
Abstract
Low-density lipoprotein (LDL) uptake is one of a number of tests used to demonstrate hepatocyte-like function after stem cell differentiation. Use of two compounds, LDL and acetylated LDL (AcLDL), has been described despite each having different mechanisms of uptake. Three primary hepatocyte cultures and three sets of mesenchymal stromal cell (MSC) cultures, derived from both adipose tissue and bone marrow, were harvested from dogs. Those cells were compared to commercially available human and mouse bone marrow-derived MSCs. LDL receptor expression was demonstrated by gene expression and immunofluorescence in all primary hepatocyte cultures, undifferentiated canine bone marrow MSCs and canine adipose MSCs. Undifferentiated human and mouse bone marrow MSCs also expressed the LDL receptor. In vitro, canine hepatocytes took up labeled LDL, but not AcLDL. All undifferentiated MSCs took up LDL, but not AcLDL. In conclusion, LDL and not AcLDL is a test of canine hepatocyte-like phenotype, but this is not tissue or species specific and, therefore, is not informative assay when testing proof of MSC to hepatocyte differentiation.
Introduction
T
The pathophysiology of many spontaneous canine diseases has been shown to be similar to human clinical conditions for example, specifically in hepatology, hepatic encephalopathy [3] and fibrotic liver disease [4,5]. The ability to culture autologous stem cells, differentiate and reimplant into a large animal model with a comparable spontaneous disease niche to human cases is highly attractive. As a result, dogs are now recognized as a useful large animal translational model in the step between laboratory disease models and human clinical cases [6]. Use of animals with spontaneous disease also allows a reduction in experimental animal use.
An additional area that would be immediately attractive is in vitro drug modeling to reduce the cost of development, animal experimentation, and failure at phase III clinical trials [7 –9]. The ability to produce in vitro canine hepatocytes with species-specific CYP profiles would allow rapid screening of candidate drugs for potential hepatotoxicity not only of the parent drug, but also the hepatic metabolic products. This would not only greatly reduce the cost of drug development for the veterinary market, but also contribute to reducing the number of animals required in preclinical testing. Furthermore, the ability to screen different breed phenotypes in vitro would hopefully avoid idiosyncratic breed reactions once the drug has been licensed, reducing the number of drugs withdrawn from the market and also reducing canine patients harmed.
Isolation and culture of primary hepatocytes was first described over 40 years ago, although culture conditions which allow mature hepatocyte division in vitro have not been achieved, and hepatocytes in culture do not multiply to any appreciable extent [10]. Furthermore, the majority of functions that would be desired of a hepatocyte, for example, protein production, CYP activity are lost rapidly, in vitro [11]. A final problem with primary hepatocyte cultures is batch variability, depending on donor source and time between death and processing [12]. As a result, the continual sacrifice of animals to supply in vitro hepatocytes is not ideal, especially at a time when much effort is being directed toward the mantra of the three R's (replace, reduce, refine) in animal research. Stem cells can be isolated without the need for animal sacrifice and have a high proliferative capacity. There has been much interest in differentiation of stem cells toward a hepatocyte phenotype [1]. This would allow stem cells to be isolated from a phenotypically diverse canine population and be differentiated toward hepatocytes as required.
Of the potential stem cell sources of hepatocyte-like cells, mesenchymal stromal cells (MSCs) hold the most immediate promise in veterinary medicine. As MSCs can be isolated from almost any tissue, differences in resultant functionality from different tissue sources can be assessed [13]. Most commonly, adipose and bone marrow are used as sources and can be expanded to large numbers [14,15].
In producing hepatocyte-like cells, there is the question of how to characterize the resultant cells. Hepatocyte functionality is complex and as a result, a wide range of tests have been described, including qualitative assays such as morphology and gene expression, as well as functional assays, including albumin production, urea synthesis, and low-density lipoprotein (LDL) uptake [16]. One difficulty is that no one test is considered specific for hepatocytes, for example, although albumin is often used, gene expression has been demonstrated in pancreas, kidney, bone and microglial cells, synthesis in bone and microglia, and human bone marrow mesenchymal stromal cell (BM-MSC) and human adipose mesenchymal stromal cell (Ad-MSC) [17 –21].
A minimum panel required for differentiated cells to be classified as a hepatocyte has been defined by Hengstler et al. [22]. As it is technically demanding to provide a complete panel of testing, demonstrating one each of storage, metabolic and synthetic function is commonly accepted as proof of hepatocyte-like phenotype [23].
One of these functional assays is LDL uptake. LDL is a transport molecule for lipids, including cholesterol and triglycerides, in extracellular fluid. Specific LDL receptors on the cell membrane bind LDL and allow endocytosis. In vivo, most LDL receptors are present on hepatocytes to supply cholesterol for bile secretion, conversion to bile acids, and production of de novo lipoproteins [24]. Commercially, two types of fluorescently tagged LDL are available, native LDL and acetylated LDL (AcLDL), which is a synthetic analogue of naturally occurring oxidized LDL. AcLDL (and oxidized LDL) is not recognized by the LDL receptor, but is taken up by scavenging receptors, present chiefly on macrophages and endothelial cells [25]. In vivo, oxidized LDL is responsible for endothelial dysfunction leading to atherosclerosis [26].
Nahmias et al. showed that human and rat primary hepatocytes took up LDL, but not AcLDL [27]. Harada-Shiba et al. [28] demonstrated that culture of wild-type mouse primary hepatocytes avidly took up fluorescently labeled LDL, however, hepatocytes from LDL receptor knockout mice failed to do so. In stem cell differentiation to hepatocyte-like cells, LDL uptake is commonly used as part of confirmatory testing, yet despite this apparent clear-cut division, there appears to be confusion. There is variation in whether LDL or AcLDL is used, with many papers describing LDL use in the abstract yet stating use of AcLDL in the materials section [29 –32].
