
Abstract
A hot issue in current research regarding stem cells for regenerative medicine is the retainment of the stemness and multipotency of stem cell. Endothelial progenitor cells (EPCs) are characterized by an angiogenic switch that induces angiogenesis and further ameliorates the local microenvironment in ischemic organs. This study investigated whether EPCs could modulate the multipotent and differential abilities of mesenchymal stem cells (MSCs) in vitro and in vivo. We established an EPC/MSC indirect Transwell coculture system and then examined the effects of EPCs on the regulation of MSC biological properties in vitro and bone formation in vivo. The in vitro studies showed that cocultured MSCs (coMSCs) display no overt changes in cell morphology but an enhanced MSC phenotype compared with monocultured MSCs (monoMSCs). Our studies regarding the cellular, molecular, and protein characteristics of coMSCs and monoMSCs demonstrated that EPCs greatly promote the proliferation and differentiation potentials of coMSCs under indirect coculture condition. The expression of the pluripotency factors OCT4, SOX2, Nanog, and Klf4 was also upregulated in coMSCs. Furthermore, coMSCs combined with fibrin glue showed improved bone regeneration when used to repair rat alveolar bone defects compared with monoMSC grafts in vivo. This study is the first to demonstrate that EPCs have dynamic roles in maintaining MSC stemness and regulating MSC differentiation potential.
Introduction
T
The microenvironment plays an important role in regulating the biological properties and activity of cells. Stem cells dwell in a special microenvironment in the bone marrow compartment, that is, the stem cell niche, which is composed of supporting cells, blood vessels, and matrix glycoproteins [6,7]. Niche signals between these elements are central to the regulation of stem cell self-renewal and multipotency. In particular, the intimate relationship between stem cells and the blood vessels contributes to tissue homeostasis and repair. As recently reported, MSCs surround the capillaries in a variety of tissues [8,9]. An ancestor of MSCs has been confirmed to be natively associated with the blood vessel wall and, more precisely, to belong to a subset of perivascular cells, that is, pericytes [10]. This finding suggests an essential role for the vascular niche in supporting the biological function of MSCs.
Progenitor cells that are able to initiate postnatal neovascularization, that is, endothelial progenitor cells (EPCs), were first described in 1997 by Asahara et al. [11]. Subsequent studies demonstrated that EPCs are derived from bone marrow, circulate in peripheral blood, and home to sites of ischemic tissues and tumor microenvironments. Phenotypically, EPCs are identified by the surface expression of endothelial markers, such as Von Willebrand factor (vWF), CD31, eNOS, Cdh-5, Tie-2, and Flk-1, and the hematopoietic stem cell (HSC) markers CD133 and CD34 [12,13]. These cells can also be functionally characterized by the uptake of acetylated low-density lipoprotein (acLDL) and Ulex europaeus agglutinin-I (UEA-I). As a progenitor cell in bone marrow, EPCs have the capacity to proliferate, migrate, and differentiate into mature endothelial cells (ECs) that participate in new blood vessel formation [14]. In vascular biology, the roles of EPCs in the angiogenesis and neovascularization of injured and ischemic tissues have been studied extensively both in vitro and in vivo. Because of the outstanding ability of EPCs to promote revascularization, these cells are currently being widely used for proangiogenic applications in tissue engineering. Proper vascularization in implants is known to distribute oxygen and nutrients to the tissue and remove the waste products, consequently improving the local microenvironment, preventing cell death, and promoting host integration [15]. Moreover, EPCs have also demonstrated the promotion of bone regeneration in host bone defects [16], possibly by osteogenic transformation of the cells in an appropriate microenvironment [17] or by inducing recruitment, proliferation, survival, and activity of skeletal progenitors via paracrine factors from the EPCs [18,19]. Hence, both EPCs and MSCs have been considered promising candidates for bone tissue engineering and large segmental bone defect therapies. Recently, some experimental investigations presented evidence that coimplantation of EPCs and MSCs significantly improves bone formation and neovascularization, highlighting the intimate connection of cellular processes in EPCs and MSCs, although the details of the regulatory mechanisms remain unclear [20,21]. The cellular cross talk between EPCs and MSCs involves paracrine mechanisms based on several cytokine factors and direct cell-to-cell communication via gap junctions [22,23]. Furthermore, the new blood vessel network initiated by EPCs in implants not only exerts its traditional blood carrier function but also releases trophic paracrine factors that may indirectly affect MSCs. These molecular and cellular processes constitute a particular microenvironment similar to the bone marrow niche. This microenvironment influences the survival [24,25] and functional activities [21] of MSCs.
Previous studies have confirmed the roles of EPCs as possible components of the stem cell niche and as regulators of MSCs. However, whether their regulatory role is actually involved in maintaining MSC stemness and multipotency is currently undetermined. Indirect coculture systems can clarify the potential paracrine effect of EPCs on MSCs and contribute to elucidation of the cellular mechanisms by which EPCs regulate the biological activity of MSCs during the bone repair process.
Materials and Methods
Isolation, culture, and characterization of MSCs and EPCs
See Supplementary Materials and Methods (Supplementary Data are available online at
MSC/EPC indirect coculture conditions
Passage 10 MSCs and primary EPCs were used for the experiments. These cells were trypsinized and centrifuged and then seeded on six-well plates and Transwell inserts (Costar), respectively. The cocultured cells were maintained in MSC medium. The experimental groups were described as follows: (1) test: MSCs were cocultured with EPCs (coMSCs) that were separated by a Transwell insert (0.4-μm pore size) in a 1:1 ratio unless otherwise stated. MSCs were placed in the lower chambers of a six-well plate, and EPCs were added to the upper chambers of Transwell inserts and (2) control: MSCs were monocultured (monoMSCs) at the same density as the coMSCs.
Flow cytometric analysis
In total, 1 × 105 cells were fixed in 2% formaldehyde solution for 30 min and then were washed twice. Next, the cells were labeled with (1) fluorescein isothiocyanate (PE)-conjugated polyclonal antibody directed against CD34 (ab187284, 1:100; Abcam) or (2) anti-rat monoclonal/polyclonal antibodies directed against CD90 (ab225, 1:500; Abcam), CD105 (ab11414, 1:200; Abcam), CD146 (ab75769, 1:80; Abcam), Sca-1 (ab124688, 1:160; Abcam), CD31 (ab64543, 1:200; Abcam), CD45 (ab33923, 1:50; Abcam), CD14 (ab157312, 1:20; Abcam), vWF (ab6994, 1:100; Abcam), VEGFR-2 (ab9530, 1:50; Abcam), α-SMA (ab119952, 1:100; Abcam), and PDGFR-β (ab69506, 1:100; Abcam) and then stained with anti-IgG-conjugated fluorescein isothiocyanate (FITC/PE). FITC- and PE-conjugated isotype-matched antibodies (Abcam) were used as controls. The labeled cells were then washed twice and resuspended in phosphate-buffered saline (PBS). Analysis was performed using a flow cytometer (FACSAria; BD).
