
Abstract
Despite many attempts, true bovine embryonic stem cells (bESC) still remain elusive. The WNT pathway has been associated with stem cell control in vertebrates and its role in pluripotency maintenance has been proven for several mammalian species, including rodents and primates. Thus, we have aimed to investigate the effect of WNT activation on pluripotency marker gene expression in the inner cell mass (ICM) and the trophectoderm (TE) and to study the derivation potential of primary bESC lines from blastocysts obtained in the presence of the glycogen synthase kinase 3 inhibitor (GSK3i). WNT activity clearly exerted a positive effect on pluripotency gene expression in developing bovine embryos, manifested by upregulation of OCT4, NANOG, REX1, SOX2, c-MYC, and KLF4 in the ICM and downregulation of CDX2 in the TE. However, the prolonged exposition of preimplantation embryos to the GSK3i resulted in reduced potential to form primary ESC-like colonies. The results of bESC derivation experiments allowed us to speculate that the derived cell lines may share features of both naïve and primed ESCs. Similar to mouse epiblast stem cells and human ESCs, the derived lines grew as flat monolayer colonies intolerant to passaging as single cells. JAK/STAT signaling was indispensable for proper colony formation and proliferation, yet LIF alone was inefficient to support self-renewal. Concomitant with the naïve state of mouse ESCs, WNT activity supported by LIF had beneficial effects on cell culture propagation, survival after passage, morphology, and pluripotency-related marker gene expression. Moreover, colonies derived in the presence of LIF and GSK3i maintained KLF4 transcription over several passages, whereas EpiSCs virtually do not express KLF4.
Introduction
A
The existing ungulate ESC lines exhibit several deficiencies ranging from short life in culture, inability to contribute to the chimeras, to the failure to achieve germline transmission. Undoubtedly, mammals share a mechanism controlling early development; however, the noted differences in embryogenesis, gene expression patterns, and lineage specification create problems still to be overcome. With our growing understanding of these processes, it is now becoming evident that the studies should concentrate on targeted elimination of differentiation-inducing signaling.
In early development, signaling pathways are critical to support cell-to-cell communication and generate a normally patterned embryo. Our study was directed to investigate the effect of WNT signaling during the preimplantation period of bovine development, aiming to explore the subsequent potential of primary inner cell mass (ICM) outgrowths to support bESC maintenance in culture. From studies done on other species, such as mouse, human, and rat, it may be concluded that the WNT pathway most likely plays the pivotal role in pluripotency maintenance in mammals.
At the early stages of embryonic development, concomitant with the increasing number of cells, two successive differentiation events lead to the segregation of the committed lineages. The first begins at the morula stage and results in the formation of the ICM and the trophectoderm (TE). The establishment of the ICM is supported by the interactions of transcription factors, such as OCT4, NANOG, SALL4, and SOX2 (review in Ng and Surani [2]). The second round of segregation divides the ICM into the epiblast (nascent embryo proper) and the primitive endoderm (PrE). The subsequent formation of the distinctive embryonic lineages is governed by the activation of the specific signaling pathways.
In murine embryos, mitogen-activated protein kinase (MEK)/extracellular signal-related kinase ERK1/2 signaling through the FGF receptor has been implicated in trophoblast differentiation and diploid trophoblast proliferation [3 –6]. Pharmacological inhibition of the MEK/ERK pathway shields epiblast cells from inductive FGF signals and blocks their differentiation. In vivo, it prevents the epiblast from diverting into extraembryonic lineages, and in vitro may be applied to promote pluripotency in embryo-derived cell lines. The reduction of ERK activity has been shown to enhance the efficiency of rodent ESC derivation by supporting the retention of OCT4-positive epiblast cells during the initial outgrowth formation [7]. Yet, ERK pathway inhibition alone is insufficient to support undifferentiated ESCs in culture, unless the cells are maintained at high densities or provided with LIF [8].
The maintenance of pluripotency in mouse and human ESCs is, however, possible through activation of WNT signaling through selected inhibition of glycogen synthase kinase 3 (GSK3) [9]. Ying et al. [8] showed that the addition of a selective GSK3 inhibitor (GSK3i, CHIR99021) restored viability and allowed efficient expansion of undifferentiated mouse ESCs in the absence of ERK signaling. Based on these findings, Buehr et al. [10] equipped with two sets of chemically defined signaling pathway inhibitors were able to establish stable rat ESC lines. The inhibition of GSK3 plays a pivotal role in these systems. The 2i system relies only on GSK3 and MEK/ERK pathway inhibition. In the 3i system, the restricted activity of GSK3 is supported by the inhibition of both FGF and MEK/ERK signaling. This system requires LIF supplementation necessary to suppress differentiation and maintain viability and growth rate when FGF/MEK-ERK signaling is reduced. Eventually, the 2i conditions proved optimal to derive rat ESCs since it produced equivalent numbers of male and female cell lines and chromosomal abnormalities were not observed.
GSK3 inactivation leads to stabilization of intracellular β-catenin. It is subsequently transported to the nucleus where it interacts with transcription factors such as lymphoid enhancer-binding factor1/T-cell-specific transcription factor (LEF1/TCF). As a result, the WNT pathway remains active and stimulates target genes important for directing developmental processes, such as pluripotency maintenance, embryonic axis formation, cell migration during gastrulation, and neurulation.
WNT signaling commonly regulates cell proliferation—WNT knockout phenotypes may often be explained by loss of cell proliferation (detailed review in Logan and Nusse [11]). Some WNT target genes encode components of cell junctions, such as L1-CAM, Nr-Cam, and connexin 43 (review in Gavert and Ben-Ze'ev [12] and Heuberger and Birchmeier [13]). WNT signaling might regulate cell adhesion as β-catenin is the main component of adherens junctions, linking the adhesive glycoprotein (E-cadherin) to actin filaments [14]. In homozygous E-cadherin (−/−) mutant mouse embryos, the adhesive cells of the morula stage dissociated shortly after compaction and their morphological polarization was disrupted [15]. Since all of the above are crucial for proper preimplantation development, evidently a strong link exists between β-catenin/WNT signaling and early development.
The establishment of stable ESC lines relies on a complex network of transcription factors and signaling pathways. Cytokines play important roles in maintaining the proliferative state of pluripotent stem cells; however, it has been shown that LIF and bone morphogenetic protein (BMP) are dispensable for mouse ESC maintenance as the 2i system proved to be sufficient to maintain the naïve state of mESCs in the absence of extrinsic signals [8].
Pluripotent cell lines may exist in two states: naïve ESCs (equivalent to cells from the early epiblasts) and primed EpiSCs (developmentally more advanced epiblast stem cells). The genome of the naïve ESCs displays an unusual open chromatin state at the genome-wide level and possesses a minimum of repressive epigenetic marks [16]. EpiSCs, on the contrary, have already activated the epigenetic machinery that supports differentiation toward the embryonic lineages [17,18]. Transition from the naïve to the primed state therefore represents a pivotal event in cellular differentiation.
Studies by ten Berge et al. [19] showed that paracrine and autocrine WNT signals were essential for self-renewal of mESCs and prevented differentiation toward the EpiSC state. WNT proteins in combination with LIF were sufficient to support ESC self-renewal and allowed ESC derivation from nonpermissive mouse strains [19]. Although both mouse and human ESCs are derived from blastocyst stage embryos, they have different biological properties. Studies suggest that the pluripotent state of hESCs corresponds to mouse EpiSCs, which includes flattened morphology, intolerance to passaging as single cells, dependence on TGFβ/Activin signaling [20], inactivation of X-chromosome in most of the female cell lines derived [21], and higher susceptibility to differentiate into primordial germ cells in the presence of BMP4 [22].
Relying on the scarce data on bovine and other ungulate ESC-like-derived cell lines, it may be speculated that these cells are poised to differentiation in a similar manner to EpiSCs. It has been shown that bovine ICM primary outgrowths expressed transcripts for receptors belonging to the TGFβ pathways (BMP4 receptor gene), Activin A, WNT (FZD receptor gene), and FGF pathway (FGF2 receptor), all present in the EpiSC [23]. Despite the species-specific differences, WNT seems to be a common pathway as studies have indicated that GSK3 inhibition was alone sufficient to support self-renewal of both mESCs and hESCs [9]. The ectopic induction of OCT4, KLF4, and KLF2 combined with LIF, GSK3, and MEK/ERK signaling inhibition enabled hESCs to be rewired into a more immature state, sharing features associated with mESCs [24].
Learning from the existing data, we believe that the initial steps of ICM outgrowth formation and the gene expression profiles retained within the ICM itself are critical for the successful derivation of stable ESC lines. Although some literature evidence exists that the WNT pathway is present and active in bovine preimplantation embryos, the actual correlation between the WNT activity and pluripotency potential of the ICM and ESC in cattle has not been looked at.
The results of our previous findings have clearly indicated the existence of lineage-specific differences in the expression patterns of key pluripotency marker genes in bovine ICM and TE, both at the mRNA and protein levels [25]. Knowing the important role of the WNT pathway for mouse and human ESC maintenance, we have aimed to study the effect of GSK3 inhibition on bovine ICM and TE marker gene expression and investigate the subsequent derivation potential of primary ESC lines. We believe that before testing the ESC culture conditions that have proven successful for the model species and switching to the combination of the inhibitors, it is important to understand the roles of the crucial signaling pathways independent of one another.
Materials and Methods
All procedures were performed in accordance with the guidelines of the National Ethics Commission for Animal Research (Ministry of Science and Higher Education, Poland). The study was approved by the Local Ethics Commission (Z.E. Madeja personal licence permit number: 142/2010).
Unless stated otherwise, all reagents were supplied by Sigma Aldrich.
In vitro production of bovine embryos
The media were made based on sterile embryo-tested water (Gibco, Life Technologies). The in vitro production of bovine embryo procedures were done as previously described by Madeja et al. [25], and the in vitro embryo culture (IVC) system was based on Holm et al. [26].
Bovine ovaries were obtained from the local slaughterhouse and within 1.5–2 h transported to the laboratory. Cumulus–oocyte complexes (COCs) were aspirated from 2- to 6-mm follicles and selected according to de Loos et al. [27]. COCs were matured in vitro for 24 h in TCM199 medium supplemented with 1 mg/mL fatty acid-free bovine serum albumin (fafBSA), 0.05 mg/mL gentamicin, 0.022 mg/mL Na-pyruvate, 2.2 mg/mL NaHCO3, and hormones (5 UI/mL hCG, 10 UI/mL PMSG, Intervet) at 39°C in humidified atmosphere with 5% CO2.
