
Abstract
The potential use of human embryonic stem cells (hESCs) in cell-based therapies points out the critical importance of epigenomic evaluation for cell-based therapies. Specifically, DNA methylation appears to be a crucial player in establishing cell fate commitment and lineage choices. In this study, we report the global changes observed on the CpG islands distributed in promoters, gene bodies, and intergenic regions and the major biochemical pathways and genes involved in methylation changes as H9-hESCs turn into a neuronal culture containing medium-sized spiny striatal neurons (MSNs). Using an ontogeny-recapitulating protocol of striatal neuron differentiation, we analyzed DNA methylation profiles during the conversion from pluripotency to neuropotency up to the acquisition of a mature neuronal phenotype. H9-hESCs changed the methylation pattern both through de novo methylation and hypomethylation of specific gene promoters. Bioinformatic analysis allowed us to identify a panel of striatal-associated genes, which were regulated by DNA methylation and differentially expressed during striatal commitment. Importantly, DNA methylation analysis revealed that H9-hESCs did not acquire methylation-based oncogenic properties after differentiation. Indeed, hypermethylation of cancer-associated genes that characterize transformed cells, such as Polycomb repressive complex-associated genes, was not detected in the neuronal cultures. However, the oncosuppressor gene, BCL2L11, became hypermethylated in H9-hESC-derived mature neurons. Whole-genome DNA methylation profiling could become a technological platform to predict the differentiative potential of hESC-derived cultures and establish further biosafety assessment quality control tools of the cell-based products.
Introduction
C
DNA methylation is one of the most extensively studied epigenetic landmarks and contributes to several biological processes, including cell identity and tissue-specific gene expression [4,6,7]. The process consists of the covalent addition of a methyl group on the 5′ position of cytosine, catalyzed by DNA methyltransferase, and occurs predominantly at CpG dinucleotides (80%) that are largely concentrated in small regions termed CpG islands (CGIs) [8]. CGIs localized in about 70% of human promoters [9] are grossly unmethylated, but tend to be methylated in specific tissues or during development [10]. DNA methylation plays an essential role during embryogenesis [11,12], and abnormal methylation severely blocks cell differentiation to certain lineage states [13]. Therefore, the effects of DNA methylation changes during development and differentiation are of considerable interest as this process still remains elusive.
Human embryonic stem cells (hESCs), derived from the inner cell mass of the preimplantation overexceeding blastocysts [14], are pluripotent cells capable of self-renewal and proliferation while retaining the potential to differentiate into cell types representative of the three germ layers [15]. hESCs can be exploited as an in vitro system to study DNA methylation and cellular identity as they are able to recapitulate the in vivo embryonic developmental programs. Specific and peculiar cytosine methylation patterns contribute to maintain the pluripotent status of hESCs [16,17], and as development proceeds, fine-tuned cell-specific changes in CpG methylation occur and contribute to the lock of cell fate decision [18]. DNA methylation during cell commitment is characterized both by temporal regulation and CpG content of specific sequences [19].
To fully take advantage of hESC potential in cell-based therapy strategies and in in vitro modeling of neurodegenerative disorders, not only efficient differentiation protocols in addition to comprehensive knowledge of the epigenetic mechanisms regulating neuronal cell commitment and biosafety are needed. It has been reported that the hESC DNA methylation profile is not directly involved in the pluripotency network, but it is critically implicated in differentiation processes by setting the stage for lineage switching [20]. DNA methylation is also an important marker to be considered to monitor possible cancer-associated modifications of pluripotent cells during differentiation as hypermethylation of peculiar loci might influence the oncogenic potential of cell progenies [21].
In this study, we compared the CpG methylation profiles of pluripotent H9-hESCs during neuronal differentiation using an established model that is able to generate striatal neurons [22]. We investigated the DNA methylation status of 27,800 CGIs in the H9-hESC line and in vitro committed cells at different time points, which represent neural induction, patterning, and terminal differentiation.