DiI-acLDL uptake
To examine whether MSCs cocultured with EPCs are capable of endothelial differentiation, a DiI-acLDL (1,10-dioctadecyl-3,3,30,30-tetramethylindocarbocyanine-labeled acetylated low-density lipoprotein) staining kit (Invitrogen) was used to detect the ability of ECs to take up and metabolize acLDL. After coMSCs were cultured for 10 days, they were incubated with complete medium containing 2.4 μg/mL DiI-acLDL for 1 h in 5% CO2 at 37°C. Then, the cells were washed thrice with PBS and stained with Hoechst 33342 (Molecular Probes) in the dark for 10 min to counterstain the nuclei. Fluorescent images of the labeled cells were acquired using a fluorescence microscope (Leica).
Cell proliferation and DNA synthesis assays
Colony formation assay
Colony-forming unit fibroblast (CFU-F) assay was performed to assess the capacity and efficiency of cell self-renewal. To evaluate the effect of EPC inoculation density on MSC behavior, the following EPC:MSC ratios were tested: 1:1 (EPCs: 50 cells/cm2 and MSCs: 50 cells/cm2), 10:1 (EPCs: 500 cells/cm2 and MSCs: 50 cells/cm2), and 100:1 (EPCs: 5,000 cells/cm2 and MSCs: 50 cells/cm2). The monoMSCs were seeded at a density of 50 cells/cm2. After cells were cultured for 14 days, they were washed in PBS, fixed in 2% formaldehyde, and stained with 0.1% toluidine blue solution for 30 min. Excess dye was removed with PBS. The colonies consisting of 50 cells or more were scored as CFUs and were counted.
EdU staining
5-Ethynyl-2′-deoxyuridine (EdU) incorporation into cellular DNA method was used to detect DNA synthesis in proliferating cells [26]. The monoMSCs or coMSCs were seeded at a density of 1 × 104 cells/cm2 and cultured for 3 days. Then, the cells were immediately incubated with serum-free α-minimum essential medium (α-MEM) supplemented with 2 μM EdU (Click-iT EdU Imaging Kits; Invitrogen) for 48 h. After the cells were labeled, they were washed in PBS, followed by fixation and permeabilization with PBS containing 2% formaldehyde and 0.5% Triton X-100 for 20 min. After the cells were extensively washed in PBS, they were stained with 10 μM Alexa Fluor 594 azide (Invitrogen) for 20 min and subsequently counterstained with Hoechst 33342 for 10 min to visualize cell nuclei. The cells were washed again, mounted in standard mounting media, and imaged by fluorescence microscopy.
Cell cycle analysis
To further determine whether EPCs can promote the cell cycle progression of MSCs, cell cycle analysis was performed. Cells were seeded at a density of 1 × 105 cells/cm2 and cocultured for 3 days. At subconfluence, coMSCs and monoMSCs were trypsinized with 0.1% trypsin–EDTA solution, collected by centrifugation (5 min at 1,000 rpm), and washed in ice-cold PBS. Thereafter, the cells were fixed in 2 mL ice-cold 70% alcohol and 1 mL ice-cold PBS and incubated at 4°C for a minimum of 30 min. Then, the fixed cells were subjected to flow cytometry (FACSCalibur; Becton Dickinson) for cell cycle analysis.
Cell differentiation assays
Passage 10 MSCs were grown in α-MEM/10% fetal bovine serum (FBS) at a density of 3 × 104 cells/cm2 under mono/coculture conditions. After the cells reached 80% confluence, the proliferative medium was replaced with specialized media according to the desired differentiation.
Osteogenic differentiation
The cultured cells were treated with osteogenic differentiation medium (ODM: 10% FBS/α-MEM, 10 nM dexamethasone, 10 mM β-glycerophosphate, and 50 μM ascorbic acid; Sigma) to induce osteogenic differentiation. The medium was replaced twice a week. After osteogenic differentiation terminated, the cells were washed in PBS and fixed in 2% formaldehyde for 20 min. Calcium accumulation was detected by Alizarin Red staining, and calcium levels were analyzed using a Calcium Colorimetric Assay Kit (BioVision) according to the manufacturer's protocol. Alkaline phosphatase (ALP) staining was performed using a BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime), and ALP enzymatic activity was analyzed using an ALP detection kit (Jiancheng Bioengineering) according to the manufacturer's suggested protocol. Real-time polymerase chain reaction (PCR) and western blot analyses were performed to analyze changes in the expression of osteogenic markers.
Adipogenic differentiation
To analyze the adipogenic differentiation potential of these cells, the proliferative culture medium was replaced with adipogenic differentiation medium (ADM) consisting of 10% FBS/α-MEM, 1 mM dexamethasone, 100 mM indomethacin, 0.5 mM methylisobutylxanthine, and 10 mM insulin (all by Sigma). The medium was changed twice a week. When adipogenic differentiation terminated, the cells were fixed in 2% formaldehyde, washed in PBS, and then stained with 0.2% Oil Red O solution (Sigma) to detect lipid droplets within the cells. To quantify the lipid content in the stained adipocytes, the dye was eluted by adding 100% isopropanol (Invitrogen), and the values of optical densities were measured using an enzyme-linked immunosorbent assay at a wavelength of 510 nm. Real-time PCR and western blot analyses were performed to analyze changes in the expression of adipogenic markers.
RNA isolation, reverse transcription polymerase chain reaction (RT-PCR), and real time polymerase chain reaction (PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen), and complementary DNA (cDNA) was obtained using SuperScript II Reverse Transcriptase (Invitrogen) according to the manufacturer's directions. Reverse transcription polymerase chain reaction (RT-PCR) was performed using an ExTaq polymerase kit (TaKaRa). The thermal cycling conditions were as follows: 5 min for 94°C, followed by 35 cycles of 94°C for 20 s, 54°C–59°C (depending on the primers used) for 20 s, and 62°C for 30 s. After amplification, PCR products were separated by electrophoresis in 1.5% agarose gels, stained with ethidium bromide, and visualized using an ultraviolet illuminator. Real time polymerase chain reaction (PCR) was performed using an ABI 7500 Real-Time PCR System (Applied Biosystems) and SYBR Premix Ex Taq™ (TaKaRa). The thermocycler parameters were as follows: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 57°C–60°C (depending on the primers used) for 1 min. The target genes and primer sequences are shown in Supplementary Table S1. The messenger RNA (mRNA) expression levels of the target genes were compared after normalization with GAPDH, and fold changes relative to monoMSCs were calculated using the comparative threshold cycle (ΔΔCT) method [27].
Western blot analysis
Western blot analysis was performed as previously described [28]. Briefly, the cells were lysed in lysis buffer on ice following a standard procedure and then centrifuged (12,000 rpm, 10 min) at 4°C to collect the cleared total cell protein. The cell protein concentrations were determined using a BCA Protein Assay Kit (Pierce) according to the manufacturer's suggestions. The proteins were subsequently denatured by boiling, resolved using 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred onto PVDF membranes by electrophoretic separation. The membranes were blocked with 5% skimmed milk in TBS-T buffer (25 mm Tris–HCl [pH 8.0], 125 mm NaCl, and 0.1% Tween 20) for 1 h at room temperature, followed by incubation with primary antibodies overnight at 4°C. The primary antibodies included rabbit anti-rat OCT4, SOX2, Nanog, and Klf4 and mouse anti-rat osteocalcin (OCN), bone sialoprotein (BSP), runt-related transcription factor 2 (Runx2), LPL, PPARγ (Abcam) at a dilution range of 1:500–1:1,000. The membranes were washed three times with TBS-T to remove the excess antibodies and then incubated with anti-rabbit/anti-mouse secondary antibodies (1:8,000) conjugated with horseradish peroxidase. The proteins were detected by enhanced chemiluminescence (Beyotime). GAPDH was used as an internal control to normalize the samples.