The aims of this article were to define if LDL receptors were present on canine hepatocytes in culture by gene expression and immunofluorescence as well as comparing uptake of LDL and AcLDL by these canine hepatocytes to define which should be used to demonstrate a hepatocyte-like phenotype when differentiating canine stem cells. Gene expression of the LDL receptor was compared between canine hepatocytes and canine bone marrow and adipose-derived MSCs. Finally LDL receptor expression by immunofluorescence as well as LDL and AcLDL uptake of undifferentiated canine, murine, and human BM-MSC and canine Ad-MSCs were examined. Canine hepatocytes were found to take up LDL and not AcLDL. Undifferentiated canine, human, and murine MSCs also demonstrated specific LDL uptake and the presence of LDL receptors by gene expression and immunofluorescence.
Materials and Methods
The methods used in this study were approved by the R(D)SVS Veterinary Ethics Review Committee (Study 70:13).
Primary cell isolation and culture
Isolation of canine hepatocytes
Hepatocytes were derived from liver tissue obtained postmortem from dogs euthanized for reasons unrelated to this study. Liver tissue was acquired within 20 min of euthanasia. A wedge of hepatic tissue with one cut surface and intact Glisson's capsule was sectioned (between 20 and 100 g), placed in William's Medium E (WME) with 10% fetal bovine serum (FBS) and 100 U/mL penicillin G and 100 μg/mL streptomycin (WME; Invitrogen), and transported to the laboratory on ice. The digestion protocol was based on the methods described by Seglen [33]. The tissue was rinsed with Earl's balanced salt solution (EBSS; Invitrogen, UK), placed in a Petri dish and 20–22 g plastic catheters (Vygon) inserted into the vessels on the cut surface. Chelating buffer [490 mL EBSS, 10 mL of pH7.4 25 mM ethylene glycol tetraacetic acid (Sigma-Aldrich)] at 37°C was perfused at ∼6 mL/min using a 20-mL syringe (BD) for 15 min. EBSS was then perfused at the same rate for 10 min. Finally collagenase buffer [50 mg collagenase type II (Sigma-Aldrich), 200 μL 1 MCaCl2, 100 mL EBSS] was perfused between 30 and 45 min until the tissue appeared spongy and digested. The liver capsule was then torn and the tissue gently agitated to release dissociated hepatocytes. The cell suspension was filtered through a 70 μm cell strainer (BD). The remaining liver tissue was rinsed with WME and this suspension also filtered. The resultant suspension was centrifuged at 350 rpm for 3 min to pellet mature hepatocytes. The supernatant was removed, the pellet resuspended, and centrifugation repeated.
Hepatocytes were plated at a density of 1 × 105/cm2 in WME on collagen-coated wells. After 3 h to allow attachment, the wells were gently rinsed with phosphate-buffered saline (PBS) at 37°C before hepatocyte culture medium (Lonza) was added. Hepatocytes were cultured for 24 h before further experiments were performed.
Canine MSC isolation
For cells derived from bone marrow, the distal femoral epiphysis from dogs was removed postmortem. A Jamshidi bone marrow biopsy needle (Baxter) was inserted into the medullary cavity through the trochanteric fossa and ∼40 mL of Dulbecco's modified Eagle's medium (DMEM) low glucose containing GlutaMAX-I with 10% FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin (all Invitrogen) (MSC media) was injected with the resultant cell suspension collected from the distal segment. This was transported on ice to the laboratory.
The cell suspension was diluted 1:1 with PBS and 20 mL layered onto 15 mL Ficoll-Paque PREMIUM (GE Life Sciences) in a 50-mL Falcon tube. This was then centrifuged at 450 g for 30 min without brake. The cell-containing interface was removed to a fresh Falcon tube, PBS added, and pelleted at 150 g for 5 min. The cells were then resuspended in 30 mL of MSC media, transferred to a T150 and incubated at 37°C and 5% CO2. After 48 h, media were removed, the flask washed with warm PBS and fresh media added. Media were changed every 2–3 days and cells passaged once confluent.
For cells derived from adipose tissue, ∼10 g of falciform fat was excised from dogs postmortem and placed in chilled MSC media. After transport to the laboratory, this was finely chopped into 2–4 mm pieces, placed in a 50-mL Falcon tube and warm PBS containing 100 U/mL penicillin G, 100 μg/mL streptomycin, and 1 mg/mL collagenase type II (Sigma-Aldrich). This was then incubated with constant shaking at 37°C for 2 h. Ten percent FBS was then added to inactivate the enzymes and the tube centrifuged at 300 g for 5 min. The cell pellet was resuspended in MSC media and filtered with 70 μm cell strainer (BD Biosciences). The cells were then repelleted, suspended in 30 mL MSC media transferred to a T150 and incubated at 37°C and 5% CO2. After 48 h, media were removed, the flask washed with warm PBS, and fresh media added. Media were changed every 2–3 days and cells passaged once confluent.
Human and murine MSC culture
Mouse and human bone marrow mesenchymal stem cells were purchased from Life Technologies (Gibco Cat. No. S1502-100 and A15652, respectively). These were thawed and processed according to the suppliers instructions. Ten milliliters of warm DMEM/F12 medium containing 10% FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin (all Invitrogen) was added to the mouse cells and 10 mL of MesenPRO RS™ added to the human cells. Media were changed every 3 days and cells passaged when 80% confluent.