While undifferentiated cells showed a prevalence of unmethylated CpGs, differentiated neurons were characterized by a predominant gain of methylation. These results suggest that the differentiation process is, at least in part, epigenetically regulated and that H9-hESCs acquire cell type-specific de novo DNA methylation marks during differentiation. A subset of genes associated with neuron development was also detected, revealing regulatory networks between DNA methylation and cell fate specification.
Methylome mapping together with a fully defined ontogeny-recapitulating protocol provides a comprehensive view of the methylation changes that occur during differentiation and enables a better understanding of the mechanism underlying neuronal subtype-specific differentiation. Importantly, H9-hESCs showed no DNA methylation hallmarks of cancer cells when compared with stem cells from glioblastoma multiforme (GBM) [23]. Overall, these data suggest that whole-genome DNA methylation profiles contribute to a better understanding of the differentiative potential of hESC-derived neuronal subpopulations and may constitute an optional quality control system to determine the safety of cell-based products.
Materials and Methods
Cell culture and striatal neuronal differentiation
The H9-hESC line was cultured and differentiated as described in Delli Carri et al. [22]. During neuronal induction, cells were plated on Matrigel (BD, Becton Dickinson) at a density of 0.7 × 105 cells/cm2 in mouse embryonic fibroblast-conditioned hESC medium supplemented with 10 ng/mL FGF2 and 10 μM Y-27632 (Sigma-Aldrich). Cell cultures were expanded for 3 days until confluent. The starting differentiation medium included knockout serum replacement medium (Life Technologies) supplemented with 500 nM LDN (provided by Evotec) or 500 ng/mL noggin (R&D Systems) and 10 μM SB431542 (Tocris), which was used until day 12. Every 2 days, the medium was replaced with fresh medium containing an increasing proportion of N2 medium (25%, 50%, 75%). Starting on day 5, 200 ng/mL SHHC-25II (R&D Systems) and 100 ng/mL DKK1 (Peprotech) were added to the culture and maintained for 3 weeks. From day 15, the entire cell population was detached and replated on poly-
Immunofluorescence
Cells were fixed in 4% formaldehyde, permeabilized with 0.5% Triton X-100, and blocked with 5% fetal bovine serum. Primary antibodies were incubated overnight at 4°C (Supplementary Table S1A; Supplementary Data are available online at
DNA isolation and sonication
Total DNA was isolated from H9-hESC-derived samples at days 0, 5, 15, and 45, using the QIAamp DNA Mini Kit (Qiagen), and the DNA concentration was determined using Nanodrop ND-1000 (ThermoScientific). DNA fragmentation was checked on 0.8% agarose gel. Five micrograms of genomic DNA was resuspended in 250 μL of 1× phosphate-buffered saline and sonicated to obtain sheared DNA that ranges from 200 to 1,000 bp in size. Two biological replicates were performed for each differentiation experiment [24 –27].
Agilent 244K meDIP-Chip DNA methylation assay
The Agilent platform contains 237,220 oligonucleotide probes across the genome covering about 60% of RefSeq genes (about 15,200 RefSeq genes) with an average number of 8 probes per CpG site and probe spacing of 100 bp apart. Probes are distributed on promoters, gene bodies (intragenic), intergenic, and unknown regions, and the locations are downloaded from the UCSC genome browser, hg18, NCBI build 26.2, March 2006, as reference assembly. Specifically, probes are associated with promoter regions when located within 10 kb upstream of the transcription start site (TSS), intragenic or gene body probes instead are placed within 10 kb downstream of the TSS, whereas intergenic regions are 10 kb downstream of the end of the gene transcription. Unknown regions mean that the probes do not fit any of the above criteria.
Five micrograms of sheared genomic DNA was immunoprecipitated using an anti-5-methylcytosine antibody (Eurogenetec). Immunoprecipitated and reference DNA were labeled with two different fluorescent dyes using the Agilent Genomic DNA Labeling Kit Plus, and then hybridized onto the microarray (Agilent Human CpG Island 1 × 244K Microarray Kit; Agilent Technologies).