Cell labeling and implantation
Cell labeling
Before performing an in vivo bone formation assay, cells from donor rats were labeled with a carbocyanine lipid cell membrane tracer (CellTracker CM-DiI; Molecular Probes) in vitro so that the cells could be traced after implantation. After the mono/coMSCs were indirectly precultured with or without EPCs for 7 days, the mono/coMSCs were collected and adjusted to a final concentration of ∼5 × 106 cells. The cells were then labeled with 50 μg/mL CM-DiI according to the manufacturer's recommendation and were prepared to mix with fibrin glue (FG) scaffold.
FG scaffold preparation
Pasteurized FG (Bolheal) was formed by mixing solution A and B in a 1:1 (volume/volume) ratio [22]. When the solutions were mixed at room temperature, a semirigid three-dimensional gel was produced by a typical clotting reaction. In total, 5 × 106 labeled cells were then mixed with solutions A and B in a 1-mL sterile EP tube to produce a polymerized scaffold with suitable dimensions (4 × 3 × 3 mm).
Surgical procedures
Eight-week-old male Sprague Dawley rats were anesthetized by intraperitoneal injection of 2% pentobarbital (40 mg/kg) and then placed supine on the operating table. On two sides of the rat's maxillary alveolar bone, a critical-sized bone defect (4 mm anteroposterior, 3 mm buccolingual, and 3 mm deep) mesial to the first molar was made using a dental drill. Under a sterile environment, the prepared coMSC/FG composition and monoMSC/FG control were implanted immediately and separately into the bilateral defects of the rats. Finally, the gingival incision was sutured to ensure the fastening of scaffold and the healing of the bone defect.
Analysis of bone regeneration
Microcomputed tomography scanning
At 0, 2, 4, and 6 weeks after implantation, the rats (n = 4) were thoroughly anesthetized by intraperitoneal injection with pentobarbital before undergoing microcomputed tomography (μCT) scanning (Siemens Inveon Micro-CT). All living samples were scanned in transverse orientation through the center of the defect site. The following CT settings were used: pixel matrix, 1,024 × 1,024; voxel size, 10 × 10 μm; slice thickness, 10 μm; 80 kV voltage, 500 μA current, and 3,000 ms exposure time. The density of the newly formed bone of implanted sites was evaluated using intensity analyzing software (Inveon Research Workplace V2.0).
Morphological and histological examination
Rats (n = 4) were sacrificed at 6 weeks postoperation for histological analysis. The entire maxilla was isolated carefully to keep the surgical areas intact. The alveolar bone defects with implants were examined grossly and imaged. The target areas for histological observation were then fixed in 4% paraformaldehyde for 48 h and decalcified in 12.5% EDTA (pH 7.0; Sigma) for 3 weeks. The specimens were then washed gently in running water for 24 h. After these steps, the specimens were embedded in paraffin and longitudinally cut into 5-μm sections. The prepared slides were stained with hematoxylin and eosin (HE; Sigma) and Masson's trichrome (MT; Sigma) as recommended by the manufacturer.
Cell tracing and immunohistochemical staining
Rats (n = 4) were sacrificed at 4 weeks postoperation, and specimens were removed, fully decalcified, embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek), rapidly frozen in liquid nitrogen, and sliced into 5-μm sections using Cryocut equipment (Leica CM3050S; Leica). The frozen sections were fixed in 10% phosphate-buffered formalin mixed with cold acetone (Merck) and permeabilized with 0.05% Triton X-100. The sections destined for fluorescence imaging of CM-DiI-labeled cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma), and fluorescence imaging was performed using a fluorescence microscope. The other frozen sections were immunostained for the osteoblast marker Runx2 and the EC-specific marker CD31. Briefly, these sections were washed in PBS and preincubated for 1 h at room temperature with PBS containing 20% normal goat serum (Gibco). Then, the sections were incubated overnight at 4°C with mouse monoclonal anti-Runx2 (ab76956, 1:500; Abcam) or mouse monoclonal anti-CD31 (ab64543, 1:50; Abcam) primary antibody. Next, the sections were incubated with goat anti-mouse IgG (ab150117, 1:1,000; Abcam) as the secondary antibody for 1 h. Images were captured using a confocal laser scanning microscope (Nikon Eclipse Ti Series; Nikon).
Statistical analysis
All experiments were performed at least in triplicate. All data are expressed as the mean ± SD. Statistical analyses were performed with Student's t-test when only two groups were compared. One-way analysis of variance with Tukey's post-hoc test was used when comparing three or more groups. Differences were considered significant at *P < 0.05 and **P < 0.01. SPSS Statistics 19.0 (IBM) was used for statistical processing and analysis.
Results
Characterization of rat bone marrow-derived MSCs and EPCs
To ensure pure MSC populations for subsequent experiments, the differentiation potential and surface immune phenotype of single-cell-derived, clonally expanded MSCs were determined as previously described [29,30]. MSCs that adhered to culture plates displayed a homogeneous population of confluent cells with fibroblastic, spindle, or rhomboid morphology (Fig. 1A). The multiple differentiation potentials of MSCs were determined by osteogenic and adipogenic differentiation assays (Fig. 1B, C). MSCs from passages 2 and 10 were immunopositive for the surface markers CD90, CD105, CD146, and Sca-1 but were negative for the hematopoietic lineage markers CD34, CD45, and CD14. Moreover, compared with passage 2, MSCs of passage 10 had lower expression levels of CD90, CD105, CD146, and Sca-1 (Fig. 1D).

Characterization of mesenchymal stem cells (MSCs).
The mononuclear cell (MNC) fraction was isolated from a bone marrow suspension using the density gradient centrifugation technology. A modified cultural method for EPC culture was used as previously described [31,32]. To avoid possible contamination of EPC cultures by hematopoietic cells and/or ECs, we used a preplating step of 24–48 h to remove all the rapidly adherent MNCs and collected the nonadherent population for further studies. Primary EPC cultures presented a serial change in cell shape similar to that described by Kahler et al. [32]. Figure 2A shows the morphological transformation of the cultured cells from an initial elongated spindle appearance to well-circumscribed monolayers with a typical cobblestone-like morphology. These cells expressed the HSC markers CD133 and CD34 and the endothelial markers CD31, eNOS, and vWF. The endothelial phenotype of EPCs was further confirmed by double-positive fluorescence staining for DiI-acLDL and FITC-UEA-I (Fig. 2B, C). These cell biological characterizations form the fundamental basis for the EPC identification studies published to date [33 –35].