Canine MSC characterization
Flow cytometry analysis
Cells were suspended in fluorescence-activated cell sorting (FACS) buffer (PBS with 1% bovine serum albumin) at a concentration of 1 × 107 cells/mL and 100 μL added to 5-mL Falcon tubes (BD Biosciences). Primary antibodies and, where required, secondary antibodies are listed in Table 1. For CD11b staining 20 μL blocking reagent: 2.4G2 [anti-Fc receptor (BD Pharmingen)] was added for 10 min at room temperature before antibody addition. After incubation at 4°C in the dark with antibody, cells were washed in FACS buffer by centrifugation at 4°C, 250 g, for 5 min three times. Samples were resuspended in 500 μL of FACS buffer and kept on ice in the dark for analysis. One microliter of SYTOX–Red (ThermoFisher) was added before analysis to allow gating of dead cells. Samples were run on a FACSCalibur and results acquired with CellQuestPro (both BD Biosciences). Postacquisition analysis was performed using FlowJo (Treestar). Three adipose-derived MSCs, three bone marrow-derived MSCs, all at passage 4 were tested. No antibody and isotype controls were run for each cell type and experiment. Canine peripheral blood mononuclear cells were used as positive control for MHCII, CD45, and CD19 and canine bone marrow-derived macrophages as a positive control for CD11b.
Canine MSC trilineage differentiation
Induction of osteogenesis and adipogenesis
Three adipose-derived MSCs, three bone marrow-derived MSCs, all at passage 3 were tested. Once 60%–70% confluence was achieved, commercially available media were used for adipogenesis and osteogenesis according to the manufacturer's instructions (STEMPRO® Adipogenesis Differentiation Kit and STEMPRO Osteogenesis Differentiation Kit, Invitrogen, respectively). Control cells were maintained in standard MSC media. Media were aspirated from the wells and the cells rinsed in PBS before being fixed in 10% formaldehyde for 30 min. For lipid staining, the wells were then gently rinsed with distilled water then 60% isopropanol added for 5 min. This was then aspirated and the Oil Red O solution added. The wells were then incubated for 5 min before the solution rinsed with distilled water until the rinse became clear. For osteogenesis, media were aspirated from the wells and the cells rinsed with PBS. Absolute ethanol was added to the wells for 30 min then aspirated and the wells allowed to dry. Alizarin red solution was then added for 5 min, removed, and the wells carefully rinsed three times with distilled water. Wells were examined grossly for staining as well as microscopically.
Induction of chondrogenesis
Three adipose-derived MSCs, three bone marrow-derived MSCs, all at passage 3 were tested. 2.5 × 105 MSC were aliquoted into polypropylene 1.5-mL microcentrifuge tubes (Fischer Scientific). These were then spun at 500 g for 5 min to pellet the cells. MSC media were then aspirated and either fresh MSC media or chondrogenic media added. Five tubes of each cell and media type were run. After 12 h, the cell pellet was gently detached from the bottom to become free floating by pipetting. Media were then changed every 2–3 days for 21 days. RNA extraction was performed on two pellets in each experiment. The three remaining pellets were fixed in 4% formaldehyde and Toluidine blue staining performed. Pellets were embedded in paraffin wax blocks and sections cut using a microtome. Dewaxing was performed using xylene for 15 min, descending concentrations of ethanol (100%, 95%, 90%, 70%, and 0%) for 10 min each. The sections were rinsed in distilled water and submerged in 1% aqueous Toluidine blue solution (Sigma-Aldrich) for 30 min at room temperature. The slides were then rinsed in one part distilled water and four parts 1% HCl in 70% ethanol for 5 s. The slides were then rinsed in distilled water, dried, and mounted using DPX Mountant (VWR).
Analysis of gene expression
Two adipose-derived MSCs, two bone marrow-derived MSCs, all passage three were tested along with three sets of freshly dissociated primary hepatocytes cultured for 24 h. The RNeasy Mini Kit (Qiagen) was used according to the manufacturer's instructions. Cell pellets were disrupted in 350 μL of lysis buffer using a QIAshredder (Qiagen). DNA digestion was performed using DNase I (Qiagen) at the recommended point in the RNA extraction protocol. Total RNA was quantified and purity checked using absorbance spectrophotometry at 260 and 280 nm (NanoDrop 1000; Thermo).
RNA was converted to cDNA using the Omniscript Reverse Transcription Kit (Qiagen) according to the manufacturer's instructions using random nanomers (Sigma-Aldrich) and RNase inhibitor (Promega). One microgram of RNA was added per reaction.
The Platinum SYBR Green qPCR Kit (Invitrogen) was used for all qPCR reactions. Reactions were performed on the Stratagene MX3000P (Agilent). Relative gene expression was performed using cDNA, diluted 1:20 and 9.5 μL of this added to each well. Each reaction was performed in triplicate and three no-template controls were also performed for each primer using 9.5 μL of nuclease-free water. Primers are summarized in Table 2. Cycling conditions were as follows: 2 min at 50°C, 2 min at 95°C, then 40 cycles of 95°C ×15 s and 60°C ×30 s. The final cycle was 95°C for 1 min then cooling to 60°C before monitoring for dissociation to 95°C. The dissociation curve produced and lack of amplification of the no-template controls were checked using MXPro Software (Agilent). Data were analyzed using Microsoft Office Excel 2003 program using the method described by Pfaffl [34] to calculate relative gene expression.
LDL, low-density lipoprotein.