Microarrays were scanned using the Agilent microarray scanner and images were analyzed with Agilent Feature Extraction software v10.7, which allows carrying out linear normalization and calculation of the log2 ratio value. Data were further analyzed by means of Agilent Genomic Workbench v5.0 software. The Agilent algorithm associates with each probe with combined z-score and P values for each probe expressing probabilities and confidence values for methylated and unmethylated probe populations. Cutoff value determination and data analysis were performed according to the methodological approach conceived by Straussman et al. [28]. X-chromosome was excluded from further analysis to eliminate gender effects.
Microarray data analysis
Differentially methylated regions (DMRs) on promoters were identified by calculating the differences in average combined z-score values between groups using a threshold of >0.10 [29,30] and false discovery rate (FDR) <0.05 in a t-test analysis [31,32] to ensure a stringent control and reduce the number of false positives, increasing the chance to identify robust DMRs. In a pairwise analysis, CGIs with a coefficient variation >0.25 were excluded to discard intersample variation [31].
Bioinformatic analysis
Bioinformatic analysis allowed to integrate DMR data and to understand the biological processes and functions in which differentially methylated genes were involved. Differentially methylated gene lists were analyzed to identify enrichment of functional annotated categories [gene ontology (GO)] using GOstat software [33], and then summarized using REVIGO [34]. Cluster analysis and heat map of relevant GO-associated genes were assessed through CIMminer resource, using the average linkage with Euclidean distance setting [35].
Pathway analysis was performed to identify over-represented signaling using Ingenuity Pathway Analysis software (IPA,
Expression analysis by quantitative real-time PCR
H9-hESC mRNA was isolated using the RNAeasy Mini Kit (Qiagen), quantified with Nanodrop ND-1000 (ThermoScientific), and retrotranscribed with Oligo(dT) primers and SuperScript II (Life Technologies). Quantitative real-time PCR (qRT-PCR) was assessed in triplicate on at least two independent replicates on Rotor-Gene Q (Qiagen) using SYBR Green (Fermentas) detection. Data were normalized to GAPDH expression and compared using the ΔΔCt method. Primer sequences are described in Supplementary Table S1B.
Results
Generation of neurons with MSN properties from hESCs
Recently, a new ontogeny-recapitulating stepwise method has been established to obtain authentic and functional striatal neurons [22]. To generate specific MSNs, rigorous work on human developmental biology was performed to define key neurodevelopmental molecules, which play a role in the specification of striatal neurons and their temporal and spatial expression [38].
Briefly, H9-hESCs were exposed to ventral telencephalic specification by inhibition of bone morphogenetic protein (BMP)/transforming growth factor beta (TGFβ) and further SHH/DKK1 treatment (Fig. 1A–J). Telencephalic progenitors, OTX2+ (Fig. 1G) and FOXG1+ (Fig. 1H), were obtained from the neuralizing H9-hESCs, respectively, at days 5 and 15. The telencephalic identity of these cells was confirmed by the expression of FOXG1 (58% of cells, Fig. 1H), while ventral specification was revealed by the presence of about 60% of cells, which were GSX2+ (Fig. 1I). At day 45 of neural differentiation, βIII-tubulin+ neuronal cells (80% of cells) and MAP2ab positivity were evident in the dish. The early postmitotic striatal marker, CTIP2, was found in the majority of the cells (>50%) and differentiated DARPP-32+ MSNs began to appear (Fig. 1I).

Neural conversion of H9-hESCs to MSNs.
At the end of differentiation, 51% of cells were MAP2ab+. A definitive 20% of neurons DARPP-32+/CTIP2+ was obtained. The use of an established differentiation protocol and the presence of marker-specific cell populations, representing subsequent developmental phases of striatal development, allowed us to follow the DNA methylation changes during defined differentiation stages.
DNA methylation profiles of hESCs exposed to a striatal differentiation protocol
We monitored DNA methylation changes using the Agilent DNA methylation array during H9-hESC differentiation into striatal neurons. The Agilent microarray consists of ∼15,200 RefSeq genes and about 27,800 CGIs distributed in promoter, intergenic, intragenic, and unknown regions. Combined z-score values for each probe were normalized ranging between −1 and 1 (where −1 is not methylated and 1 is fully methylated), resulting in a bimodal pattern of methylation.