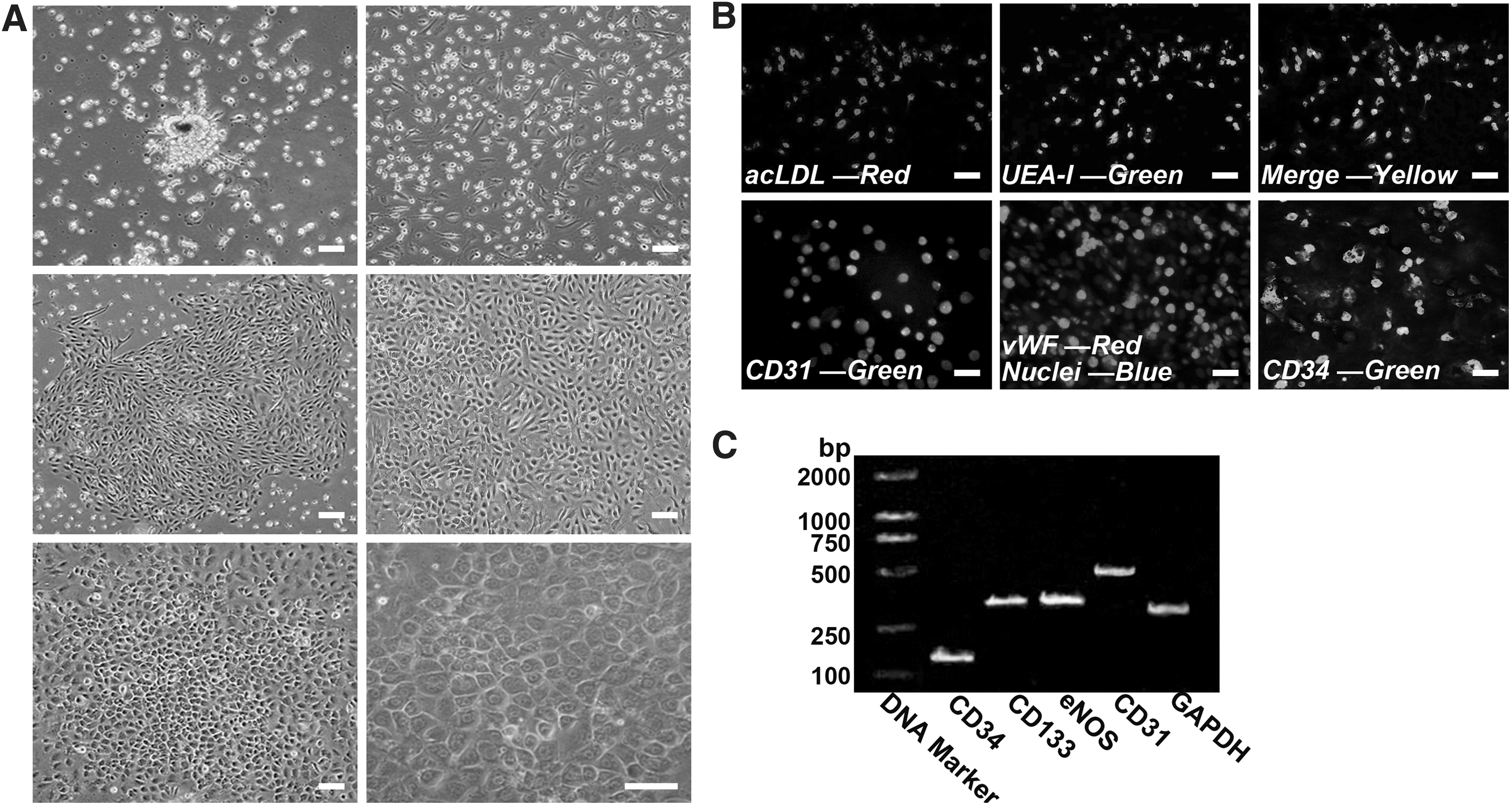
Characterization of endothelial progenitor cells (EPCs).
Effects of EPCs on cell fate determination of MSCs
The microenvironment plays crucial roles in stem cell differentiation partly through soluble factors secreted from neighboring cells [36]. To determine whether EPC indirect contact can change the biological features of MSCs or whether the EPC niche can induce MSC differentiation into endothelial-like cells [37] or pericyte-like cells [38], we introduced an indirect coculture system into our study and analyzed the biological behavior of MSCs under this coculture condition. We found no or minimal observable morphological changes in the coMSCs compared with monoMSCs after 10 days of indirect coculture (Fig. 3A). Then, coMSCs were fluorescently labeled with DiI-acLDL, and their uptake behavior was compared to that of the positive control EPCs. The results indicated that the fluorescent dyes were rapidly incorporated into the EPCs but were not present in the cellular compartments of coMSCs (Fig. 3B). To validate whether indirect coculture with EPCs can trigger endothelial differentiation or advocate pericyte-like cell differentiation for MSCs, we compared surface markers for coMSCs and monoMSCs by flow cytometric analysis. The expression levels of the endothelial-specific markers CD31 and vWF and the pericyte surface markers α-SMA [39] and PDGFR-β [40] were very low in the coMSCs, while MSC surface markers CD90 and CD105 are expressed at high levels in the coMSC groups, even more than those in the monoMSC groups (Fig. 3C). In addition, gene expression analysis of coMSCs indicated negative gene expression profiles for EC surface markers (CD31, eNOS, and vWF) and pericyte surface markers (NG2 [41], α-SMA, and PDGFR-β) but positive gene expression profiles for MSC markers, such as CD90 and CD105 (Fig. 3D).

The effects of EPCs on the MSC phenotype under indirect coculture conditions.
Effects of EPCs on the proliferative ability of MSCs
A series of cell proliferation assays were used to determine whether the paracrine factors from EPCs have an impact on the proliferative ability of MSCs. The self-renewal efficiency of the cells was assessed by CFU-F assay. We found that both CFU-F numbers and the mean colony size were broadly larger in coMSCs than in the control group (Fig. 4A). Notably, EPC enhanced MSC colony formation in a density-dependent manner. EdU incorporation assay after coculture for 3 days demonstrated that EPCs had a significant effect on cellular DNA synthesis in proliferating MSCs. The percentages of proliferating coMSCs and monoMSCs were 71.49% (±13.04%) and 33.61% (±9.99%), respectively (Fig. 4B). Furthermore, cell cycle analysis by flow cytometry indicated a higher percentage of coMSCs in S and G2/M phases, while a lower percentage of coMSCs in G0/G1 phase compared with the control cells (Fig. 4C). This result suggested that under indirect coculture conditions, EPCs accelerated the normal quiescent MSCs engaging in S and G2/M phases, while the vast majority of monoMSCs remained in G0/G1 phase of the cell cycle. Taken together, these data revealed that the EPC microenvironment greatly influenced the proliferative ability of MSCs.