Insemination was done with bull sperm supplied by the Centre for Animal Breeding and Reproduction at a concentration of 1 × 106/mL. Cryoprotectants were removed from the sperm by repeated centrifugation (2×) in standard IVF-Sperm-Talp medium supplemented with 4 mg/mL fafBSA. Sperm capacitation was induced by PHE (penicillin, hypotaurine, epinephrine). After 20 h of gamete coincubation at 39°C in humidified atmosphere with 5% CO2, the cumulus cells were mechanically removed by pipetting. The presumptive zygotes were transferred in groups of 20–25 in 35 μL IVC drops covered by embryo culture-tested mineral oil. IVC was carried out at 39°C in humidified atmosphere with 5% CO2, 5% O2, and 90% N2. The basic IVC medium consisted of synthetic oviduct fluid (SOF) culture medium supplemented with essential (BME) and nonessential amino acids (MEM) and 4 mg/mL fafBSA. At 3 days postinsemination (dpi), the embryos were subjected to selection. Only cleaved embryos of no less than three to four cells were placed in fresh IVC medium and were left in culture until days 8–9 (hatched blastocyst stage [HBl]). This timing represents the time needed for bovine blastocysts to hatch, which is variable [28]. For the purpose of these experiments, zygotes were placed either in SOF or in SOF supplemented with 3 μM GSK3i (CHIR99021; Axon Medchem) prepared in DMSO. A group of control embryos was cultured with SOF+DMSO.
Gene expression analysis—material selection
The selected material (embryos or cells) was washed with 0.25% polyvinylpyrrolidone in phosphate-buffered saline (PBS) and placed in 1.5-mL tubes (low binding, Eppendorf) in minimal volume, snap-frozen in liquid nitrogen, and stored at −80°C. Gene expression analyses on hatched blastocysts 8–9 dpi were performed as previously described by Madeja et al. [25]. The blastocysts were microsurgically dissected into the ICM and the TE, as presented in Supplementary Fig. S1 (Supplementary Data are available online at
RNA extraction and cDNA synthesis
Total RNA (embryos or cells) was extracted with the High Pure miRNA Isolation Kit (Roche Diagnostics) according to the manufacturer's protocol. This kit is routinely used by our group to isolate RNA from oocytes, embryos, and small copy number samples [25]. RNA concentration was measured on NanoDrop (Thermo Scientific). For each sample, reverse transcription was performed from 100 ng of total RNA. cDNA synthesis was performed with the Transcriptor High Fidelity cDNA Synthesis Kit (Roche Diagnostics) according to the manufacturer's protocol. The samples were stored at −20°C.
Quantitative real-time polymerase chain reaction analyses
The experiments were performed on the Roche LightCycler 2.0 instrument. The calculations were based on relative gene expression data analyses using real-time polymerase chain reaction (PCR) and the 2−ΔΔCT method [30,31]. To establish the differences in gene expression levels between the ICM and the TE, the samples containing whole blastocysts served as reference points for relative gene expression level calculations. HBl was referred to as a calibrator. Gene expression levels were normalized to the expression level of the 18S rRNA gene that served as a reference. Each sample was analyzed with all of the primer sets chosen for the experiment and for the reference gene. The reactions were repeated in triplicates. The primers were designed to span the introns (Table 1). The reactions were carried out in 10-μL capillaries (Roche Diagnostics) and the PCR mix comprised 1 μL of LightCycler FastStart DNA master SYBR Green (Roche Diagnostics), 5 mM of MgCl2, 0.3 μM of primers, and 1 μL of cDNA. Real-time PCR reaction conditions included initial polymerase activation at 95°C for 10 min, followed by 42 cycles of denaturation at 95°C for 10 s, annealing at 58°C for 10 s, and elongation at 72°C for 20 s. Product specificity was confirmed by melting analysis and agarose gel electrophoresis.
Qualitative gene expression analysis
Samples were collected from bESC-like lines at the time of cell passage and subjected to RNA extraction and cDNA synthesis as described above. The reactions were performed on Roche LightCycler 2.0 in the same reaction conditions as described for the quantitative analyses. The appropriate primer sets are listed in Table 1.
Immunofluorescent protein localization
This part of the experiment, in total, utilized about 260 bovine blastocysts 8–9 dpi cultured in SOF, SOF+GSK3i, or SOF+DMSO and primary ESC lines derived under culture conditions described in the bovine ESC derivation section. Immunofluorescent protein labeling and appropriate control experiments were done as described by Madeja et al. [25]. The control groups underwent the same staining protocol as the experimental group, with the exception of primary antibody. The embryos and cell lines were processed as follows: briefly washed in PBS (pH 7.3) and fixed with 4% paraformaldehyde (PFA) in PBS with 0.1% Triton X-100 and 0.1% Tween-20 (pH 8.0) for 15 min at 39°C.
Immunofluorescent protein detection was initiated by permeabilization with 0.55% Triton X-100/PBS for 20 min; blocking of nonspecific antibody binding with 10% fetal calf serum in PBS, 60 min at room temperature (RT); three washes in 0.1% Triton X-100 in PBS (PTX); overnight incubation with primary antibodies (ABI) diluted 1:50 in blocking buffer at 4°C; three washes in PTX; blocking for 40 min at RT; secondary antibody (ABII) incubation at 1:200 dilution; three washes in PTX; and embryo mounting on 18-well μ-slides (tissue culture treated, Ibidi Gmbh) in 40 μL drop containing antifade solution with DAPI (Vectashield mounting medium; Vector Laboratories). The slides were stored at +4°C.
To avoid cross-reactivity between the AB hosts, the embryos were double labeled with various sets of ABI and ABII. The ABI set included anti-CDX2 mouse monoclonal AB (ab15258; Abcam); anti-CDX2 rabbit polyclonal AB (ab88129); anti-OCT4 rabbit polyclonal AB (ab18976; Abcam) or goat polyclonal AB (ab27985; Abcam); and anti-NANOG rabbit polyclonal AB (500-P236l; PeproTech). The ABII set consisted of Santa Cruz Biotechnology antibodies
Bovine ESC derivation
Day 8–9 hatched blastocysts were plated on the layer of feeder cells—mouse embryonic fibroblasts (MEFs) in the bESC culture medium according to the experimental setup presented in Fig. 1. MEFs were obtained in collaboration with The Institute of Human Genetics, Polish Academy of Sciences, Poznan, Poland (Department of Reproduction and Stem Cells), according to the standard protocol from day 13.5 mouse embryos and inactivated with mitomycin-C (1 mg/mL).
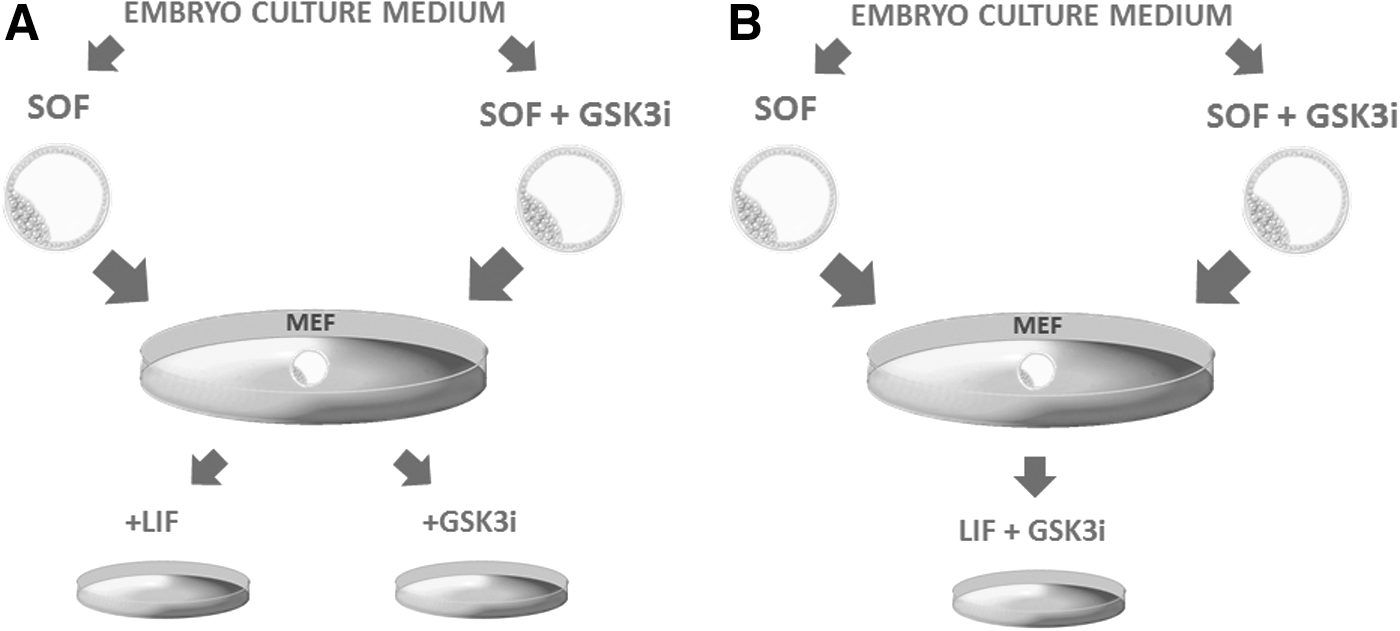
Experimental setup for bESC derivation experiments.
This part has been divided into two experiments. For Experiment 1, we have used SOF-obtained blastocysts (SOF-HBl) and blastocysts obtained in the presence of CHIR99021 (SOF+GSK3i-HBl) and plated them on inactivated MEFs in the ESC derivation medium [referred here as basic Dulbecco's modified Eagle's medium (DMEM)] supplemented either with LIF (10 ng/mL, human recombinant) or with CHIR99021 (3 μM) as presented in Fig. 1A. In Experiment 2, SOF-HBl and SOF+GSK3i-HBl have been cultured in basic DMEM in the presence of both LIF and CHIR99021 (10 ng/mL and 3 μM, respectively) according to the diagram in Fig. 1B. The basic DMEM consisted of 83% DMEM culture medium (Life Technologies, Gibco) containing 15% knockout serum (Life Technologies, Gibco), 1 mM L-glutamin, 1% essential amino acids MEM, and 1% antibiotic–antimycotic solution. For the purpose of ESC derivation, basic DMEM was supplemented with LIF (10 ng/mL), GSK3i (3 μM, CHIR99021), or with LIF+CHIR99021 (10 ng/mL and 3 μM, respectively).