DNA methylation changes were monitored at day 0 (pluripotent status), day 5 (neural induction), day 15 (neural patterning), and day 45 (mature neurons). A global hypomethylation of CpG sites was observed in H9-hESCs at the pluripotent stage (18% of methylated CpG), whereas gain of methylation resulted at day 5 (28% at neural induction) and day 45 (32% with generation of striatal neurons, Fig. 2A). A substantial decrease of methylation occurred at day 15, reaching a level close to the pluripotent state. This pattern indicates the dynamicity or epigenomic flexibility of hESCs throughout the differentiation processes.
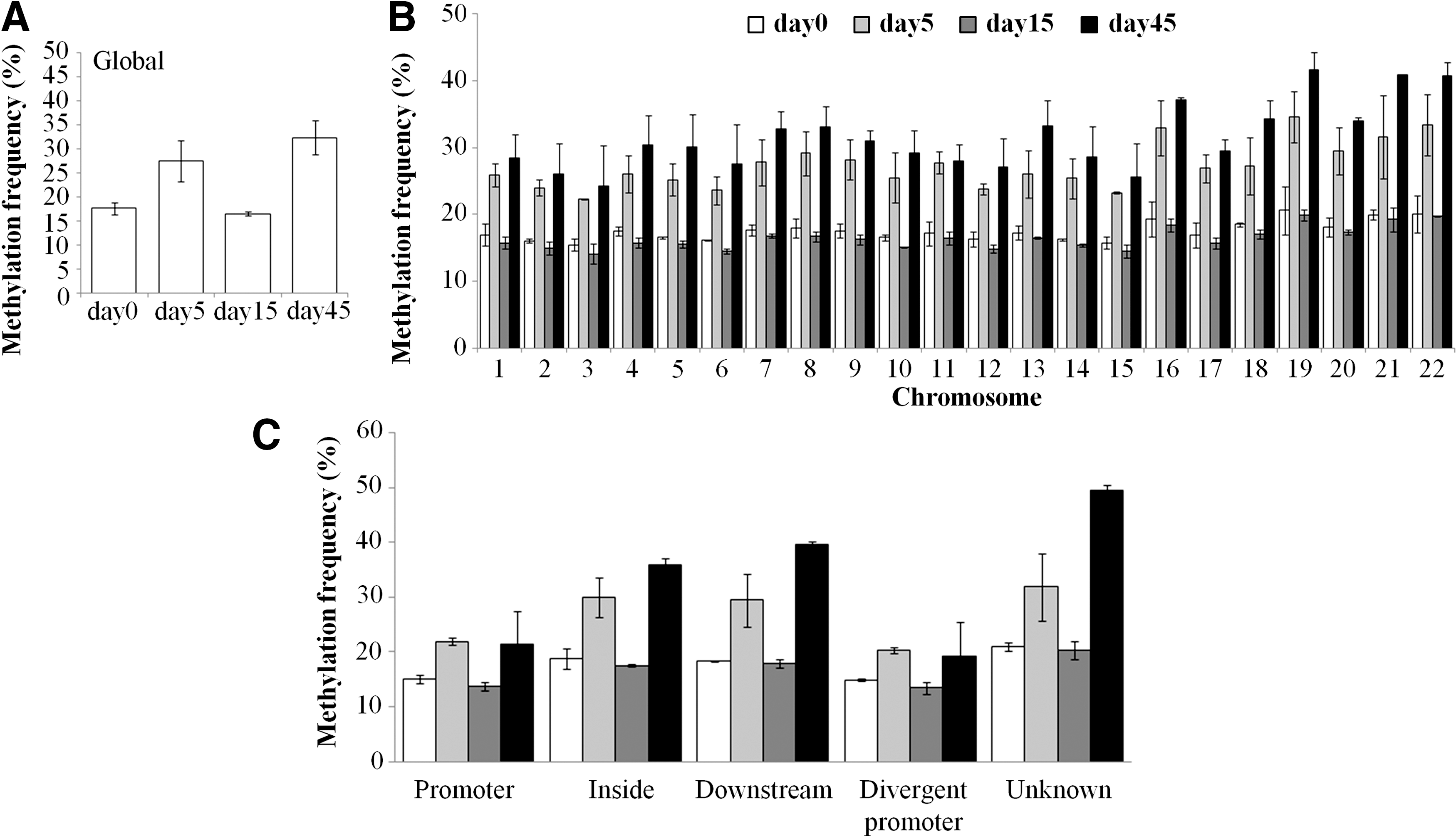
Global methylation frequency.