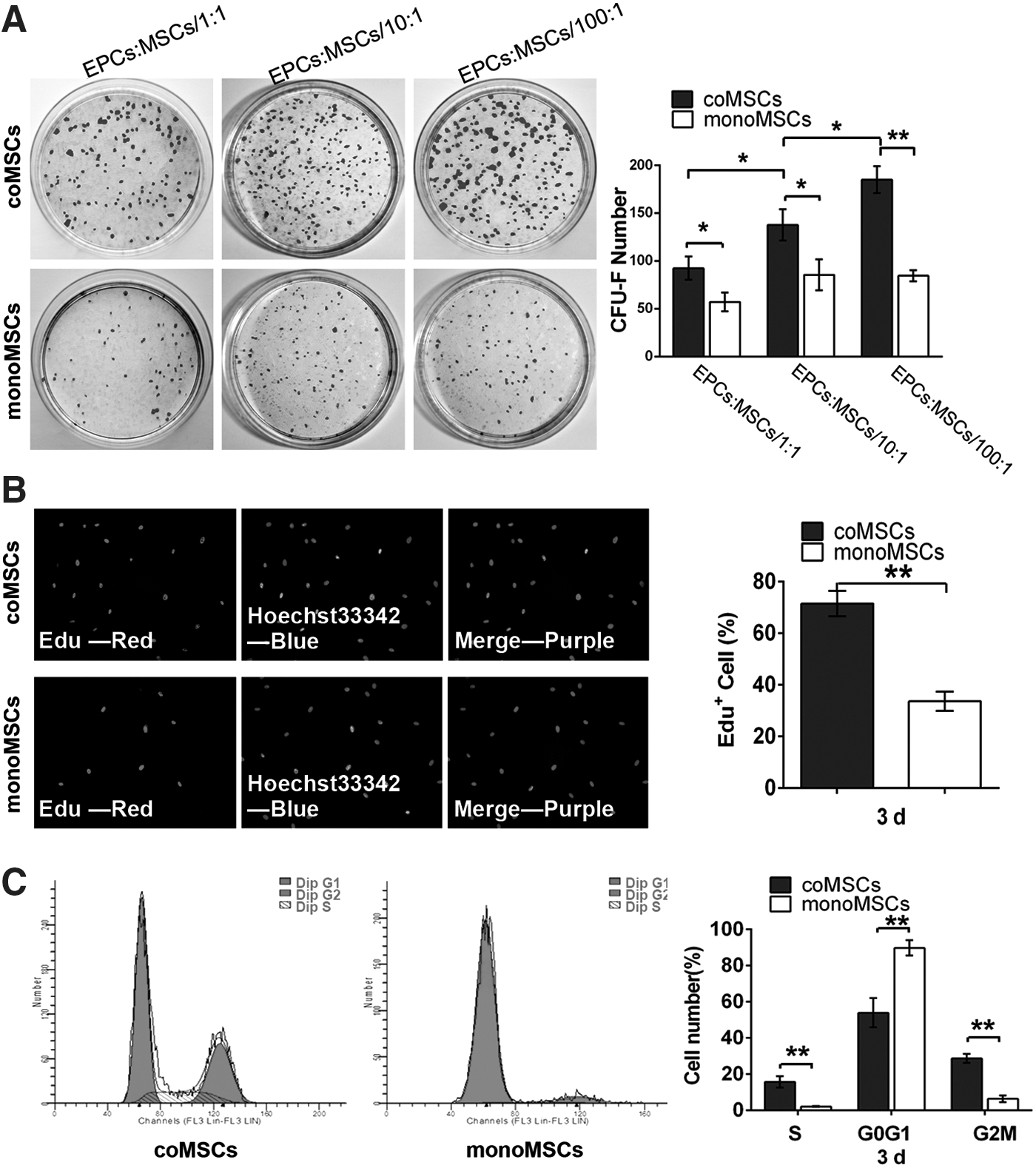
The effects of EPCs on the proliferative ability of MSCs after indirect cell–cell contact.
Increased multipotent gene expression after coculture with EPCs
Maintenance of the multipotent state of multipotent stem cells is known to be associated with a characteristic expression pattern of the pluripotency factors OCT4, SOX2, Nanog, and Klf4 [42,43]. Over many self-renewing divisions in vitro, stem cells may gradually lose their multipotent properties [23]. In our study, RT-PCR results demonstrated high positive expression of OCT4, SOX2, Nanog, and Klf4 genes in monoMSCs at passage 2. However, when the cells were expanded to passage 10 in vitro, the expression levels of these genes decreased, with much lower levels of OCT4 and SOX2 (Fig. 5A). Then, the mRNA expression levels of these genes were examined for the two cell groups at passage 10 using real-time PCR. After being cocultured in the Transwell system for 3, 7, or 10 days, the levels of gene expression in the coMSC groups remained relatively high level compared with those in the monoMSC groups (Fig. 5B). To further confirm that the EPC microenvironment affected the maintenance of MSC pluripotency, we compared the protein levels of the four analyzed genes in these two distinct groups using western blot analysis. As shown in Fig. 5C, the protein expression profiles showed increasing trends similar to the mRNA expression profiles of the examined pluripotency-associated genes. These data indicated that MSC pluripotency decreased after continuous culture and passage in vitro, while EPCs showed a considerable antidecline effect on MSC pluripotency-associated gene levels through indirect cell–cell interactions.

The expression levels of OCT4, SOX2, Nanog, and Klf4 on MSCs were regulated by indirect coculture with EPCs.
Osteogenic and adipogenic induction potentials
To examine the effect of EPCs on MSC differentiation capacity, MSCs were cultured in osteogenic and adipogenic induction media after exposure to an indirect coculture system. The results showed that after being cultured in osteoblastic induction cocktails, coMSCs had a higher osteoblastic differentiation potential than did monoMSCs, which was evidenced by ALP staining and Alizarin Red staining (Fig. 6A, B). Upregulated ALP activity and increased mineralized nodule accumulation were found in coMSCs compared with monoMSCs (Fig. 6C, D). As shown in Fig. 6E, the transcription levels of the osteoblastic marker genes OCN, BSP, and Runx2 were all significantly upregulated in the coMSC groups compared with the monoMSC groups. This upregulation of the osteogenic differentiation capacity of MSCs caused by EPCs was further confirmed by western blot analysis. The results indicated that the OCN, BSP, and Runx2 protein levels were increased in the coMSC groups compared with the monoMSC groups after 14 and 21 days of osteogenic differentiation (Fig. 6F).