Depending on the culture conditions, the primary outgrowths formed between days 3 and 6. Upon reaching 60%–70% confluency, the colonies were passaged onto the fresh layer of MEFs (in proportion 1:2). Because enzymatic methods of cell detachment are often associated with chromosomal abnormalities and we have noted that trypsinization impaired colony recovery after passage, we have relied on mechanical disaggregation for cell passaging. The colonies were cut out and separated from MEFs by an ophthalmological scalpel and disaggregated by intensive pipetting through narrow glass pipettes. The culture medium was changed every 2 days.
Alkaline phosphatase activity
For alkaline phosphatase (AP) staining, the colonies were cultured at least for 3–4 days before staining. The AP test was performed with the Alkaline Phosphatase Detection Kit (Millipore) according to the manufacturer's protocol. Before AP activity detection, the colonies were briefly washed in PBS and fixed in 4% PFA (in PBS) for 2 min at RT. Next, the colonies were washed in TBST buffer (20 mM Tris-HCl, 0.15 M NaCl, 0.05% Tween-20, pH 7.4) and subjected to AP staining for 15 min in the dark. Finally, the colonies were washed with TBST and covered with PBS to prevent drying of the specimen.
Embryoid body formation
The embryoid bodies (EBs) were obtained from the bESC-like colonies derived from SOF-HBl plated on MEFs in basic DMEM devoid of LIF and CHIR99021. The colonies were removed from the feeder cells and mechanically disaggregated by intensive pipetting through a narrow glass capillary and diluted in culture medium to the concentration of 1 × 104/mL. Hanging drops were formed from 25 μL of the cell suspension on the cover of a 30-mm cell culture dish (ThermoScientific—Nunc). First, cell aggregates became visible around day 2 and EBs have been formed by day 4. After 6–7 days in culture, the EBs have been fixed and prepared for immunofluorescent antibody staining according to the same protocol as described for the blastocysts. The primary antibodies consisted of rabbit polyclonal anti-CRIPTO-1 (ab19917; Abcam) and mouse polyclonal anti-BRACHYURY (BRY), (ab167337; Abcam) and the secondary ABs were selected from the panel listed for embryo immunofluorescence.
Fluorescent in situ hybridization
The cells were collected during passage from the colonies that were of correct ESC-like morphology. A part of an expanding colony was cut away for the purpose of fluorescent in situ hybridization (FISH) analysis. The colony subjected to FISH was trypsinized in 0.25% trypsin-EDTA solution; fetal bovine serum was added to neutralize the action of trypsin. Next, cell suspension was centrifuged at 500 g for 10 min, washed twice in PBS, and finally suspended in 30 μL of PBS. A small volume of cell suspension was placed on a clean microscopic slide (Menzel) and left to dry. Subsequently, the cells were fixed on microscopic slides in 3:1 solution of methanol:acetic acid at −20°C for 24 h.
In situ hybridization and posthybridization treatment
Two-color FISH was performed with probes corresponding to the locus-specific regions of the bovine chromosome pairs 12, 23 and X, Y. Each bESC sample collected in the first step was analyzed with probes for chromosomes, 12 and 23, and in the second step with probes for chromosomes, X and Y. The probes for the 23 and X chromosomes were labeled with Spectrum Green (Abbott Laboratories) and for the 12 and Y chromosomes with Spectrum Orange (Abbott Laboratories). Microscopic slides with fixed bESCs were denatured in 70% formamide at 74°C for 2 min and passed through series of cold ethanol (70%, 85%, 100%). The hybridization mix (10 μL) comprised two precipitated locus-specific probes (12, 23 or X, Y at 200 ng each), 50% formamide, 2× saline sodium citrate buffer (SSC), and 10% dextran sulfate. The mix was denatured at 74°C for 10 min on thermoblock, applied on each slide, and covered with a 24 × 24 mm coverslip. Slides were incubated overnight in a humidified dark chamber at 37°C. After 24 h of incubation, the coverslips were carefully removed and the slides were washed in 2× SSC and incubated in 0.7× SSC/0.3% Tween-20 at 72°C for 2 min. Finally, the slides were washed in 2× SSC, dried, and mounted with 0.2 μg/mL DAPI/Antifade (Vectashield mounting medium; Vector Laboratories). Microscopic analysis was performed using a fluorescence microscope with DAPI/FITC/Rhodamine filter sets (Zeiss Axiovert 200).
Statistical analysis
A comparison of the experimental groups was performed using IBM SPSS Statistics 22.0. The differences in relative transcript abundance were calculated using the nonparametric Mann–Whitney test. All data (before computing) were subjected to testing for normal distribution using the KS test (Kolmogorov–Smirnov).
Results
GSK3 inhibition significantly increases classical pluripotency marker gene expression within bovine ICM and TE lineages
To establish whether inhibition of the GSK3 exerts an effect on transcript abundance of pluripotency and lineage-specific markers, in vitro obtained bovine blastocysts were cultured in the presence of the specific inhibitor (CHIR99021). Embryos cultured without the inhibitor served as control. Before the gene expression studies, we have evaluated the possible effect of DMSO on embryo quality and gene expression profiles. Because no statistically significant differences in the expression levels of the analyzed genes were noted between SOF and SOF+DMSO (Fig. 2A) culture conditions, in the following experiments SOF blastocysts served as controls.

Inhibition of the GSK3 causes changes in transcript abundance of lineage and pluripotency-related genes in bovine inner cell mass (ICM) and trophectoderm (TE).
mRNA expression profile
To build the basis for this experiment, we have first evaluated the relative transcript abundance of pluripotency-related genes among the ICMs and the TEs of the GSK3i-obtained embryos in relation to the control group. In general, the GSK3i treatment resulted in upregulation of pluripotency and lineage-related transcripts both within the ICM and the trophoblast cells of bovine blastocysts as presented in Fig. 2B. On close examination, the data indicated a highly significant increase in OCT4 (6.7-fold, P < 0.0001), NANOG (4.72-fold, P < 0.05), c-MYC (8.02-fold, P < 0.005), REX1 (4.37-fold, P < 0.01), GATA6 (13.31-fold, P < 0.0001), and KRT18 (12.65-fold, P < 0.0001) mRNA levels within the ICM of the GSK3i embryos compared with the control group (Fig. 2B). No statistically significant differences were noted for the FN1 and the CDX2 transcripts. However, we have observed an increasing trend in the mRNA expression levels for these genes within the ICM lineage of the GSK3i blastocysts. We have also compared the specific gene expression levels between the ICM and the TE of the control embryos, and similar to our previous findings [25], we have noted a significantly higher mRNA level for NANOG (P < 0.0001) and OCT4 (P < 0.0001) in the ICM compared with the TE of blastocysts obtained without the inhibitor. It is important to note that in the control group, the CDX2 gene expression level was significantly higher in the TE (P < 0.0001) than in the ICM where 8 of 12 samples were negative for the CDX2 transcript.
A strong evidence for the positive influence of the GSK inhibition on the expression of pluripotency-related factors comes from the trophoblast lineage of the GSK3i-treated embryos (Fig. 2B). Within this tissue, we have noted a drastic increase in the OCT4 expression level (27.3-fold, P < 0.0001). As a result, in the GSK3i embryos, there was no difference in the OCT4 transcript abundance between the ICM and the TE. Furthermore, the GSK3i treatment also caused an upregulation of FN1 (P < 0.012), GATA6 (P < 0.0001), and KRT18 (P < 0.0001) transcripts in the trophectoderm cells. An effect of the GSK3 inhibition has also been manifested by a 2.36-fold downregulation of trophoblast-specific transcription factor, CDX2 (P < 0.046), within the TE lineage of the GSK3i blastocysts (Fig. 1B).
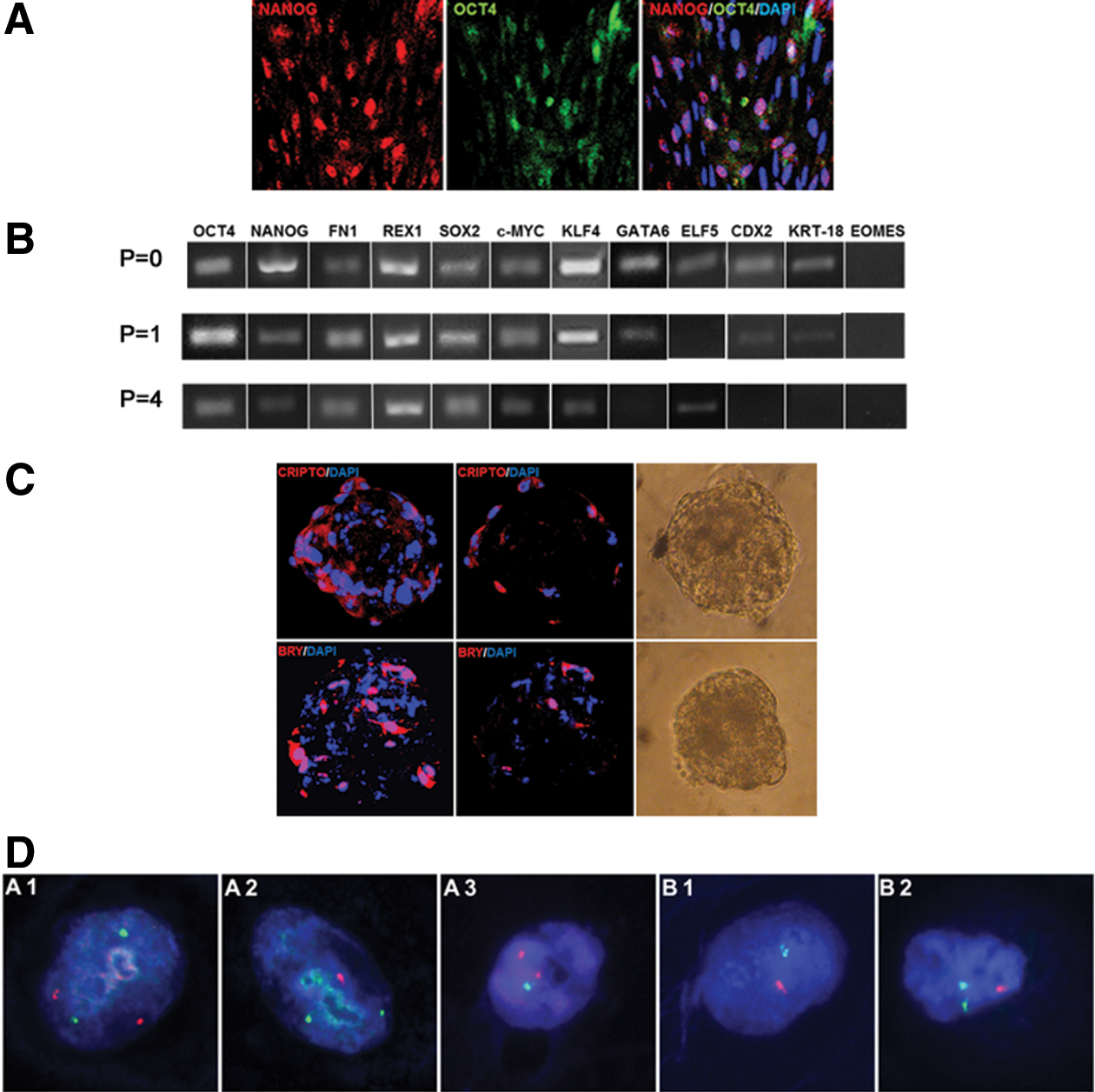
Protein distribution
To further investigate the biological effect of the applied inhibitor, we have subjected day 9 bovine blastocysts to immunofluorescent protein labeling. The obtained results complement the mRNA data. GSK3 inhibition altered OCT4 and CDX2 expression (Fig. 3A). As indicated by confocal images (concomitant with CDX2 transcript downregulation), CDX2 protein expression was suppressed in the TE of GSK3i embryos in 92.5% of the analyzed blastocysts (37/40) (Fig. 3A) in relation to the control group (Fig. 3B). Within the group of the GSK3i cultured embryos, 26 displayed a total loss of CDX2 and the remaining 14 blastocysts presented some residual signal in a small proportion of cells (Fig. 3C). The signal, however, lost its characteristic expression pattern presented in control (Fig. 3C-a). The observed loss of CDX2 expression was accompanied by upregulation of OCT4 protein both within the ICM and the trophoblast cells (Fig. 3A, D). Within the ICM cells, OCT4 expression retained its nuclear localization as presented on a cross section through the ICM (Fig. 3D-a), whereas in the trophoblast cells, it was predominantly located in the cytoplasm (Fig. 3D-b).