The mammalian genome consists of CpG-rich (high CpG content, HCpG) and CpG-poor (low CPG content, LCpG) regions [39,40], and very little is known regarding the functional regulation of the LCpG regions. A trend toward higher levels of 5-methyl-cytosine (5mC) in LCpG regions with respect to HCpG was seen in terminally differentiated H9-hESCs (Supplementary Fig. S1). However, the LCpG regions showed a higher variance in their methylation status and thus HCpG regions seemed to own a more stable methylation status. The percentage of 5mC was uniformly distributed among all the chromosomes (Fig. 2B), although a higher methylation frequency was identified in chromosomes with a greater gene density (such as chromosomes 16 and 19–22) and maintained during the differentiation process, both in HCpG and LCpG regions (Supplementary Fig. S2).
The Agilent annotation platform defines four probe categories: those located in the promoters (within 10 kb upstream of the TSS); intragenic probes located in transcribed regions or gene bodies (within 10 kb downstream of the TSS), intergenic regions (within 10 kb downstream of the transcription end), and unknown regions. Globally, the methylation pattern was conserved among all the genomic regions, although nonpromoter regions appeared to be more influenced by methylation changes when raw methylation percentages are considered (Fig. 2C). Indeed, at days 5 and 45, the fold changes in the methylation pattern were higher in other genomic regions than those in the promoter and divergent promoter regions (Supplementary Fig. S3).
Analysis of methylation changes in the promoter regions during striatal commitment
We searched for possible methylation signatures as H9-hESCs were progressing toward striatal specification by focusing on the main promoter regions since the functional role on other genomic regions is still debated and no univocal mechanism has yet been proposed. Indeed, changes in the methylation status of promoter regions are associated with functional changes in gene expression. In any case, a global analysis on changes in the DNA methylation of inside and downstream regions is included in Supplementary Fig. S4.
DMRs at promoters were identified through a pairwise comparison between different time points, using an FDR correction in a t-test analysis with methylation changes greater than 10%. DMR differences ranged from 0.9% to 17.7% (Fig. 3A and Supplementary Table S2), most occurring at day 45 when compared with the pluripotency stage (day 0, 17.7%) and rosette stage (day 15, 7.5%). The epigenomic dynamics during exposure of H9-hESCs to the striatal differentiation protocol is appreciated in Fig. 3B, where strong prevalence of hypermethylated DMRs appeared during differentiation: at days 5 and 15, the same percentage of methylation occurred when compared with day 0 and sustained at day 45. On the other hand, serially comparing each differentiation stage to the previous one revealed hyper- and hypomethylation switches between differentiation steps (Fig. 3B).