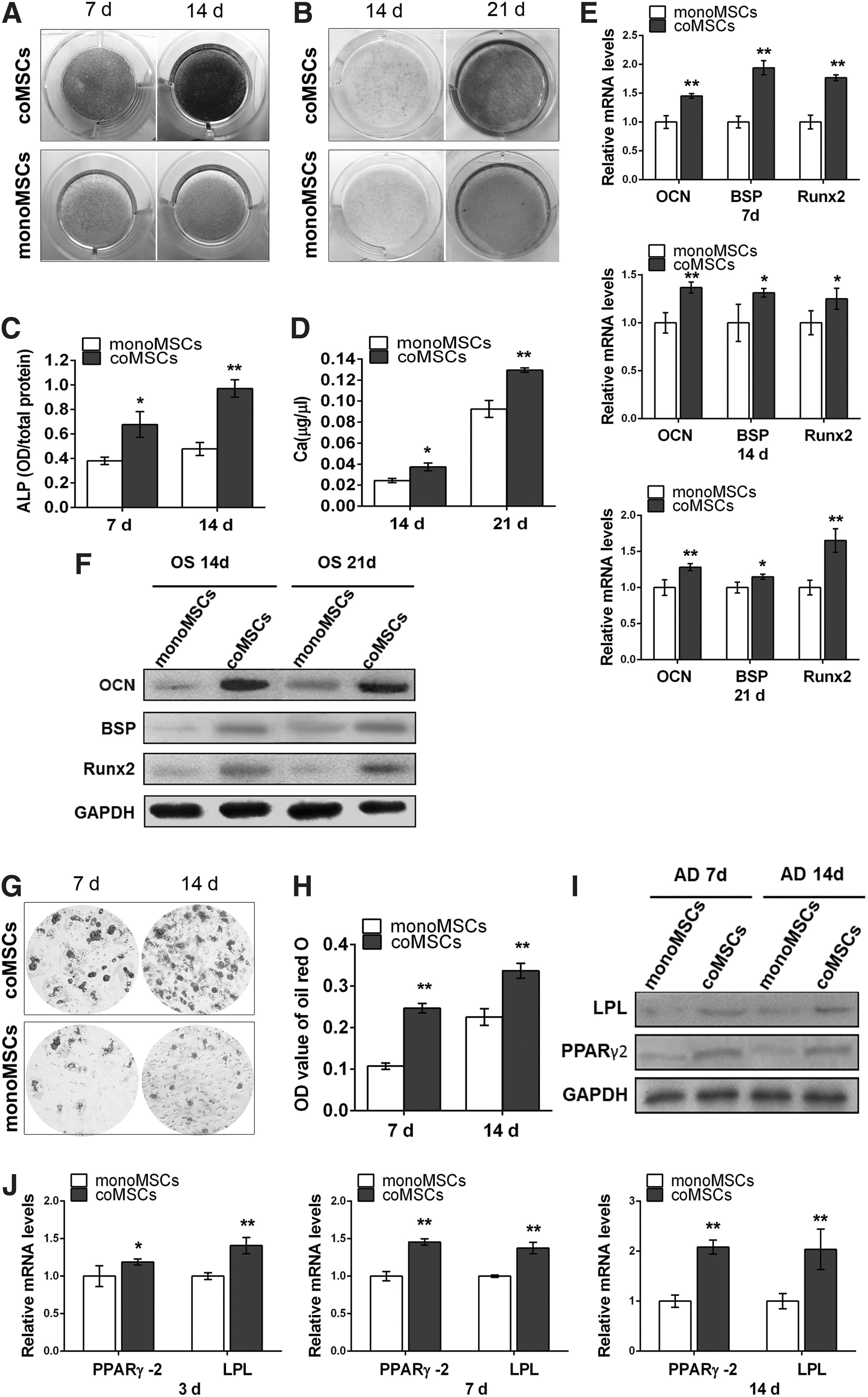
The effect of EPCs on the osteogenic and adipogenic differentiation capacities of MSCs in vitro after being exposed to an indirect coculture system.
We next detected the effects of EPCs on the adipogenic differentiation capacity of MSCs. After adipogenic induction, the adipogenic differentiation capacity of coMSCs increased as confirmed by Oil Red O staining and staining quantification (Fig. 6G, H). Both protein and mRNA expression levels of adipogenic marker genes, that is, LPL and PPARγ2, were examined, and the results demonstrated a relatively higher level in the test groups compared with the control groups (Fig. 6I, J). Therefore, EPCs have positive effects on MSC adipogenic and osteogenic differentiation.
In vivo alveolar bone regeneration
All rats exhibited good health without any inflammation or complications during the postoperative period. The healing conditions of the maxillary alveolar bone defects in the two groups were grossly observed, and the samples were evaluated by μCT analysis and histological examination.
μCT analysis
At 4 and 6 weeks postoperation, the coMSC/FG group exhibited larger areas of calcification and rapid healing of the bone defects compared with the monoMSC/FG group, which exhibited smaller areas of bone formation and a slower healing rate (Fig. 7A). To quantify the alveolar bone repair ability more precisely, the bone mineral density (BMD) of the newly formed bone in defect sites was calculated. The results indicated that significantly greater BMD values were found in the coMSC/FG group, whereas lower BMD values was found in the control groups (Fig. 7B).
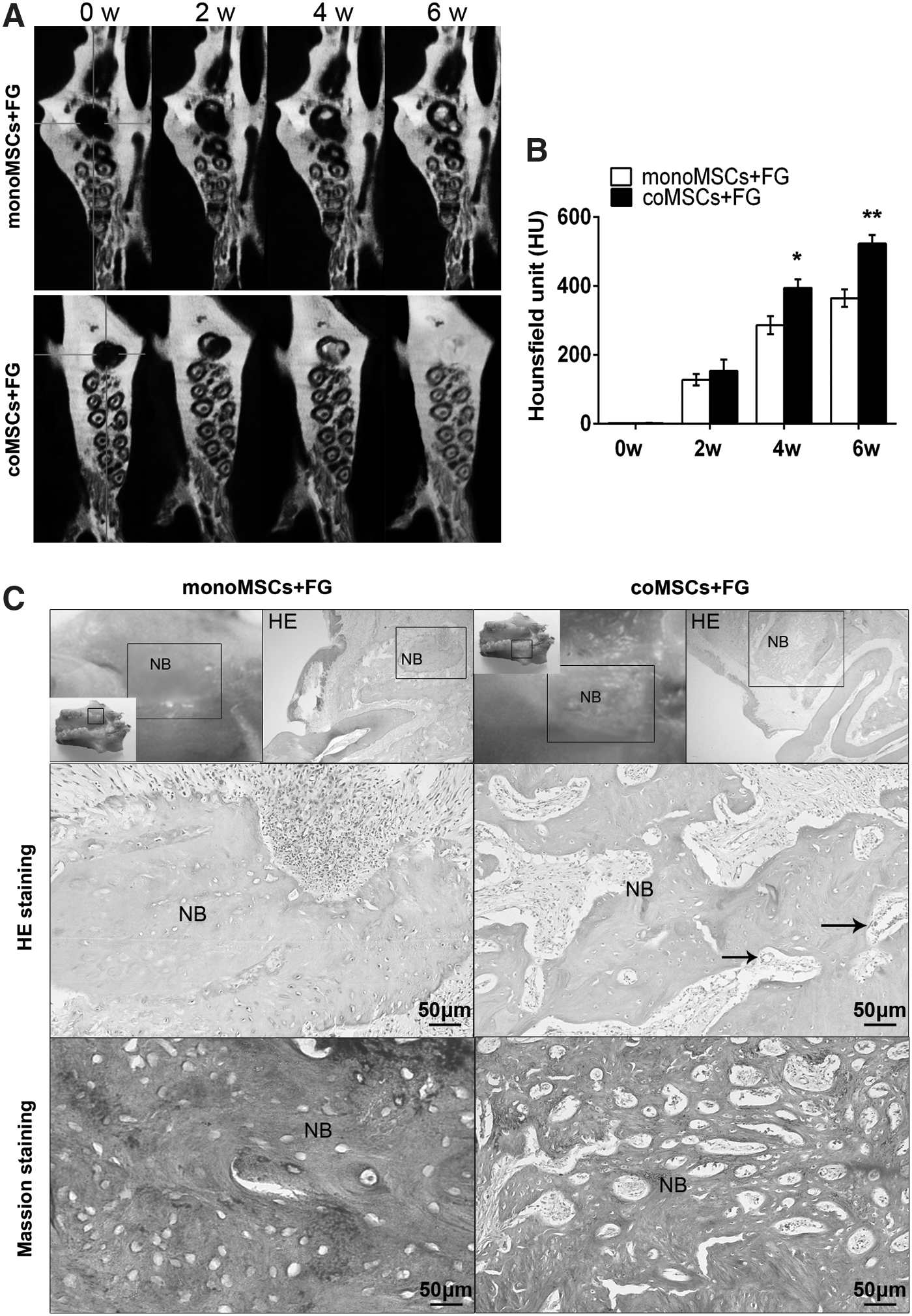
Rat alveolar bone regeneration after cells were implanted into the bone defect.