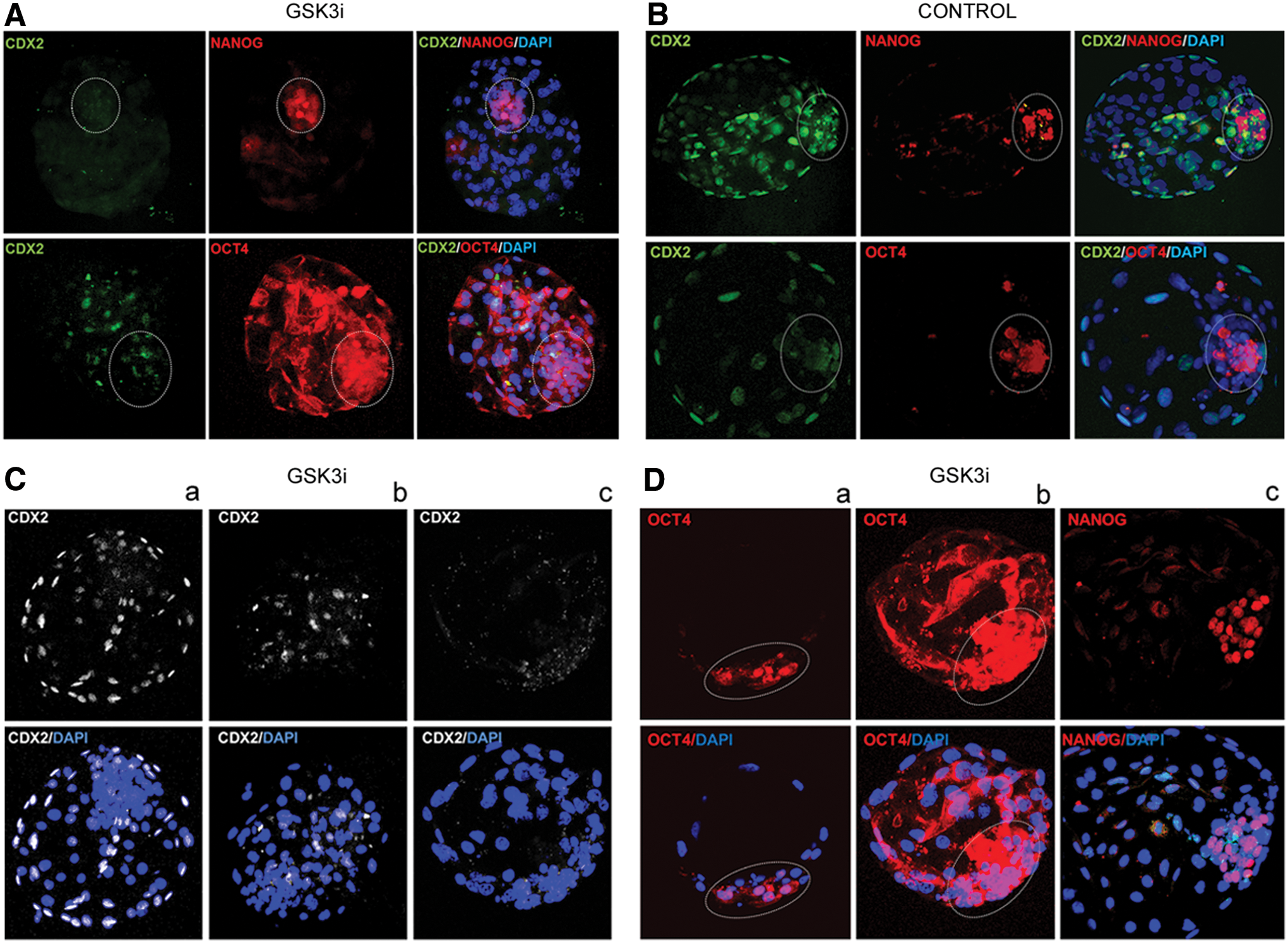
Pluripotency-related protein distribution in GSK3i-treated bovine embryos.
The inhibition of the GSK3 did not alter the NANOG distribution pattern as the protein maintained its ICM-specific localization (Fig. 3D-c). The average number of NANOG-positive cells did not differ significantly between the studied and the control groups (Table 2). On average, the ICM of control embryos comprised about 66 cells, of which ∼34 were NANOG positive. In case of the GSK3i embryos, ∼78 cells made up the ICM and 32 cells expressed NANOG. In all of the analyzed cells, NANOG location was nuclear (Fig. 3D–c).
GSK3i, glycogen synthase kinase 3 inhibitor; HBl, hatched blastocyst; ICM, inner cell mass; SOF, synthetic oviduct fluid.
GSK3i inhibition supports the primary colony formation rate; however, the prolonged exposition of preimplantation embryos to the GSK3i does not have a positive effect on bESC derivation
The results of the gene expression studies (mRNA and protein) allowed us to conclude that the GSK3 inhibition throughout the period of bovine preimplantation development exerts a biological effect and alters pluripotency marker gene expression. Thus, in the next step of our study, we have tested whether the inhibitor-dependent maintenance of the WNT signaling during early development supports the establishment of primary bESC-like colonies.
Experiment 1
We have evaluated whether the prolonged admission of the GSK3i promotes the initial ICM outgrowth formation and changes the blastocyst adhesion rate to the feeder cell layer (MEF). The ICMs were not dissected from the trophoblast as we were also interested whether the CDX2 downregulation in the GSK3i embryos has a direct effect on the origin and the morphology of the primary colonies. The blastocysts were obtained in an inhibitor-supplemented culture system (SOF+GSK3i) and in standard SOF medium (w/o the inhibitor). Only day 8/9 hatched blastocysts were used, plated on MEFs, and cultured in basic DMEM supplemented with either LIF or GSK3i as illustrated in the diagram (Fig. 1A).
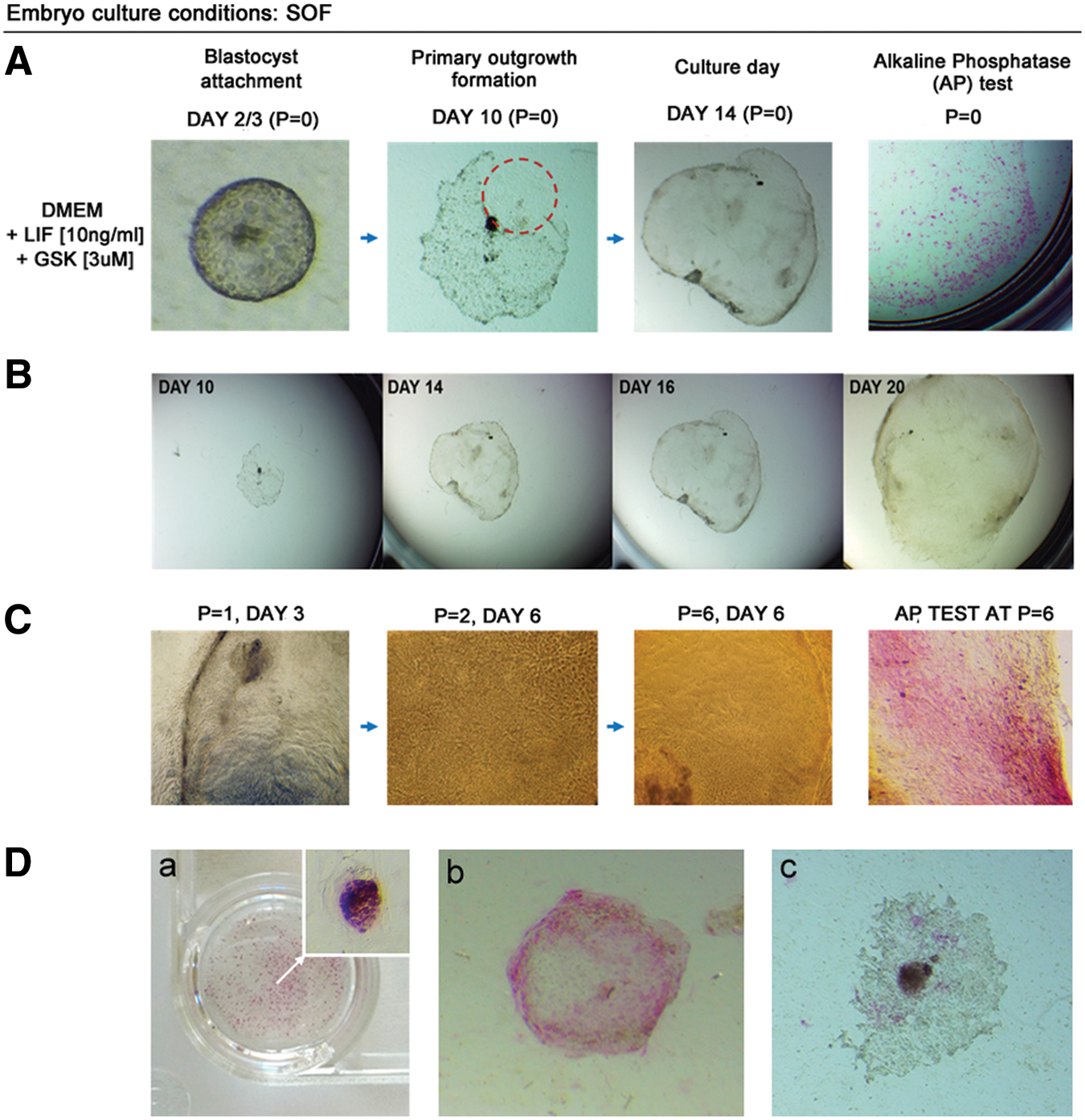
In case of SOF-obtained blastocysts (SOF-HBl), the primary outgrowths were formed from the majority of blastocysts (Table 3). In case of LIF media supplementation, the initial outgrowths were observed between days 6 and 7 of culture (Fig. 4). The GSK3i treatment resulted in an early outgrowth formation (day 3/4), which was consistent for nearly all of the embryos forming the primary colonies (n = 21/22).