LDL receptor immunofluorescence
Three canine adipose-derived MSCs, three canine bone marrow-derived MSCs, as well as human and mouse MSCs were tested along with a canine transitional cell carcinoma cell line. Cells were cultured in four chambered slides (BD Biosciences) until they were ∼50% confluent. Three sets of canine primary hepatocytes were cultured on collagen type I-coated plastic at a seeding density of 1 × 105/cm2 for 48 h before staining. Media were removed, the cells washed with PBS, and then fixed with 4% paraformaldehyde for 30 min. The cells were washed with PBS three times, permeabilized with 100% ethanol for 5 min before repeating the wash step. Blocking buffer [PBS containing 10% goat serum (Invitrogen) and 0.1% Tween 20 (Sigma-Aldrich)] was added for 1 h at room temperature. This was then aspirated and rabbit anti-human LDL receptor antibody with known cross-reactivity with the dog (ABIN672111, antibodies-online) was diluted to 1:500 with PBS containing 1% goat serum and 0.1% Tween was added. This was then incubated at 4°C overnight and then washed three times in PBST. Fluorescently tagged goat anti-rabbit secondary antibody (AlexaFluor 594 goat anti-rabbit IgG; Life Technologies), diluted 1:1,000 in PBS containing 1% goat serum and 0.1% Tween was added and incubated at room temperature in the dark for 1 h. Nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI) was performed as described previously. Secondary only and no antibody controls were also performed for each cell type. For each sample tested, 500 cells were examined and the percentage of positive staining was calculated.
DiI-LDL and DiI-AcLDL uptake
Sets of three canine adipose-derived MSCs, three canine bone marrow-derived MSCs, human and mouse MSCs, three canine primary hepatocytes cultured for 48 h were tested along with a canine transitional cell carcinoma cell line. DiI-LDL and acetylated DiI-LDL (both Invitrogen) were diluted to 20 μg/mL in appropriate culture media and added to cell culture wells for 3 h. Wells were then washed with PBS three times and fixed with 4% paraformaldehyde for 30 min. Nuclear staining with DAPI was performed as described previously. The slides were then washed with PBST and examined using fluorescent microscopy. No LDL controls were performed. For each sample tested, 500 cells were examined and the percentage, positively taking up the compound, was calculated.
Results
Canine MSC can be derived from bone marrow and adipose tissue and demonstrate trilineage differentiation