Morphological and histological analyses
The 36 mm3 bone defects in adult rats were larger than previously reported critical-sized defects [44,45], which means that these defects are unable to heal by themselves. At 6 weeks postimplantation, bone tissue regeneration was observed (Fig. 7C). Gross observations showed only slight bone reconstruction and obvious defects remaining in the monoMSC/FG group. In contrast, the bone defects in the coMSC/FG group were nearly completely repaired. The borders of the bone defects in the test groups were indistinct and undistinguishable from the primary alveolar bone compared with the control groups. Histological results showed that the defects in the control group were only filled with osteoblasts or osteogenetic cells and extracellular matrix. However, in the coMSC/FG group, the defect sites presented better healing conditions, with a large amount of bone marrow stromal cells and osteogenetic cells in the center of the defects, contributing to new bone regeneration. The trabecular pattern in the new bone of the coMSC/FG group was clear and had a high density. More importantly, blood vessel structures were also observed in the area of the newly formed bone.
Fluorescence staining analysis
Four weeks after transplantation, CM-DiI-positive cells were observed in the newly formed tissue areas, and the cell tracer results showed a greater number of CM-DiI-positive cells in the coMSC/FG groups than in the monoMSC/FG groups, indicating that the survival of the EPC-pretreated MSCs was higher than that of the non-pretreated groups (Fig. 8A, B). At 4 weeks postoperation, the repaired bone tissues were processed for immunohistochemical staining. The immunohistochemical results demonstrated larger amounts of CM-DiI and Runx2 double-positive cells in the defect areas from the coMSC/FG sections, which verified that more newly formed bone tissues were regenerated by implanted coMSCs compared with monoMSCs (Fig. 7C, D). We further analyzed the angiogenic function of EPC-pretreated MSCs in newly formed tissue at bone defect areas. As shown in Fig. 8E and F, the numbers of CD31(+)/CM-DiI(−) cells and vascular structures significantly increased in the EPC-pretreated coMSC group compared with the monoMSC group. This result suggested that EPC-pretreated MSCs could markedly improve angiogenesis in bone defects not by promoting neovascularization of the implanted MSCs but by stimulating the angiogenic function of the surrounding angiogenic cells.
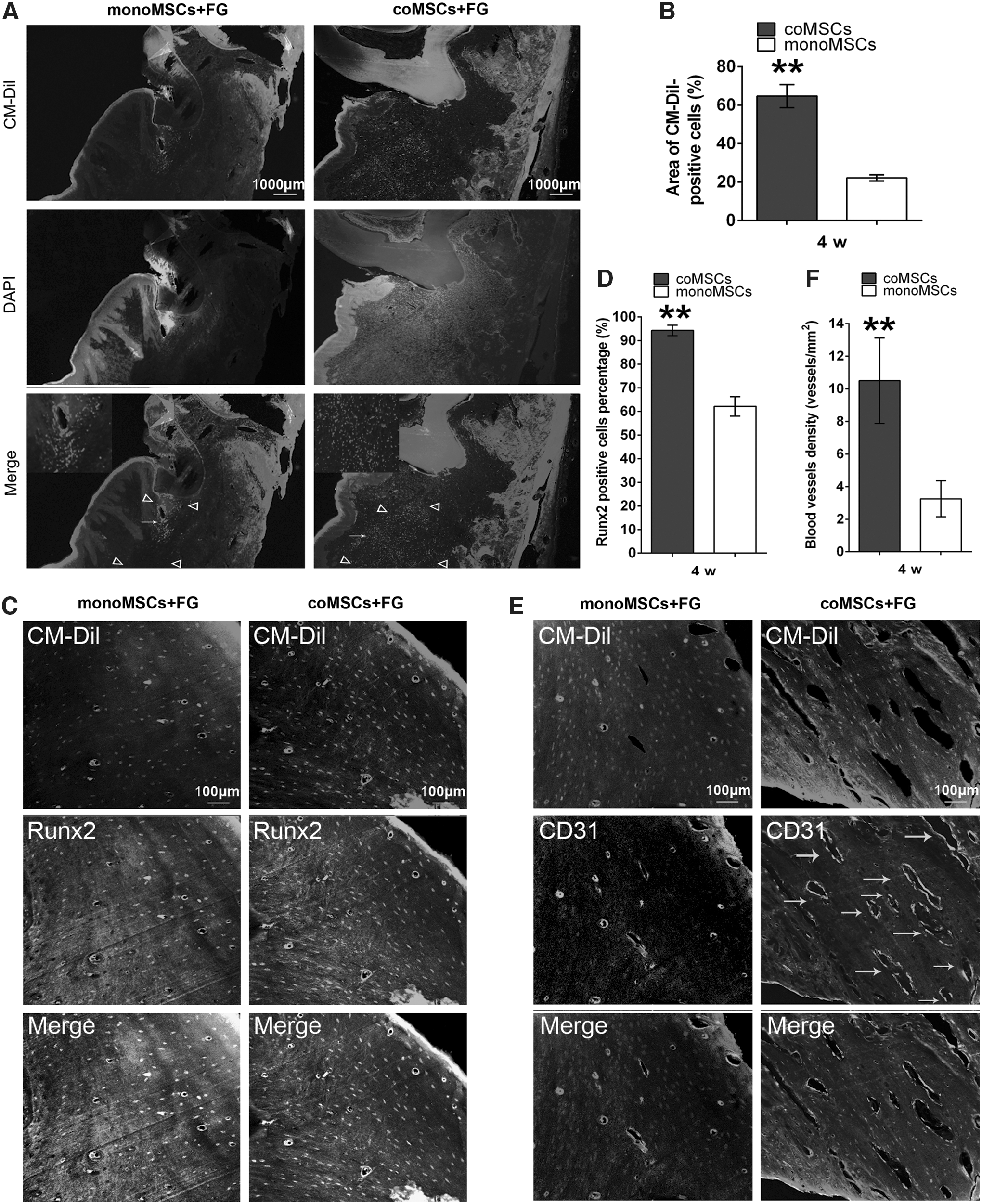
Fluorescence staining analysis of the newly formed tissue.
Discussion
In the past decade, the discovery of EPCs has opened a new research field that may have a potential value and practical application in tissue engineering and angiogenic therapy. However, current controversies regarding whether and how we can obtain true EPCs in in vitro culture have limited their use [32,46,47]. In this study, we successfully isolated an EPC population from rat bone marrow by a preplating step culture method. Based on the immunocytochemical and PCR results, we demonstrated that these cobblestone-like cells obtained from the bone marrow MNC fraction presented characteristic markers and abilities that are relevant to the biological properties of EPCs. We also investigated whether the classical MSC culture medium or ODM and ADM can affect the EPC phenotype. The results indicated that these media might accelerate the transition of early EPCs to late EPCs but might not induce osteogenic or adipogenic differentiation for EPCs in differentiation media (Supplementary Data).