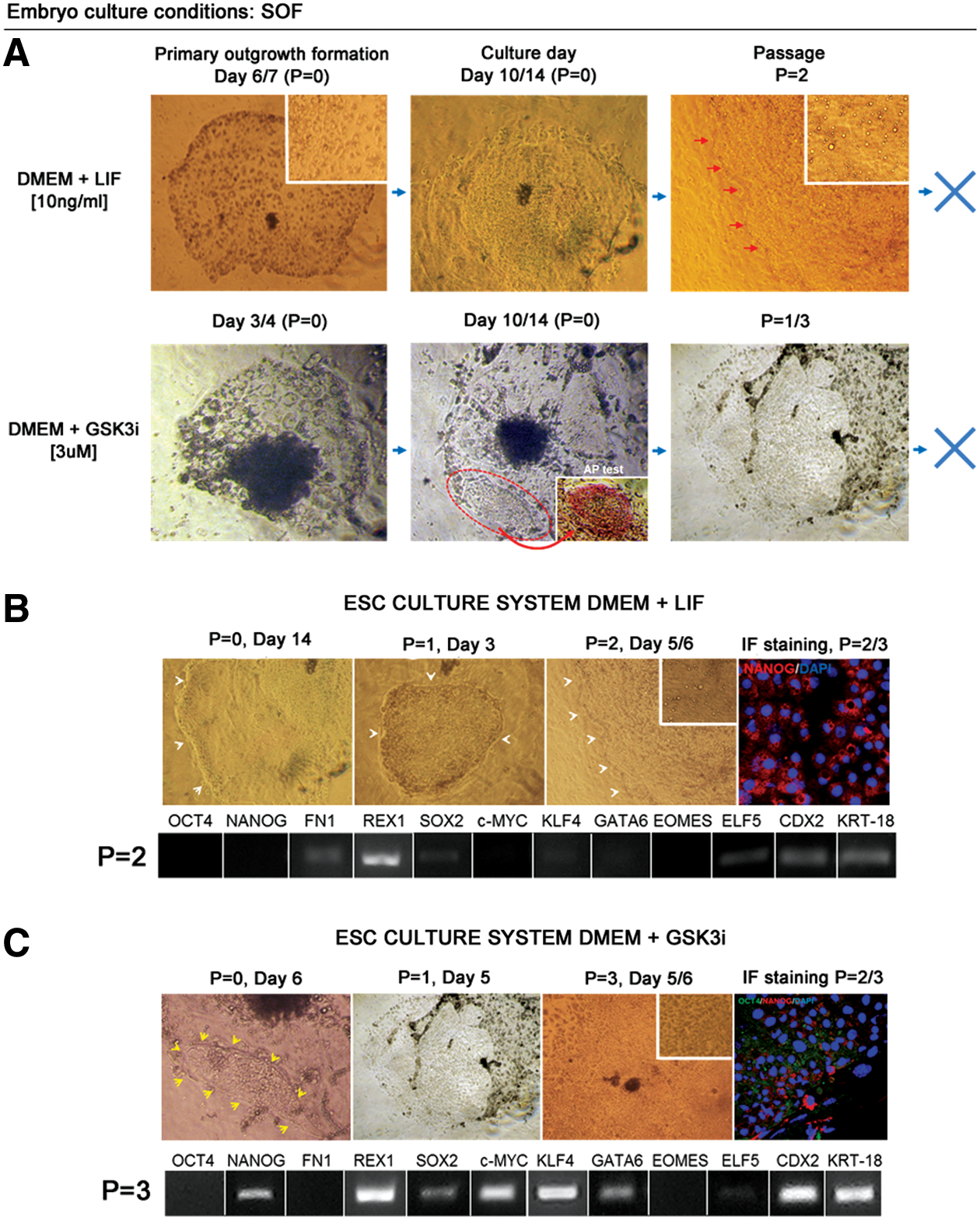
Derivation of primary ESC-like colonies is not supported by independent actions of LIF or GSK3i. Embryos were obtained in vitro in SOF culture medium.
bESC, bovine embryonic stem cells; DMEM, Dulbecco's modified Eagle's medium.
In DMEM+LIF and DMEM+GSK3i culture conditions, the first observed outgrowths consisted predominantly of the trophoblast cells (Fig. 4A). These cells disappeared during the consecutive culture days; however, the rate and the overall outgrowth morphology were different for the two systems. LIF supplementation resulted in a total loss of any trophoblast-like cells around culture days 10–14, and the colonies obtained a characteristic stem cell morphology with a clearly pronounced colony boundary (Fig. 4B). After passage, monolayer colonies with clear margins were formed, maintaining ESC-like characteristics. This, however, was not preserved over the second or the third passage. The clear colony boundary was lost (as indicated in Fig. 4B) and over time vacuole-like structures began to be visible within the colony. In culture systems, vacuolarization is attributed to senescence that arises as stress response. Therefore, it may be related to autophagy resulting from the lack of important growth factors or nutrients (we have ruled out contamination). At this stage (P = 2/3), the immunofluorescent staining did not detect OCT4 protein, and NANOG-positive signal was detected only within the cytoplasm of the cells (Fig. 4B). In agreement with these data, qualitative PCR gene expression analysis did not detect OCT4, NANOG, c-MYC, and GATA6 mRNA in these colonies. FN1, SOX2, and KLF4 transcripts remained weakly maintained at P = 2 (Fig. 4B). At the same time, REX1, ELF5, CDX2, and KRT18 transcripts were detected, which may support the interpretation that the colonies have already differentiated toward the extraembryonic lineages. ELF5, CDX2, and KRT18 are trophoblast lineage-specific transcripts, and REX1 has been reported to be identified in trophoblast-derived tissues of mouse egg cylinder [32]. Following that the colonies no longer recovered after passage.
The GSK3i-supplemented culture system produced a different primary colony growth pattern. The first outgrowths were formed early (day 3/4); however, the trophoblast cells persisted in culture. On average, around day 6, small ESC-like colonies began to be visible within the outgrowth and these have tested to be positive for AP (Fig. 4A). After passage, some colonies still partially maintained trophoblast-like morphology and partially consisted of clearly separated monolayer of cells with a clear boundary, displaying a less compact morphology than in the DMEM+LIF cultured colonies (Fig. 4C). The colonies were still formed after one to three passages, but during the subsequent days in culture, differentiated at the colony borders and lost clear margins (Fig. 4C). Similar to DMEM+LIF culture conditions, after passage, the colonies did not express nuclear OCT4 and NANOG was noted only in the cytoplasm of a small proportion of cells as indicated in Fig. 4C. mRNA was detected for the whole panel of the analyzed genes, except for OCT4, FN1, and EOMES (Fig. 4C, bottom panel). Interestingly, although the bESC culture conditions were evidently suboptimal, the vacuoles were not observed in this system.