Both canine Ad-MSC and BM-MSC had a similar level of positive staining for CD44 (23.6% and 26.0%, respectively) and CD90 (44.6% and 40.0%, respectively) (Fig. 1 and Table 3). Both cell types were negative for MHCII, CD11b, CD19, and CD45 by flow cytometry (Fig. 1 and Table 3). On flow cytometry, both Ad-MSC and BM-MSC were negative for CD105 and STRO-1, which have previously been reported to be expressed by canine BM-MSC using immunocytochemistry [35]. Both Ad-MSC and BM-MSCs appeared strongly positive for both these markers with immunocytochemistry with a negative control demonstrating no staining (Supplementary Data 1; Supplementary Data are available online at

Expression of cell surface molecules in canine MSCs by flow cytometry, showing live cell gating and representative histogram demonstrating canine adipose and bone marrow MSC cell surface molecule expression. The shaded area represents the negative isotype control and open line represent sample labeled with the indicated cell surface molecule. MSC, mesenchymal stromal cell.
The mean percent positive from three sets of Ad-MSC and BM-MSC with standard deviation. Both MSC cells types were positive for CD90 and CD44 and negative for CD11b, CD19, CD45, MHCII, STRO-1, and CD105.
Ad-MSC, adipose mesenchymal stromal cell; BM-MSC, bone marrow mesenchymal stromal cell.
Multiple Oil Red O positively staining vacuoles were detected in the cytoplasm of both Ad-MSC and BM-MSC after adipocyte differentiation (Fig. 2A, B). Alizarin red staining demonstrated calcification and osteogenic differentiation in both cell types (Fig. 2C, D). Metachromatic staining suggestive of cartilage formation was shown after chondrogenic differentiation (Fig. 2E, F). Gene expression analysis revealed upregulation in aggrecan, but not Sox9 expression (Supplementary Data 2).
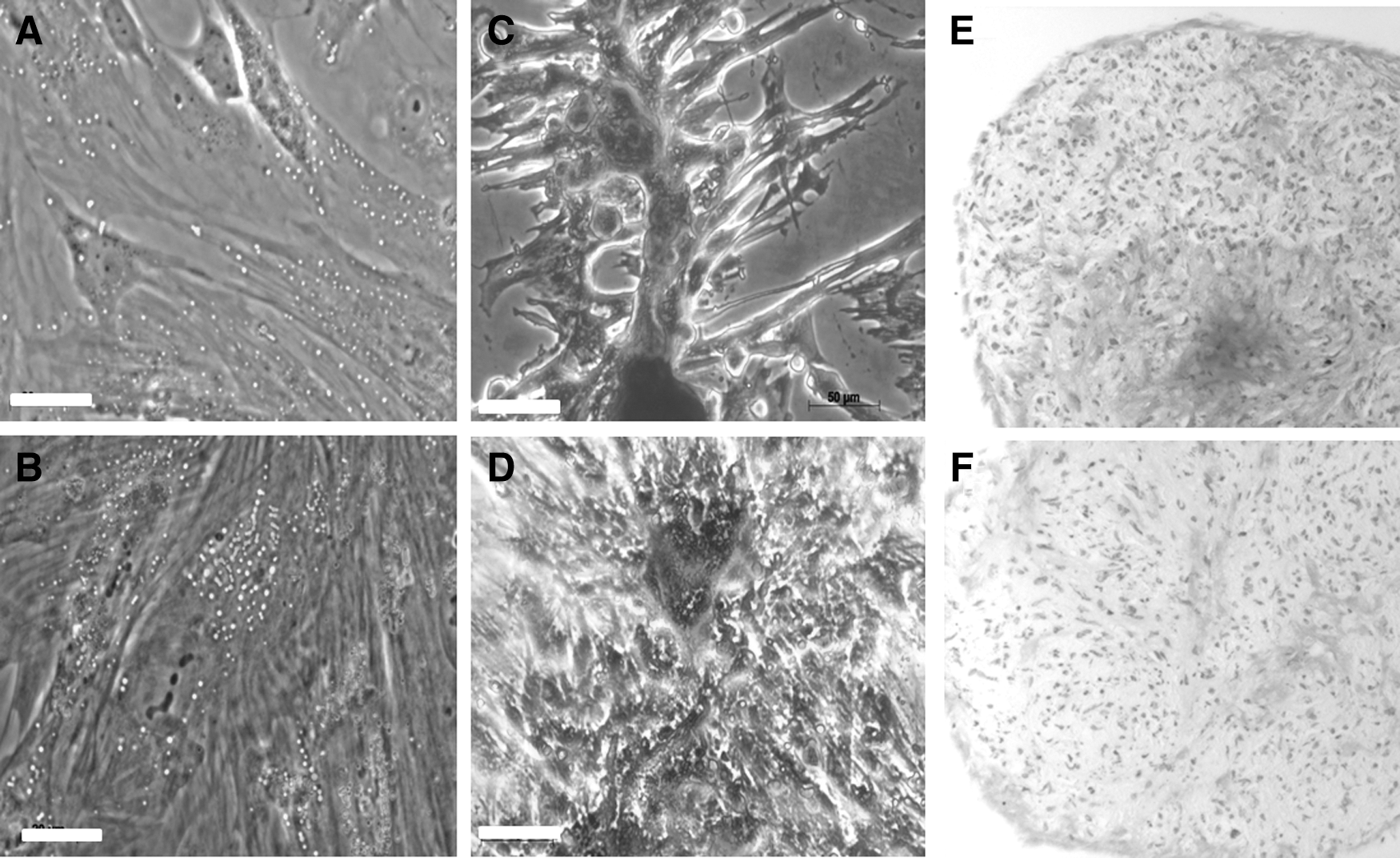
Adipogenic, osteogenic, and chondrogenic differentiation of canine MSCs. Oil Red O after adipogenic differentiation of BM-MSC
Canine primary hepatocytes and MSC demonstrate LDL receptor expression by real-time PCR and immunofluorescence
Gene expression relative to the three reference genes demonstrated LDL-receptor expression in canine BM-MSC and Ad-MSC of a similar magnitude to canine primary hepatocytes (Fig. 3). Of the three primary hepatocyte cultures, relative expression varied from 0.03 to 0.3. These were hepatocytes in the first 24 h of culture.
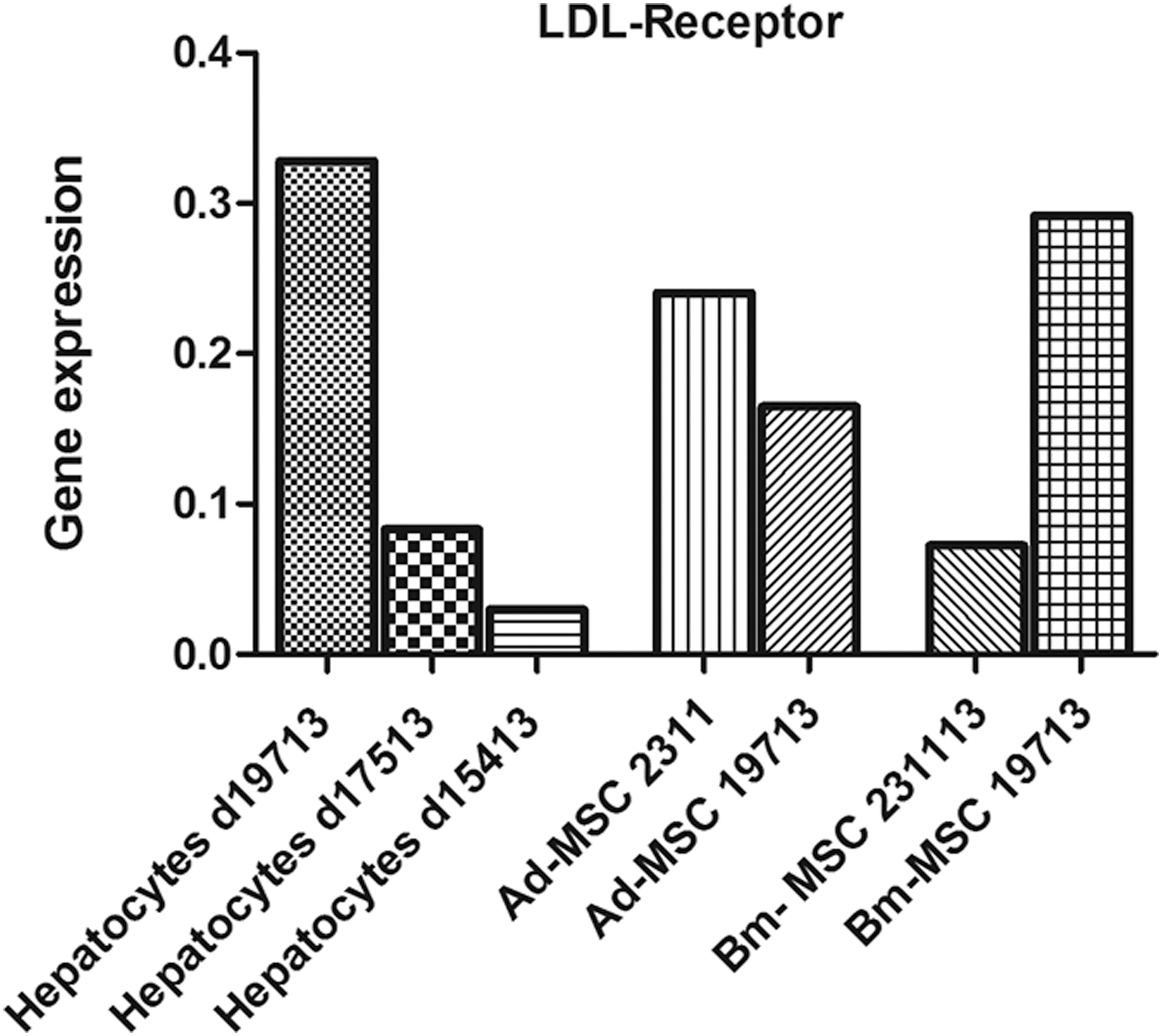
LDL-receptor gene expression in canine primary hepatocytes and Ad-MSC and BM-MSC. Relative gene expression of LDL receptor normalized to three reference genes (B2MG, RPL8, HPRT). Three sets of fresh cultured canine primary hepatocytes and two each of Ad-MSC and BM-MSC cultures. Three replicates per cell type were performed. LDL, low-density lipoprotein.