Niches play important roles in cell fate determination. The ability of MSCs to differentiate into multiple lineages is controlled by various stimulators with specific niches [48,49]. Both EPCs and MSCs reside in the bone marrow and share their origin in the bone marrow niche through interactions with each other [50]. Previous studies found that direct EPC–MSC contact can induce an endothelial phenotype and angiogenesis in MSCs [37] and also can advocate MSC differentiation toward a pericyte-like phenotype in vitro [38]. Another interesting report has shown that endothelial differentiation of MSCs only occurred in MSC–EC direct coculture conditions but not in indirect coculture conditions [51]. In the present study, we established an EPC/MSC indirect Transwell coculture system to only allow soluble factor penetration and demonstrated for the first time that coMSCs can maintain their initial biological properties as shown by continuous expression of stem cell genes but negative expression of an endothelial phenotype and pericyte surface markers. Our data demonstrated that the soluble factors released from EPCs do not induce endothelial differentiation or pericyte-like differentiation of MSCs. These findings confirmed that cell–cell contact signaling is the key determinant of the fate of MSCs, while the paracrine signals generated by EPCs seem to be insufficient to trigger this process [51,52]. More interestingly, Fig. 3C shows increased CD90 and CD105 expression in the coMSCs, which partly indicates positive regulation of MSC stemness by EPCs.
Stem cell stemness is described as self-renewal capacity and multipotency. Maintaining stemness in vitro and in vivo has been a large challenge in the field of regenerative medicine. The present study is an attempt to investigate whether the EPC niche can affect the stemness and pluripotency of MSCs. In our preliminary experiments, we monitored the stemness and differentiation capacities of early passage (P1 and P2) MSCs under indirect coculture with EPCs. We observed that cocultured early passage MSCs had higher, but not significantly different, proliferation and differentiation capabilities compared with those of monocultured cells (data not shown). This finding suggested that EPCs only slightly affect the upregulation of the high pluripotency of early passage MSCs. However, the expected positive ability of EPCs to maintain MSC stemness and differentiation potential was enhanced at prolonged passages of MSCs. Our data demonstrated that passage 10 MSCs exhibited higher proliferation and differentiation capabilities via indirect coculture with EPCs. We demonstrated that indirect contact with EPCs promotes MSC entry into the cell cycle and increases MSC self-renewal potential. Furthermore, the higher differentiation capabilities of coMSCs were confirmed by a significant upregulation of osteogenic and adipogenic differentiation during exposure to induction medium. In our model culture system, EPCs may activate MSC proliferation and differentiation through a paracrine mechanism. EPCs have been shown to produce multiple growth factors that have paracrine effects, such as VEGF, insulin-like growth factor I (IGF-I), stromal cell-derived factor 1 (SDF-1), monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein 1a, and PDGF [53,54]. Various growth factors, that is, VEGF, IGF, and PDGF, can promote cell proliferation, regulate cell survival, and maintain normal cell biological motility [55]. VEGF and its receptor have been reported to exert a mitogenic effect on MSCs [56], thereby activating cells and inducing their proliferation. Furthermore, a number of studies have demonstrated that VEGF has other important roles in stimulating cell survival, promoting osteogenic differentiation of stem cells, and recruiting and migrating major bone-forming cells [57 –59]. Other authors have reported that PDGF isoforms and IGFs enhance the proliferation and differentiation of osteoprogenitors [55]. Taken together, these findings imply that indirect contact with EPCs might partly modulate MSC proliferation and multilineage differentiation capabilities through a variety of paracrine signals. EPCs are known to be able to create a local nutritional support environment primarily caused by an indirect effect from producing various growth factors and a direct effect by initiating neovascularization activity. This well-described supporting effect of EPCs may be another factor that sustains the self-renewal and multilineage differentiation potentials of MSCs in our experiments.
In microenvironments of EPC indirect coculture systems, the pluripotency factors OCT4, SOX2, Nanog, and Klf4 were found to be significantly elevated within the coMSCs compared with monoMSCs. These four genes were confirmed to act as core regulators of cell stemness [60,61]. The downregulation of these transcription factors leads to the gradual loss of pluripotency and self-renewal and to the beginning of terminal differentiation [61]. This phenomenon was also found in our result; lower gene levels led to decreases in the self-renewal capacity and the multipotency of monoMSCs. All these results suggest that the high proliferation and differentiation potentials of coMSCs may be mediated, at least in part, by maintaining cell stemness in connection with the higher expression of these pluripotency genes. Therefore, we reasoned that the EPC microenvironment could considerably repress the declining pluripotency of MSCs in vitro.
Based on the in vitro studies, we further investigated and confirmed the effect of coMSCs combined with FG on rat alveolar bone regeneration by means of gross morphological observation and μCT analysis. HE- and MT-stained slices of repaired tissues showed that the coMSC/FG groups could not only stimulate regeneration of new bone but also regenerate a larger amount of cancellous bone defect, while only osteoblasts or osteogenetic cells and extracellular matrix were observed in the monoMSC/FG groups. In addition, CM-DiI and Runx2 double fluorescence-labeled implanted cell numbers increased in the defect areas in the coMSC/FG group, which suggested that indirect EPC contact could enhance MSC promotion of bone regeneration. Our in vivo data together with the in vitro results indicated that the coMSCs with higher self-renewal and differentiation potentials could greatly enhance high-quality alveolar bone regeneration. More specifically, in the fluorescence staining result, we also found that the survival of EPC-pretreated MSCs in the newly formed tissues was higher than that of the monoMSC group, which is another probable reason that indirect EPC contact enhances MSC promotion of alveolar bone regeneration. Furthermore, we observed that the number of CD31(+)/CM-DiI(−) cells and vascular structures in the newly formed tissue increased in the coMSC/FG group compared with monoMSC/FG group. This observation indicated the higher efficiency of indirect EPC contact in prompting coMSCs to release paracrine signals that stimulate the angiogenesis of local tissues. Numerous studies have demonstrated that EPCs provide both instructive (release of proangiogenic cytokines) and structural (vessel incorporation and stabilization) functions that contribute to neoangiogenesis initiation [12,62]. EPC-induced neoangiogenesis of tissue engineering bone is very important for vascularization during the early period of bone healing. Recent reports suggested that EPCs might be responsible for turning on the angiogenic switch in neoangiogenesis [63,64]. Therefore, from the above observations, we speculated that the paracrine signal released from EPCs might influence the biological function of MSCs and indirectly turn on the angiogenic switch in the regenerated bone, thus effectively enhancing alveolar bone healing. However, additional experiments are needed to further verify this hypothesis in vitro and in vivo.
Conclusion
Several lines of evidence in our study suggest that EPCs, a minor subpopulation of the MNC fraction in bone marrow, play important roles in maintaining the stemness and enhancing the differentiation potential of MSCs by indirect coculture. Moreover, higher survival of implanted cells and enhanced capacity of bone tissue regeneration in the repaired area were observed in the coMSC/FG groups, which indicated that indirect EPC contact greatly improves MSC promotion of alveolar bone regeneration with FG scaffold. More interestingly, paracrine signaling from EPCs might influence the biological function of MSCs before implantation and then indirectly turn on the angiogenic switch in the repaired area as partly demonstrated by CD31 immunofluorescence staining assays. Although further studies are needed, all these results suggested that an important paracrine signaling occurs between EPCs and MSCs by indirect contact. Further insights into this paracrine signaling mechanism will be a valuable tool for understanding cell and tissue dynamics in vitro, ultimately leading to the design of more rational tissue engineering strategies for in vivo applications.
Footnotes
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (no. 81170982 and no. 81100627) and the National High Technology Research and Development Program of China (no. 2013AA032201).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.