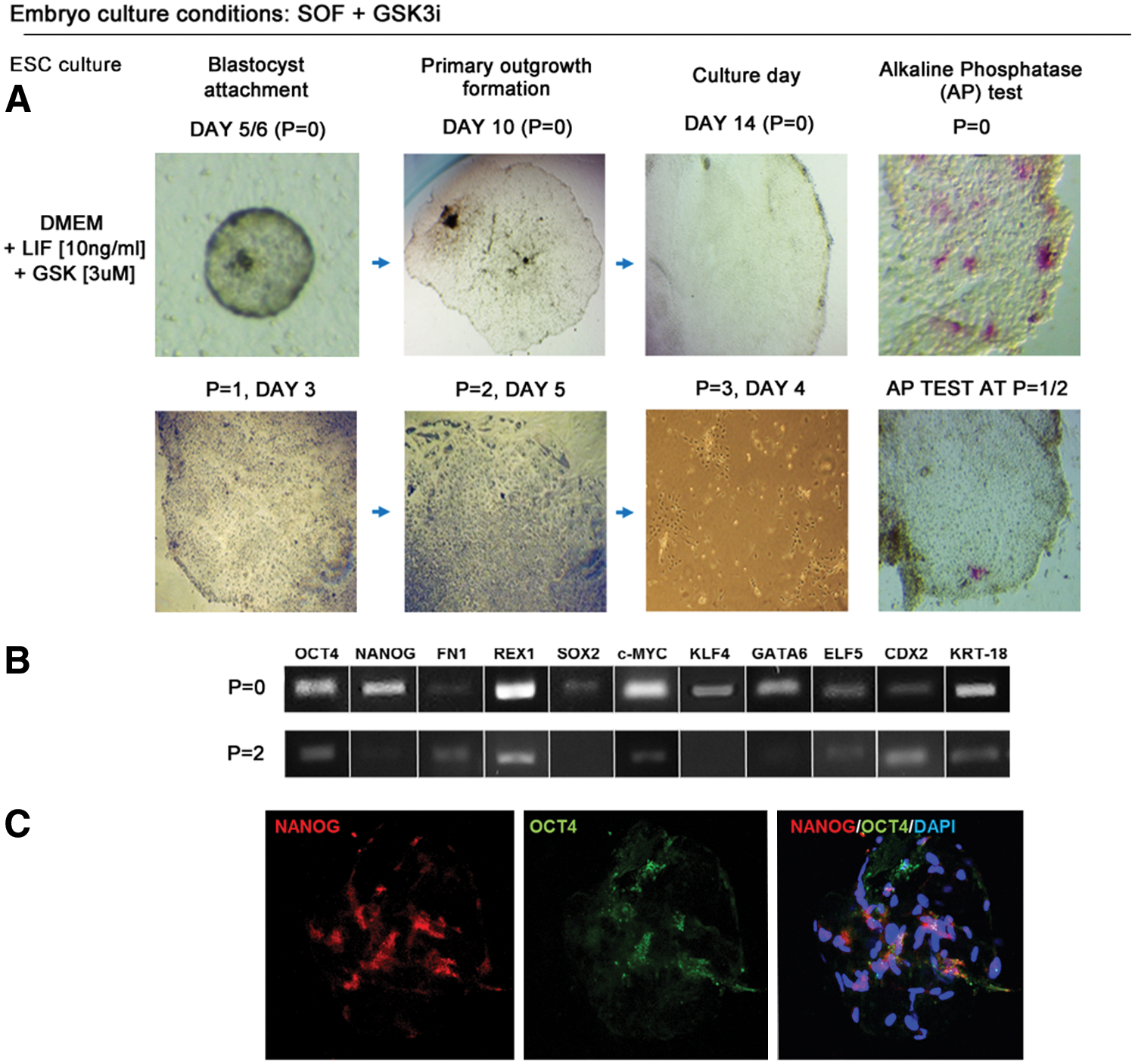
The primary outgrowth formation rate for SOF+GSK3i-obtained blastocysts (SOF+GSK3i-HBl) was also high, so the changes in pluripotency gene expression patterns for OCT4 and CDX2 in GSK3i-obtained blastocysts did not alter embryo adhesion ability and outgrowth formation (Table 3). The first outgrowths were detected between day 5/7 for LIF and day 3/4 for GSK3i (Fig. 5). In contrast to SOF-HBl embryos, the SOF+GSK3i-HBl, when plated on MEFs in the DMEM+LIF medium, did not form colonies with clearly pronounced margins. The colonies did not present a uniform morphology and did not form compact structures; however, the trophoblast cells were not visible at this stage. After several days in culture, clear-cut boundary colonies emerged, but these were able to survive only one or two passages. The morphology was different to one presented by colonies obtained from SOF-HBl in the presence of LIF. The colonies were not as compact and uniform, with dark inclusions in the cytoplasm. Although some colonies were formed after the second passage, the cells at the colony margin differentiated having a large nuclei–cytoplasm ratio (Fig. 5). The cells within the colony center still remained small, but the immunofluorescent staining revealed only residual NANOG expression in the perinuclear region of a small proportion of cells, OCT4 was not detected.
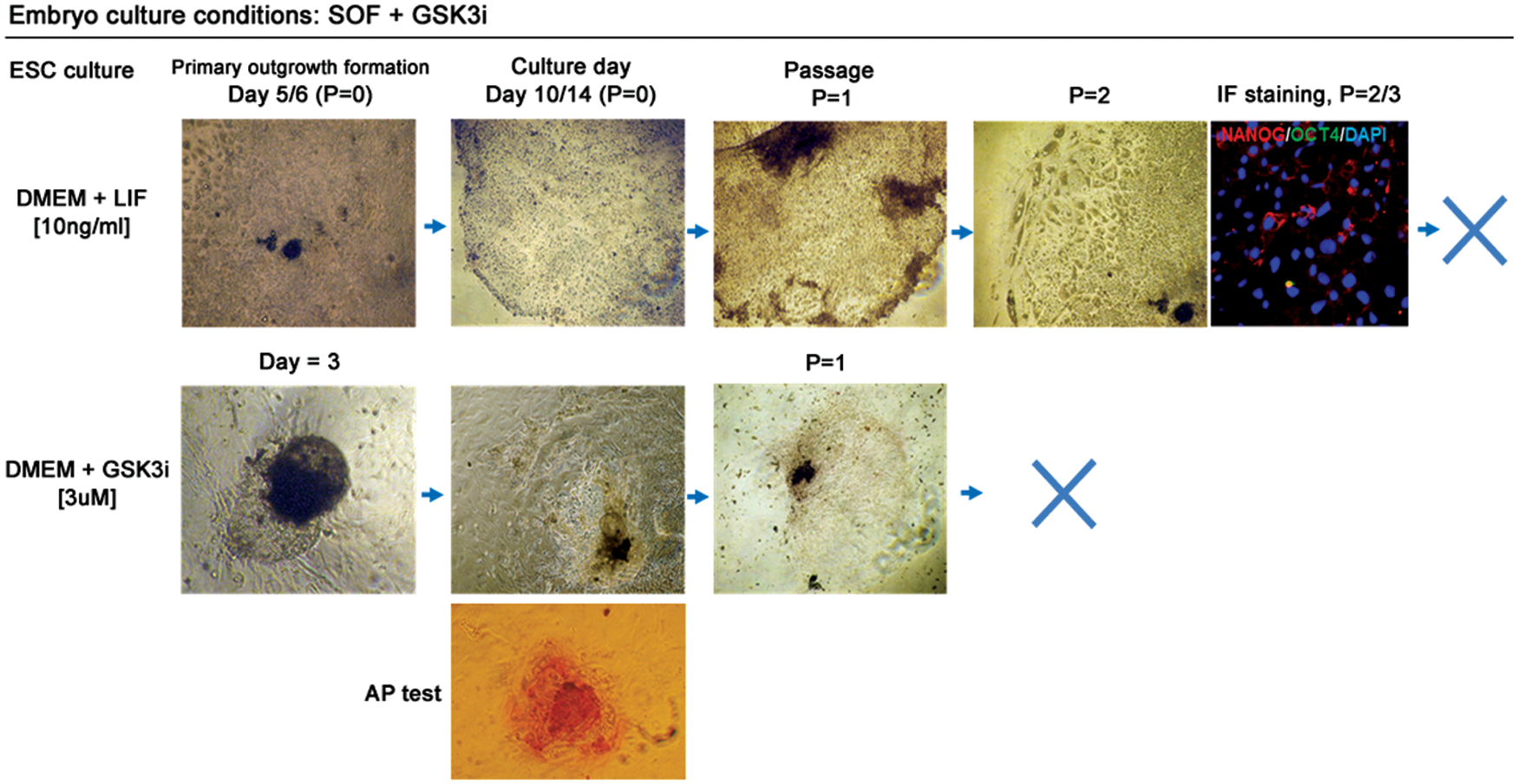
Activation of WNT signaling during bovine embryo development does not sustain bESC derivation. Top panel indicates colony growth and morphology in culture media supplemented with LIF over two passages (P). Immunofluorescence (IF) indicated a few NANOG-positive cells with cytoplasmic localization. OCT4 was absent. DAPI marked chromatin. The bottom panel represents colony formation after culture in GSK3i-supplemented medium. The explants initially displayed a characteristic ESC-like morphology and tested positive for AP, but quickly differentiated after P = 1. X indicates that colonies were no longer formed after passage. Color images available online at
A notable difference was observed for the colonies obtained in the DMEM+GSK3i culture medium. Initially, for the majority of the primary outgrowths (n = 17/21 attached HBl), no trophoblast cells were visible (Fig. 5). The outgrowths expanded in culture with colonies partially presenting a compact ESC-like morphology and partially signs of differentiation at the boundary. However, the undifferentiated cells had uniformly transparent cytoplasm without signs of degeneration. These colonies were mostly AP positive, but did not survive beyond passage 1, thus we have omitted the gene expression analyses.
Experiment 2
From Experiment 1, we have concluded that despite upregulation of several pluripotency-related factors, the prolonged exposition of preimplantation embryos to the GSK3 inhibition does not have a positive effect on bESC derivation from SOF+GSK3i blastocysts. Neither morphology and growth rate nor passaging potential has been significantly enhanced. In addition, the suppression of the CDX2 and an apparent lack of the trophoblast cells at the initial stages of colony formation did not improve colony derivation from the SOF+GSK3i cultured embryos. However, we have observed that in case of all embryos (regardless of the IVC system), supplementation of bESC derivation media with either LIF or GSK3i exhibited some positive effect on the outgrowth formation. Thus, in the second experiment, we have tested the potential of LIF and GSK3i acting together. Again, we have used embryos derived either in SOF or SOF+GSK3i in vitro culture media and we have plated them on MEFs in DMEM+LIF+GSK3i culture conditions (as presented in Fig. 1B).
In case of SOF-HBl, the emerging colonies were first observed around day 2/3 and the initial outgrowths consisted partially of the trophoblast cells and partially formed uniform ESC-like colonies, which continued to expand in culture (Fig. 6A). At this stage, the results of the AP test indicated only partial staining (in some regions of the expanding outgrowth). However, the colonies continued to grow in culture for 20 days until 60%–70% confluency was reached (Fig. 6B). After passage, the uniform single layered colonies of tightly packed flat cells were formed with clear margins and almost ubiquitous AP expression (Fig. 6C). These colonies were further passaged and maintained typical bESC-like morphology with round-shaped cells and no signs of differentiation. An example of colony recovery is shown in Fig. 6D, where numerous small colonies positive for AP emerged after passage (P = 4, day 5). Colonies from the parallel experiment were left in culture, further passaged at lower density (1:4), and left to expand for 7 days. The colonies grew larger, maintained compact morphology, and retained AP activity (Fig. 6D). In contrast, LIF withdrawal resulted in the loss of uniform appearance. Larger cells with dark inclusions in the cytoplasm emerged and AP activity was not detected.
Embryos were obtained in vitro in SOF culture medium and the ESC-like colonies were derived in basic DMEM with LIF and GSK3i.
Immunofluorescent stainings at passages 2–6 revealed nuclear localization of OCT4 and NANOG throughout the colonies (Fig. 7A). All of the analyzed transcripts were detected at P = 0 (including TE lineage-specific ELF5, CDX2, and KRT18), but CDX2 was not detected after P = 1 (Fig. 7B). KRT18 and GATA6 transcripts were expressed over the next passages and the colonies maintained the expression of pluripotency markers: OCT4, NANOG, c-MYC, FN-1, KLF4, REX1, and SOX2. Trophoblast-specific CDX2 and KRT18 transcripts were not detected at P = 4. However, the specific transcripts for the ELF5 gene, which is pointed as a gatekeeper possessing the capacity to reinforce TE lineage commitment, have been detected at later passages. Mesoderm marker EOMES transcripts were absent at all of the analyzed passages.