LDL receptor expression was documented by immunofluorescence in canine hepatocytes, canine, murine, and human BM-MSCs as well as the canine Ad-MSCs (Fig. 4). All cell types demonstrated immunofluorescence. The three primary canine hepatocyte cultures' median percentage of positive cells was 96.8% (range 94.8%–98.2%). Canine Ad-MSCs' median percentage of positive cells was 98.8% (range 98.2%–99.2%). The canine, human, and murine BM-MSCs' percentage of positive cells were: 98.6% (range 97.8%–99.9%), 99.5% (range 99.4%–99.8%), and 99.6% (range 99.4%–99.8%), respectively. Negative controls demonstrated a low level of background immunofluorescence in the canine primary hepatocytes and canine BM-MSCs. The transitional cell line demonstrated no binding.
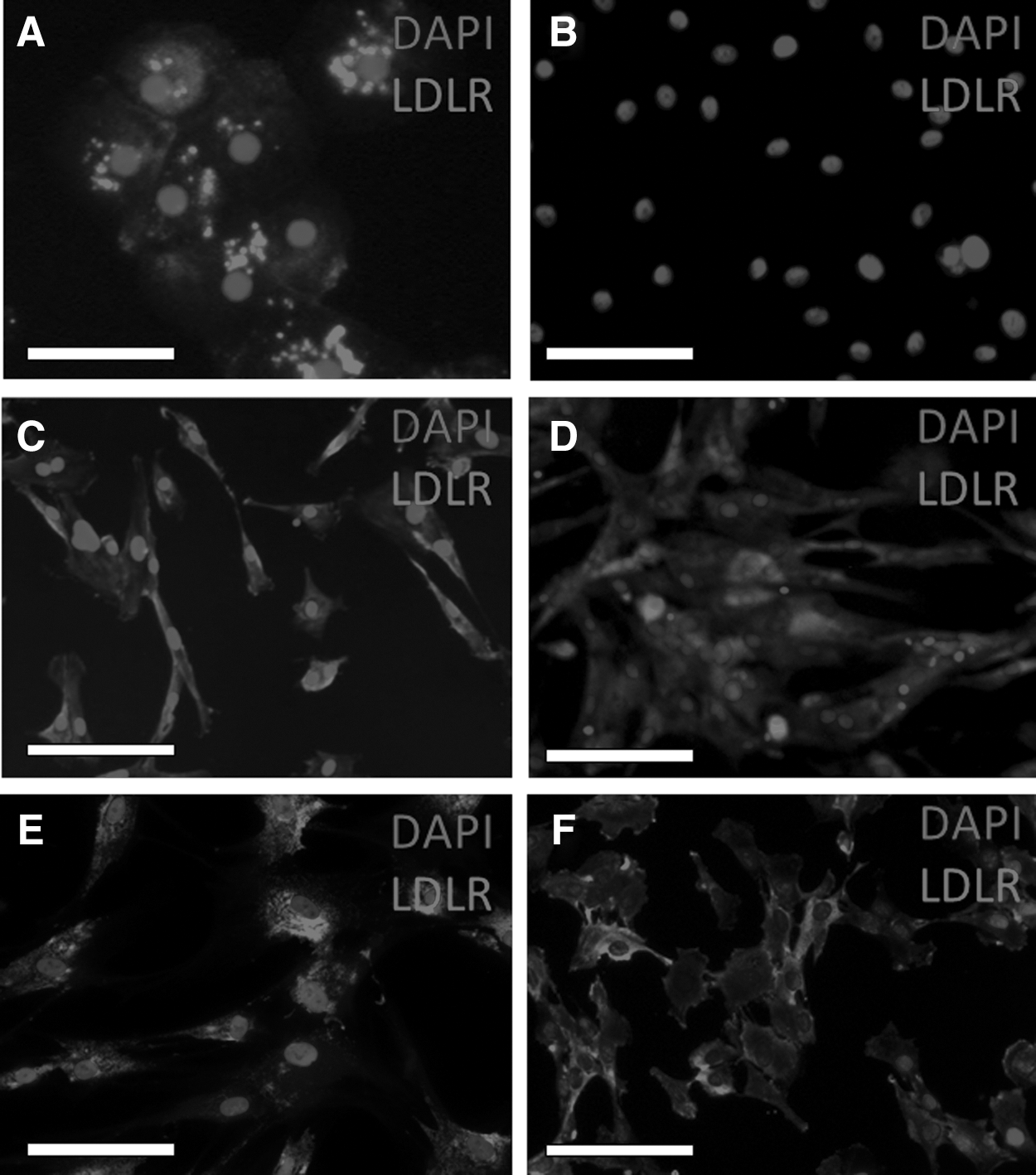
LDL receptor immunofluorescence. Canine hepatocytes
Hepatocytes and MSCs take up LDL and not AcLDL
After incubation with DiI-LDL, all canine hepatocytes, BM-MSC and Ad-MSC, as well as murine and human MSC demonstrated uptake of this compound while the transitional cell carcinoma cells demonstrated no uptake (Fig. 5). After incubation with AcLDL, none of the canine, murine, or human MSCs was positive, although some fluorescent extracellular debris was noted (Fig. 6). Within the canine hepatocyte cultures, although the vast majority of cells showed no uptake, there were sporadic individual cells, which demonstrated avid uptake of AcLDL (Fig. 6A demonstrates a cluster of these cells). Overall, a median of 4.5% of the canine primary hepatocyte culture cells was positive for AcLDL uptake (range 3.2%–6.7%).

