provide improved stoichiometry of reprogramming factors [187], enable complete epigenetic resetting, and either eliminate or alleviate the stochastic nature of reprogramming. These efforts were recently reviewed by Tsang et al. [188].
Screening hiPSC lines before modeling
Screening for biomarkers that could predict the developmental potential of hiPSCs
Studies suggest that current methods to generate hiPSCs produce lines with widely varying lineage bias. In such cases, it could be useful to screen these lines for the expression of biomarkers that could predict the neural differentiation potential of the line. For example, the Meissner lab has proposed that before starting neural differentiation, multiple hiPSC lines could be subjected to a high-throughput “scorecard assay” to identify and discard (or select) lines that have a skewed developmental potential (Fig. 4A′) [189]. Examples of potential lineage-specific biomarkers in iPSCs, include Wnt3 as a predictor of dopaminergic differentiation [190], and miR-317-3 as a predictor of neural differentiation potential [191]. Such screening could ensure that all hiPSC lines used in a given disease modeling experiment have similar propensity to generate neural cells. However, employing preselection of iPSCs might also unintentionally select against lines that would best model a disease or recapitulate a particular disease phenotype. This is especially true for neurodevelopmental disorders, but may not be an issue for late-onset diseases. On the other hand, prescreening for selected biomarkers may not necessarily identify “suitable” lines for modeling because of reprogramming event-associated differences in their genetic background.

Strategies to overcome interline and inter-individual variability between hiPSC lines. Before neural differentiation, multiple hiPSC lines could be screened for,
Epigenetic screening to identify hiPSC lines with desired properties
It is clear that epigenetic aberrations in hiPSC lines can significantly influence their developmental potential. Because these modifications may impact genes that are not expressed in iPSCs, gene expression analyses of the iPSCs might fail to fully predict developmental potential. Therefore, to ensure that the correct gene regulatory networks are recruited during differentiation, it might be advantageous to screen and select lines by examining their patterns of DNA methylation or other epigenetic marks. These could then be compared to DNA methylation patterns known to be important for neural differentiation in vivo (Fig. 4A′′), and further validated through in vitro developmental assays [114,189].
RNAi screening of difficult lines to identify barriers to efficient neural conversion
Recently, a genome-wide RNAi screen was performed to identify barriers to reprogramming [192]. A similar approach might be useful to identify roadblocks to efficient neural differentiation in hiPSC lines with low neurogenic potential (Fig. 4A′′′). These pathways could then be manipulated either using shRNA-mediated knockdown or small molecule inhibitors. RNAi-based screening could also be applied to disease-relevant neural cell types to identify molecular mechanisms underlying disease pathology. Specifically, siRNAs targeting multiple genes and signaling pathways could be screened on neural cells to determine mechanisms critical in producing or alleviating disease phenotypes.
Applying gene editing to improve disease models
Using isogenic lines to circumvent interline and inter-individual variability
A promising approach for modeling monogenic and Mendelian diseases is to use genetic complementation experiments to show that differences between patient and control lines are due to differences in genes rather than from inherent, disease-unrelated variation between iPSC lines [75,76]. Currently, TALENS, Zinc Finger nucleases (ZFNs) and CRISPR/Cas9 are the most commonly used tools for modifying the genome of iPSCs [193 –198].
Gene-editing tools can be used to introduce disease-associated mutations at specific loci. In this way, iPSC lines that share the same genetic background but differ in the modified locus can be compared (Fig. 4B′). Several studies have utilized isogenic lines to complement the traditional control versus patient approach of disease modeling [199 –201]. In future, this strategy might also be applied to a test set of highly characterized lines with known developmental potential and defined epigenomes and genomes to reliably model diseases and dissect their mechanism.
An alternative approach is to enlist gene editing to correct disease-causing mutations in patient-specific iPSCs and ensure that the disease-related phenotypes are no longer evident (Fig. 4B′′). Diseases such as Huntington's disease, spinal muscular atrophy, and tauopathies have been investigated by correcting disease-specific mutations [49,56,145,202 –205]. Rescuing mutations in patient-specific iPSCs has an advantage over recapitulating mutations in “healthy” iPSCs because protective modifier alleles in the nonpatient genetic background might mask relevant phenotypes that can only be seen in the genetic background of the patient exhibiting disease symptoms [76]. Such an approach could simplify the study of neurologic diseases that are associated with unknown genetic modifiers, such as Mowat–Wilson syndrome [206] and Huntington's disease [207].
Identifying mechanisms of polygenic diseases
In case of complex neurologic disorders associated with multiple mutations, gene editing can be useful in systematically dissecting the contribution of individual mutations to disease phenotype [20]. This can be accomplished by introducing specific mutations [72] sequentially, and evaluating the resulting phenotype in a step by step manner. This was illustrated in a recent review by Sterneckert et al. [30].