A representative image of bESC-like colonies obtained from SOF blastocysts in the presence of LIF and GSK3i immunostained (IF) for OCT4 and NANOG
Because the lines derived from SOF-HBls in DMEM+LIF+GSK3i presented typical ESC-like morphology, expressed several ICM-specific pluripotency markers, and grew well after passage, we were interested to see whether they will be capable of forming the EBs. The disaggregated cells of ready to be passaged colonies were cultured in hanging drops. After 2 days in culture, first cell aggregates began to be visible and were left to expand for another 6–7 days. At this point, the EBs were immunostained for the primitive streak markers, BRACHYURY and CRIPTO, which are known to play a role in the determination of the epiblastic cells that subsequently give rise to the mesoderm (Fig. 7C). The confocal sections revealed that at this stage, CRIPTO was predominantly located in the outer layer of cells, whereas BRY signals were differentially distributed within the EB cells.
The chromosomal stability of cell lines obtained from SOF-HBls was tested by FISH with a set of locus-specific probes to bovine chromosomes 12, 23 and X, Y (Fig. 7D). The preliminary analysis revealed that all of the bESC-like lines derived presented some level of chromosomal instability (Table 4). We have observed that chromosomal abnormalities (especially nullisomies, monosomies, and trisomies) of autosomes (12, 23) occurred in the range from 1.9% to 6.4% for DMEM+GSK3i and from 1.7% to 9% in DMEM+LIF+GSK3i cell lines. We have compared only these two culture systems as we were interested whether proper ESC-like morphology versus colonies of variable morphology may correspond to chromosomal stability of the early outgrowths. The results showed no such correlation. bESC-like colonies obtained in the same system, but derived from different embryos, showed a variable range of autosomal abnormalities. However, lower percentage of chromosomal abnormalities −1.9% for line S-G2 (DMEM+GSK3i) and 1.7% for line S-LG4 (DMEM+LIF+GSK3i) corresponded to proper sex chromosome number. Overall analysis did not suggest sex bias. However, it must be noted that due to the small size of the growing colonies (cell count) and the number of chromosomes analyzed, we were only able to evaluate from 49 to 122 cells (Table 4). Thus, these results should be treated as preliminary, yet indicating the importance of cytogenetic analysis for evaluation of ESC stability.
bESCs, bovine embryonic stem cells.
The blastocysts derived in SOF+GSK3i embryo culture medium did not preserve such potential (Fig. 8). The initial colonies were formed around day 5/6; the outgrowths formed compact structures with clear colony boundaries, but cells did not maintain typical ESC-like morphology. Within the cells, dark inclusions were visible in the cytoplasm. The results of the AP test at P = 0 were similar to the results obtained from the colonies derived from the SOF-HBl, but this has not changed, and after passage, the AP signals became scarce. At P = 1, the colonies displayed a less compact morphology with larger cells and dark inclusions in the cytoplasm. After passaging, most of the new colonies lost their clear colony boundaries, and the cells differentiated at the colony margins and did not recover after passage. Initially, mRNAs for the whole panel of the analyzed genes were detected, but already at passage 2, NANOG, SOX2, KLF4, and GATA6 transcripts were lost (Fig. 8B). Immunofluorescent protein labeling revealed some NANOG and OCT4 signal; however, both of the proteins have lost their specific nuclear localization (Fig. 8C).
Embryos were cultured in vitro in SOF culture medium supplemented with the GSK3i, and the ESC-like colonies were derived in basic DMEM with LIF and GSK3i.
Discussion
WNT signaling affects lineage and pluripotency-specific gene expression in bovine blastocysts
Studies demonstrate that WNT signaling is present in preimplantation bovine embryos and may be involved in the regulation of development; however, the precise role is still not clear [33]. The expression of 16 WNT genes and other genes involved in the WNT signaling was documented for bovine morula stage embryos [33]. Furthermore, 22 genes of the WNT pathway and 47 genes important for the MAPK signaling were expressed in bovine oocytes and blastocysts [34]. Both of the GSK3 isoforms (GSK3A and GSK3B) were detected from the two-cell stage up to the blastocyst in cattle [35]. Phosphorylation of both isoforms increased as development progressed, suggesting that the subsequent inhibition of the GSK3 accompanied by the WNT signaling activity is associated with normal embryo development.
In our system, there was no significant difference in the percentage of embryos reaching the blastocyst stage at 8 dpi (data not shown); however, the total number of cells within the ICM of the GSK3i-treated embryos was on average 17.5% higher than in the control group. The choice of the specific inhibitor may be crucial for the outcome of the signaling pathway activation. Treatment of bovine zygotes with CHIR99021 resulted in a significant increase in the proportion of embryos reaching the blastocyst stage accompanied by an increase in the cell number of blastocysts at 8 dpi [35].The ICM-to-TE ratio was also numerically affected by IVC media supplementation with AMBMP (2-amino-4-(3,4-(methylenedioxy)benzylamino)-6-(3-methoxyphenyl) pirimidine), a WNT agonist that activates canonical signaling [33]. However, at high concentrations of AMBMP, the ICM appeared disorganized.
Inhibition of GSK3 with lithium chloride at 20 mM significantly reduced the proportion of 8-cell embryos at day 3 and inhibited blastocyst formation, whereas CHIR99021 at 3 μM increased the percentage and quality of bovine blastocysts [35]. CHIR99021 has been described as the most selective GSK3i [36] and has been successfully applied to derive stable ESC lines from recalcitrant mouse strains, rats, and in nuclear reprogramming of somatic stem cells.
Quantification of accumulated β-catenin may serve as an indicator of canonical WNT signaling, but it does not confirm its translocation to the nucleus and transcription of the target genes. This may be directly evaluated by TCF reporter assays or indirectly by assessing the target gene expression levels. In the mouse, these consist of pluripotency factors, such as c-Myc, Sox2, Sox9, Sox17, Sall4, Nanog, Oct4, Fn1, Rex1, Bmp4, Bry, Cdx1, and E-cad (for reference: The WNT Homepage
The biological effect of WNT pathway activation became evident at the gene and protein expression levels. Under standard culture conditions (w/o the inhibitor), OCT4 displayed a significant upregulation only in the ICM. In SOF+GSK3i embryos, OCT4 transcripts increased to such levels that no difference in the mRNA abundance was noted between the ICM and the TE. In parallel to this observation, OCT4 protein was detected within both lineages; however, in the ICM, it retained its nuclear localization, and in the trophoblast cells, it was located predominantly in the cytoplasm. This may be explained by the fact that β-catenin is capable of forming a complex with OCT4 and modulating its activity, suggesting a mechanism through which β-catenin maintains pluripotency independent of TCF [37]. We have previously reported a unique distribution pattern of OCT4, which, in the ICM cells of early blastocysts (7 dpi), was located at the nuclear periphery and, in the TE cells surrounding the ICM, was present in the cytoplasm. At 8 dpi, OCT4-positive blastomeres were predominantly located in the ICM; however, some positive signal was still detected within the TE [25].
The results of numerous studies show that the mechanism of lineage allocation is gradual as the processes of cell fate decisions are governed by active interactions between the core pluripotency factors [38,39]. Due to the developmental differences between bovine and mouse or human embryos, the TE lineage development in these species may have different requirements.
Mouse and human embryos begin implantation soon after the blastocyst formation at 3.5–4.0 and 7–8 dpi, respectively. Cattle embryos, before their attachment to the endometrium (21–22 dpi), float in the uterus undergoing gastrulation and early neurulation. TE proliferation commences around 13 dpi, and differentiation into binucleate cells (equivalent of mouse giant cells) starts at 15 dpi. Therefore, the observed increase in the OCT4 transcripts may not exert a direct effect on the early stages of bovine TE lineage formation. However, it must be noted that in the GSK3i-treated embryos, the global increase in OCT4 expression was accompanied by a decrease in the CDX2 transcription and distorted CDX2 protein expression, with some embryos completely lacking the positive signal.
Studies by Berg [40] indicated that the bovine OCT4 locus does not contain the cis-acting regulatory region necessary for extinguishing transcription in the trophoblast. Hence, OCT4 may not interact with CDX2 in the same manner as in the mouse, where the TE lineage formation is initiated by the activation of TEAD4 [41]. It may be anticipated that at least at the early stages, factors essential for bovine TE lineage formation act independently of the OCT3/4-CDX2 signaling, allowing giving rise to morphologically correct blastocysts in the presence of OCT4 (stimulated by elevated WNT activity). Additionally, in the TE of our GSK3i-treated blastocysts, we have also noted a significant increase in the transcript level of cytoskeletal protein KTR18, which together with KRT8 builds the intermediate filaments and in cattle has been suggested as a trophoblast-specific factor [42]. In mouse embryos, a targeted deletion of KRT18 leads to trophoblast fragility and early embryonic lethality [43]. Studies on mice suggested that CDX2 is rather required to maintain the TE identity in already specified cells [44] as even CDX2-null mice were capable of producing blastocysts with distinctive trophoblast lineage. Factors acting upstream of or parallel to the CDX2, such as TEAD4, YAP1, TCFAP2C, ELF5, ETS2, and GATA3, may still be expressed and initiate TE differentiation. Adequately, TEAD4, YAP1, GATA3, and ELF5 transcripts have been detected in bovine trophoblast cells [42].
Another important player in the embryonic lineage specification and pluripotency maintenance is NANOG. The establishment of the ICM and the epiblast relies on the action of several transcription factors. Using chromatin immunoprecipitation, Rodda et al. [45] showed that OCT3/4 and SOX2 bind to the NANOG promoter in mouse and human ESCs. SOX2 acts synergistically with OCT3/4 to activate SOX2 enhancers, which in turn regulate the expression of NANOG, OCT3/4, and SOX2 [46]. At the gene expression level, we saw NANOG transcript upregulation in the ICM of the GSK3i blastocysts. Upon the WNT pathway activation, intercellular β-catenin increases. It maintains the physical ability to associate with OCT-3/4 and in turn upregulate NANOG in an OCT-3/4-dependent manner [47]. In development, NANOG acts at a later stage than OCT4. It antagonizes GATA6 and promotes epiblast differentiation. NANOG-null blastocysts differentiate into parietal endoderm-like cells in vitro [48]. Both NANOG and OCT4 are required to suppress CDX2 expression in the ICM; however, in the presence of NANOG, the ICM cells still commit to the TE lineage in OCT4-null mouse embryos [49]. These observations allowed us to conclude that the activation of WNT signaling in the developing bovine embryo may exert a positive effect on pluripotency maintenance. This could prove beneficial at the early stages of bESC derivation and possibly direct the cells toward a more naïve state.