DiI-LDL uptake in canine primary hepatocytes and MSCs. Canine hepatocytes
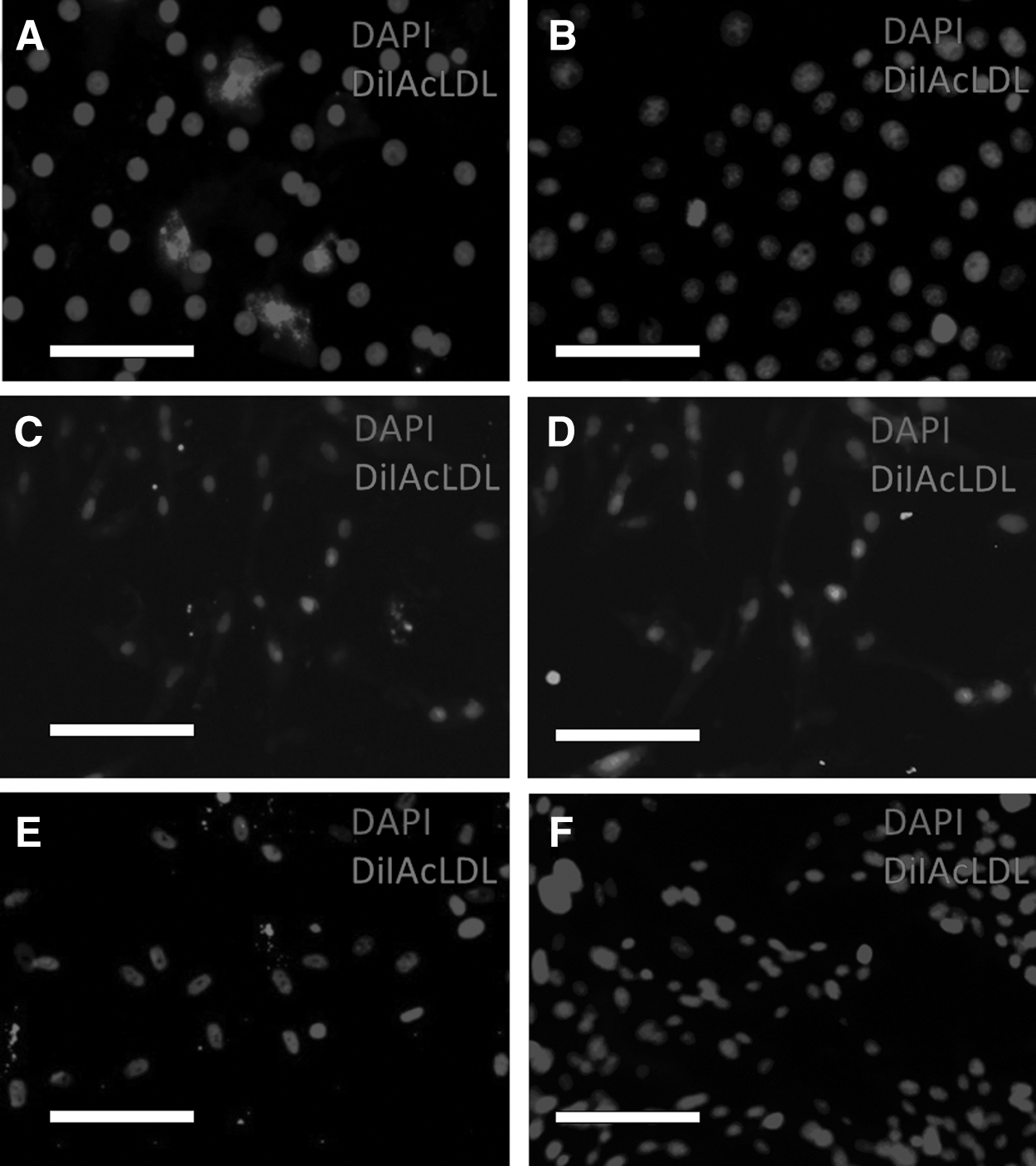
DiI-AcLDL uptake in canine primary hepatocytes and MSCs. Canine hepatocytes
Discussion
From these results, canine primary hepatocytes in culture have LDL receptor expression as demonstrated by gene expression and also immunofluorescence. Comparing uptake of LDL and AcLDL in the hepatocyte culture demonstrates that LDL uptake is mediated by this receptor as only LDL and not AcLDL is taken up by the hepatocytes. Therefore, uptake of LDL and not AcLDL should be used as part of the demonstration of hepatocyte-like function for canine cells. This is consistent with the previously reported function of human and mouse primary hepatocytes [27,28]. It is of note that sporadic cells in the hepatocyte culture plates took up AcLDL avidly and it is likely that these are either Kuppfer cells or endothelial cells, which were isolated along with hepatocytes from the liver digestion process. Both these cell types are reported to contain the scavenging receptors, which endocytose acetylated or oxidized LDL [25,36]. This is consistent with the report by Babaev et al. [37] who found that human primary hepatocyte cultures almost uniformly took up LDL with only 5% of cells in culture taking up modified LDL. These were identified as macrophages or endothelial cells based on their ability to take up tagged formaldehyde-treated albumin and carboxylated microspheres. In the present study, only 4.5% of cells in the primary hepatocyte cultures took up AcLDL, a similar number to the previous study.