Lineage selection and tracing
Gene editing has also been used to generate cell type-specific reporter hiPSC lines [208] that allow for quick, high-throughput characterization of differentiated neural cells, and facilitate lineage tracing experiments (Fig. 4B′′′). For example, an unresolved question in cortical development is how do early progenitors generate the neuronal diversity seen in the cortex? Is it because there are independent subtype-specific NSCs at the start of cortical development, or because the same population of NSCs progressively acquires the ability to produce different neural subtypes during corticogenesis [136]? Reporter-labeled hiPSCs can also enable purification of desired neuronal subtypes using fluorescent proteins or selectable markers.
Deriving and differentiating multiple control and patient lines
Currently, isogenic models are feasible predominantly for monogenic and familial diseases; for sporadic, polygenic, or modifier-influenced disorders, we are still heavily reliant on control versus patient approaches to disease modeling. In such cases, comparing multiple control and patient iPSC lines from age and sex-matched individuals, might be necessary to ensure reliability of data (Fig. 4C) [76,209]. Also, it might be necessary for multiple labs to independently corroborate new findings using different differentiation protocols and modeling methods. This has been reported for Huntington's disease [209 –212] and Rett Syndrome [35,84,199]. Reducing interline variability, applying appropriate statistical tests and reproducing findings in multiple independent experiments are crucial to the success of these studies.
hiPSC-Based Cell Therapy and Personalized Medicine
To fully realize the potential of reprogramming-based approaches in cell replacement therapy, one would require large numbers of physiologically indistinguishable and disease-relevant neurons that are nonimmunogenic, genomically stable and lack tumorigenicity. To this end, several labs have noted that transplanting terminally differentiated or lineage-committed hiPSCs is more immunogenically tolerable than undifferentiated hiPSCs [213 –215]. Several studies have used animal models to demonstrate the feasibility of an ESC/iPSC-based cell transplantation approach [43,211,216,217]. The Vanderhaeghen group provided one of the first examples of efficient integration of hiPSC-derived neurons in the rodent brain [218]. Recently, Mu et al. transplanted hiPSC-derived neurons into immune responsive rodent models of Huntington's disease, and demonstrated functional recovery [219]. Similarly, hESC-derived dopaminergic neurons were shown to induce long-term survival and restore motor function when transplanted into rat models of Parkinson's disease [220]. In another study, hiPSC-derived interneurons reduced seizures and behavioral abnormalities in epileptic mice [221]. Clinical trials for iPSC-based cell therapy for macular degeneration, spinal cord injury, and Parkinson's disease have either been attempted or are in planning stages [222]. More importantly, promising results were obtained in two recent studies where hESC-derived cardiomyocytes (for infarct repair) [223] and hiPSC-derived stromal cells (for new bone formation) [224] were transplanted into nonhuman primates (monkeys), either in the presence or absence of immune suppression. Ultimately, iPSCs derived from individual patients could be a source of autologous disease-relevant cells that may be used for transplantation therapy without eliciting an immunogenic or teratogenic response. Due to the economic and logistical hurdles associated with this approach, scientists have suggested creating HLA-matched iPSC banks that could provide nonimmunogenic cells for therapy to a large percentage of the human population [225,226]. Another interesting prospect is using genome engineering to generate patient-specific neural cells with corrected disease mutations, which can then be used for autologous cell-based gene therapy.
Conclusion
While reprogramming-based technologies, especially iPSCs, offer enormous potential in studying and dissecting the biology of neurologic diseases in a human-specific model system, several key issues need to be addressed before such a strategy becomes robust, reliable, and reproducible. Future advancements might be targeted toward deriving high quality iPSCs with fewer genomic/epigenomic aberrations and consistent developmental potential, identifying methods to generate specific neural subtypes with high specificity and efficiency, recreating entire regions of the brain in a three-dimensional setting, and using gene-editing tools to correct disease mutations and dissect mechanisms of polygenic disorders. Despite current challenges, the remarkable pace of progress in these fields suggests that reprogramming-based approaches will play increasingly important roles in developing new therapies for rare as well as common neurologic diseases.
Footnotes
Acknowledgment
We would like to thank the following for funding support- National Institute on Deafness and other Communication Disorders (DC012592 to K.K.B.), National Institute of Mental Health (MH102698 to K.K.B.), California Institute for Regenerative Medicine (RB3-02186 to K.K.B., TG201165 to A.N.), the Baxter Family, Norris and Del Webb Foundations (K.K.B.), and the Dorris Neuroscience Center (K.K.B.). We would like to thank Dr. Valentina Lo Sardo, Dr. Jennifer Hazen, William Ferguson, Sohyon Lee, Rachel Tsunemoto, Dr. Sandeepa Dey and Dr. Suvasini Ramasamy for their critical comments on the manuscript.
Author Disclosure Statement
No competing financial interests exist.