Bovine ESCs may represent an intermediate state between mouse ESCs and mouse EpiSCs or human ESCs
WNT signals are required to inhibit the differentiation of mESCs into EpiSCs. Mouse ESCs mutant for β-catenin show morphology and marker expression patterns similar to that of EpiSCs [50]. Thus, in the following experiments, we were interested in testing the ability of the GSK3i-obtained bovine blastocysts to support pluripotency. Unlike what we have speculated, overactivation of WNT signaling in blastocysts has not proven to be beneficial for bESC derivation. However, the positive effects of GSK3i administration upon primary ESC-like outgrowth formation have been noted for the control embryos. This indicates that maintaining a specific balance between the signaling cues is critical for pluripotency.
Independent of the embryo culture conditions, we have noted that WNT activation supported blastocyst attachment to the fibroblast layer and primary outgrowth formation. In culture media supplemented either with GSK3i or GSK3i+LIF, over 90% of blastocysts attached and formed colonies after 3–4 days in culture. The positive effect of β-catenin on the blastocyst adhesion rate could reflect its dual function, where apart from acting as a transcription regulator in the WNT pathway, together with γ-catenin, it makes a component of the adherens junctions. It links cadherins at the plasma membrane to α-catenin, and as a complex with E-cadherin and α-catenin, it forms a dynamic link to the cytoskeleton (review in Brembeck et al. [51]).
Overactivation of WNT signaling might jeopardize pluripotency. Studies on human ESCs suggest that chemically defined medium (containing high concentrations of specific GSK3i) could drive ESCs to differentiate toward the definitive endoderm and generate cells of the mesodermal lineage [52]. In our system, the GSK3i-obtained embryos independent of the culture conditions did not produce colonies that could be efficiently maintained after passage. The initial outgrowths derived in LIF failed to form compact colonies. After 10/14 days in culture, the clear-cut colonies have emerged, but comprised large flat cells and failed to grow beyond one to two passages. At this stage, OCT4 and NANOG expression was lost.
GSK3i media supplementation initially gave the best results. The primary outgrowth was free of any TE cells and formed a compact colony of tightly packed round cells. These colonies were initially positive for AP, but failed to expand in culture after passage 1. The positive effects have been, however, noted for embryos derived w/o the inhibitor. In terms of the initial morphology, LIF supplementation resulted in properly defined ESC-like colonies with clear colony boundaries, without any signs of differentiation. This has been lost over the consecutive passages. NANOG and OCT4 transcripts were absent at passage 2, but the colonies still expressed REX1, SOX2, and FN1. Positive signal for NANOG has been noted only in the cytoplasm of the derived colonies, OCT4 was absent.
Although LIF is essential to maintain the undifferentiated state of mouse ESCs, it is only able to sustain stable mESCs in the presence of serum. It does not prevent the differentiation of hESCs and additional factors are required [9]. At this point, we were interested to find the proof for JAK/STAT signaling activity. Although LIF alone was not able to cause bovine ICM to develop into stem cells, it clearly had a positive effect on cell proliferation and growth. Ozawa et al. [53] showed that several molecules of the JAK/STAT pathway were present in bovine ICM. In terms of gene expression profiles that were conserved after passage 3, the +GSK3i system produced better results as NANOG, REX1, SOX2, c-MYC, and KLF4 expression was still maintained. The morphology has, however, declined over the consecutive passages with large trophoblast-like cells still visible at the colony boundary. This has been reflected by the presence of CDX2, KRT18, and ELF5 transcripts.
In the following experiments, we have tested the potential of GSK3i and LIF acting together. Similar to the results described above, embryos derived w/o the inhibitor proved to possess higher potential to maintain the undifferentiated state in subsequently derived cell lines. These cell lines were successfully passaged over six times and maintained proper morphology. The blastocysts attached early (days 3–4) and although initially the primary outgrowths consisted of TE-like cells, small colonies of the ICM derivatives appeared around days 8–10. During the following days, these colonies continued to grow, reaching 60%–70% confluency.
What is important to note, at P = 0, AP displayed a heterogeneous expression pattern as the signals were noted only in some cell clusters. After the subsequent passages, AP was uniformly distributed within the colonies, suggesting that the applied culture conditions might preferentially select cells of high pluripotency status to survive the passage. This has been further supported by the expression of all of the core pluripotency markers (OCT4, NANOG, FN-1, REX1, SOX2, c-MYC, and KLF4). Furthermore, the immunofluorescent stainings at passages 2–6 revealed nuclear localization of OCT4 and NANOG in the derived colonies.
NANOG is suggested as the key repressor of the PrE marker, GATA6, as NANOG binding motif was identified in the GATA6 promoter [48]. In mESCs, NANOG repressed GATA6 expression, but LIF withdrawal resulted in a rapid differentiation toward the extraembryonic endoderm [54]. Evidence for this may also be found in our system, where bESC lines cultured only in the presence of GSK3i maintained GATA6 expression, whereas in the LIF-supplemented system, GATA6 transcripts were lost after P = 1. The trophoblast lineage-specific transcription factors, such as ELF5, CDX2, and KRT18, have also been downregulated, although some ELF5 transcripts could still be detected at P = 4. This may, however, reflect the unstable state of these cells as ELF5 has been recognized as the gatekeeping factor that is methylated in mESCs and unmethylated in TS cells [55].
To further test the stability status of the derived bESC-like lines from SOF-HBl, we have analyzed the stability of chromosomes from pairs 12, 23 and X and Y chromosomes. In cattle, chromosome 12 is recognized as a medium-sized gene-poor chromosome (about 3.5 coding genes/Mb), containing the CDX2 locus, and chromosome 23 is a small-sized gene-rich chromosome (about 12.5 coding genes/Mb), containing the OCT4 locus. Therefore, the analysis of these pairs could provide representative data on chromosomal stability. The cell lines were analyzed at early passages (P = 0; P = 2) as we were interested in the initial stages of the bESC derivation. Because enzymatic methods of cell detachment are often associated with chromosomal abnormalities, we have relied on mechanical disaggregation during cell passaging.
Cell lines derived in +LIF+GSK3i conditions were confronted with colonies obtained only in the presence of the GSK3i. Our results show that the obtained lines, despite the presence of the specific pluripotency markers, possess a certain level of chromosomal instability.
We speculate that the observed variation in chromosomal abnormalities could reflect the state already present in the embryo (embryo effect) or could be attributed to the small number of analyzed cells. The two XY chromosome stable cell lines showed the least number of aberrations in the studied chromosome pairs (on average 1%). Only recently, the presence of chromosomal abnormalities in mouse and human ESCs started to be described as previously it has been simply assumed that the ESCs are cell lines with normal karyotype. Trisomies 8 and 11 are frequent in mESCs and trisomy 12, 17, and 20p amplifications are typical for hESCs. All these changes have been reported to confer growth advantage at the expense of cell differentiation as a result of unbalanced dosage of key genes involved in self-renewal [56].
Recent studies have indicated that regardless of culture or the chromosomal stability, all cell lines preserved different potential. There was no safe passage number preserving chromosomal integrity [57]. More importantly, all of the analyzed mESCs retained the expression of OCT4, SOX2, and REX1 and were able to form the EBs. This was also true for our system as, from the lines obtained with LIF and GSK3i, we have obtained EB-like structures expressing both CRIPTO and BRACHYURY factors, which are important for mesoderm differentiation and primitive streak formation, respectively.
Final conclusions
In naïve mouse cell lines, WNT signaling induces self-renewal and prevents differentiation into the EpiSC state. This is supported by LIF signaling through the JAK/STAT3 pathway. Mouse EpiSCs may be reverted to ESCs by exposure to LIF in the presence of key reprogramming factors: KLF2, KLF4, c-MYC, and NANOG. Human ESCs share many molecular features with mouse epiblast stem cells (mEpiSCs), including flattened morphology and intolerance to passaging as single cells. Naïve cell lines grow as packed dome-like colonies, present high cloning efficiency, and may be easily propagated at the single cell level. Although we have not investigated the X-chromosome activity to distinguish between the XaXa status of the naïve cells and the XaXinactive state of mEpiSC/hESC, several other features allowed us to speculate that the derived bESC-like cells may share some features of both stem cell types.
(1) WNT signaling clearly exerted a positive effect on pluripotency gene expression in developing bovine embryos and cell lines (manifested by upregulation of OCT4, NANOG, REX1, SOX2, c-MYC, and KLF4). Overexpression of KLF4 may revert mEpiSCs back to an ESC-like state [58]. KLF4 is one of the reprogramming factors and is an integral component of the mESC self-renewal network [59 –61]. EpiSCs virtually do not express KLF4, but bovine cell lines derived in +LIF+GSK3i culture conditions maintained KLF4 transcripts over several passages.
(2) JAK/STAT signaling was active at the initial stages of bESC derivation as it managed to maintain proper morphology and growth for two to three passages; however, LIF alone was inefficient to support self-renewal.
(3) Similar to mESCs, WNT activity supported by LIF had beneficial effects on cell culture propagation, survival after passage, morphology, and pluripotency-related marker gene expression. (4) The derived bESC-like colonies shared features of mEpiSC and hESC morphology and grew as flat monolayer colonies intolerant to passaging as single cells.
Footnotes
Acknowledgments
The authors would like to thank Dr. N. Rozwadowska (The Institute of Human Genetics, Polish Academy of Sciences, Department of Reproduction and Stem Cells, Poznan, Poland) for collaboration and preparation of MEFs; Prof. D. Cieslak (Head of the Biotechnology of Reproduction Group, Poznan University of Life Sciences, Poland) and Dr. Berenika Plusa (Faculty of Life Sciences, the University of Manchester, United Kingdom) for comments on the manuscript; and the Department of Animal Physiology and Biochemistry (Poznan University of Life Sciences, Poland) for making accessible their Confocal Microscope Facility. This work was funded by the NCN (National Science Centre) grant number: 4429/B/P01/2010/39.
Prior conference presentations:
Madeja Z, K Hryniewicz and P Pawlak. (2014). The effect of Wnt/β-catenin signaling on bovine preimplantation development—prospects for bovine ESC derivation. Joint Meeting of the British Societies of Cell Biology and Developmental Biology, 16–19 March 2014, Warwick University, United Kingdom.
Madeja ZE, K Hryniewicz and P Pawlak. (2013). The influence of Wnt/β-catenin signaling on pluripotency marker gene expression in bovine blastocysts (original title in Polish: Wplyw sciezki sygnalizacyjnej Wnt/β-katenina na ekspresję markerow pluripotencji w blastocystach bydla). IV Polish Genetics Congress, 10–13 September 2013, Poznan, Poland.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.