The isolated and cultured canine BM-MSC and Ad-MSCs demonstrated adipogenic and osteogenic potential. We have previously demonstrated isolation and characterization of canine BM-MSC, including the demonstration of chondrogenic differentiation based on Toluidine blue staining, and increased collagen type II gene expression and Sox9 immunostaining [35]. In the present study, Toluidine blue metachromatic staining of cartilage pellets, although present, was less dramatic than that demonstrated by Requicha et al. [38]. Both cell types also demonstrated significant increases in aggrecan gene expression, the major proteoglycan in articular cartilage produced by chondrocytes [39]. Interestingly, both cell types showed no increase in Sox9 expression, which is the master-regulator of chondrogenesis. As one of the functions of Sox9 is to bind to the aggrecan promoter and upregulate aggrecan expression, this appears as an unusual result [40].
One possible explanation for this apparent paradox is that gene expression during differentiation is dynamic and Sox9 gene expression is shut off during final maturation of chondrocytes into hypertrophic chondrocytes [41]. Therefore, it is possible that upregulation of Sox9 was missed in these samples. Another possibility is that chondrogenesis is dysregulated and stimulation of gene expression is downstream of Sox9 expression. From the literature there appears to be a variation in the Sox9 expression. Reich et al. detected an increase in Sox9 with BM-MSC, but a decrease with Ad-MSC [42]. This corroborated with more convincing cartilage formation in BM-MSC by histology and upregulation of Collagen 2A1 only in BM-MSC.
Other authors have demonstrated convincing chondrogenic differentiation of both canine Ad-MSC and BM-MSC with Sox9 expression and also convincing chondrogenic differentiation with no Sox9 upregulation [38,43,44]. However, other studies have failed to demonstrate chondrogenesis in canine MSCs [45,46]. These disparate results need to be viewed with knowledge that a donor age-related reduction in differentiation ability of MSCs has been demonstrated, along with variation in ability according to donor site and passage number [43,47 –49].
In this study, both Ad-MSC and BM-MSc demonstrated no expression of CD105 or STRO-1 by flow-cytometry yet appeared positive on immunocytochemistry. Canine BM-MSC has previously been reported to be positive for CD105 and STRO-1 by immunocytochemistry [35], however Screven et al. found both canine Ad-MSC and BM-MSC to be negative on flow cytometry, but positive for CD105 gene expression by real-time PCR [50]. It is possible that the antibody is not suitable for flow cytometry; however, further investigation will be required to define if CD105 expression is a useful marker for canine MSC's.
Both undifferentiated canine BM-MSC and Ad-MSC demonstrated LDL receptor gene expression of a comparable magnitude to canine primary hepatocytes. Furthermore, undifferentiated canine, mouse, and human BM-MSCs and also undifferentiated canine AD-MSCs stained for the presence of the LDL receptor by immunocytochemistry. Functionality of the receptor was confirmed by LDL uptake and not AcLDL uptake. Therefore, it would appear that for differentiation studies producing hepatocyte-like cells, confirming LDL uptake is not specific for a hepatocyte phenotype and in addition it should not be used for MSC to hepatocyte differentiation studies.
It has been demonstrated that mouse BM-MSC express LDL-oxidized receptors (LOX-1) and take up DiI-oxidized-LDL (the biological analogue to acetylated-LDL) and also express LOX-1 by gene expression and western blot, which is in contrast to the present study, where all MSCs tested (canine Ad-MSC and canine, murine, and human BM-MSC) were found not to take up AcLDL [51,52]. These apparent differences may be an artefact of culture conditions rather than a fundamental cellular difference as Liesveld et al. showed that human BM-MSC took up DiI-LDL in standard McCoy's media with 10% FCS, whereas culture in McCoy's with 25% serum and 1 μM hydrocortisone, this reduced, and DiI-AcLDL uptake increased [53]. Both the mouse and human BM-MSCs were cultured under the conditions recommended by the supplier and produced identical results to the canine MSCs, that is, no AcLDL uptake, but avid LDL uptake.
In line with the theory that MSC are fibroblast populations [54], it has been demonstrated that fibroblasts express the LDL receptor, take up LDL, but not modified LDL [55 –58]. Therefore, in this respect, the presence of LDL receptors and specific uptake of LDL on MSCs is not surprising.
In summary, LDL receptor is present on canine primary hepatocytes in culture. LDL, and not AcLDL uptake, is a function of canine primary hepatocytes. LDL receptor expression and LDL uptake is not specific to a hepatocyte phenotype and undifferentiated MSCs express the LDL receptor and endocytose LDL. Assessing the effect of culture conditions on LDL receptor expression and the cells' ability to take up LDL and AcLDL would be useful to assess if canine MSC demonstrates the same plasticity as human BM-MSC with regard to LDL and AcLDL uptake.
Footnotes
Acknowledgments
Thank you to Neil MacIntyre of Easter Bush Pathology for performing the Toluidine blue staining and Irene McGuinnes for assistance with flow cytometry. A.G.G.'s PhD studentship was funded by the Biotechnology and Biological Sciences Research Council.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.