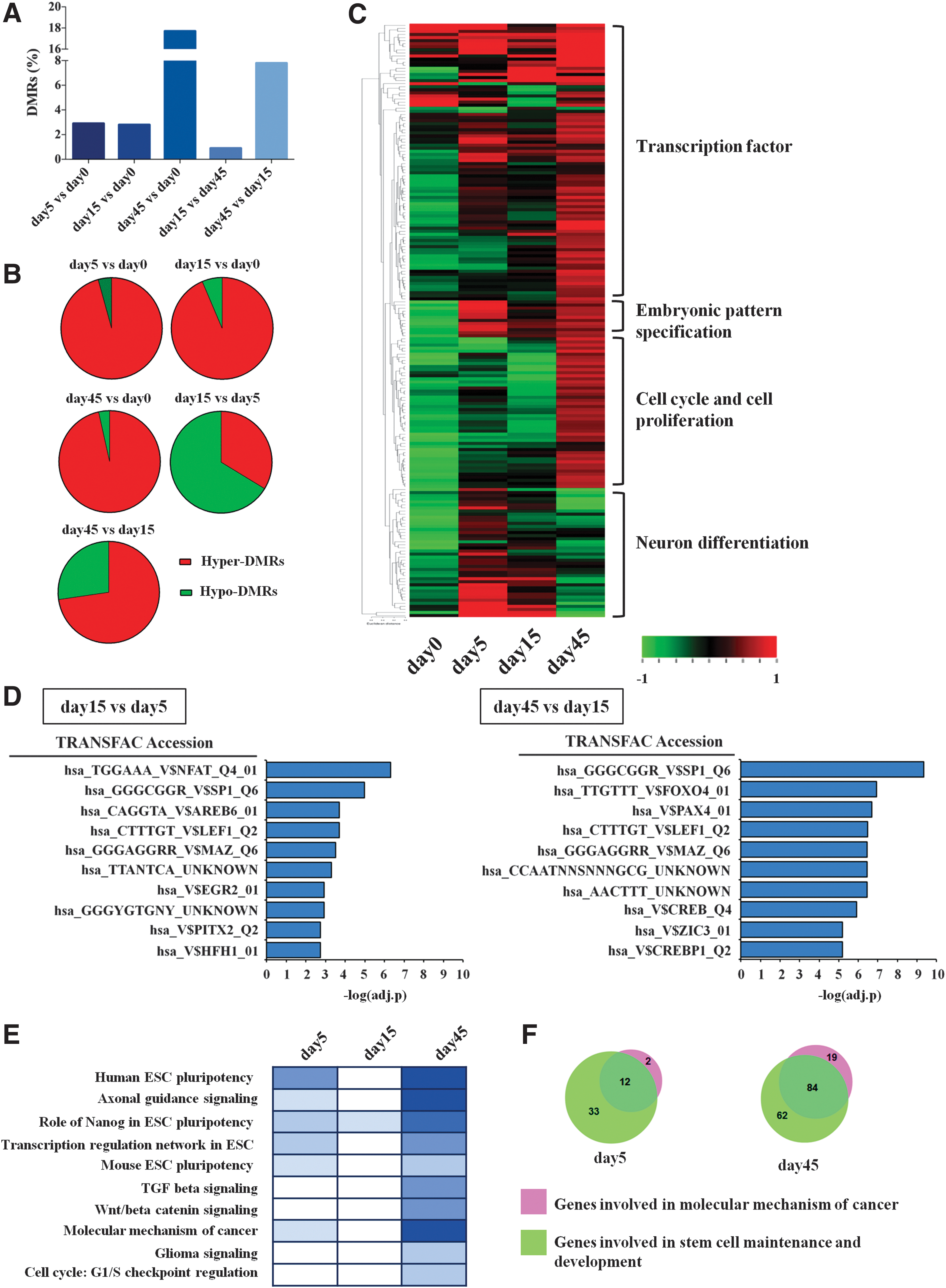
Identification of DMRs at promoters and functional analysis.
Functional relevance of promoter-associated DMR genes
Supplementary Table S3 describes the functional enrichment analysis of the biological roles associated with DMR genes. Cluster analysis of selected genes (based on their functional profile) showed distinctive patterns of methylation changes. During sequential stages of cell commitment, these changes were associated with specific biological processes, such as neuronal differentiation, embryonic patterning, or cell cycle and proliferation (Fig. 3C). Besides, to obtain a deeper insight into the role of differentially methylated genes, we investigated if hypomethylated DMRs were targeted by specific TFs. We identified top 10 TF families that showed binding site enrichment in the hypo-DMRs (Fig. 3D).
At day 15, NFAT, AREB6, and PITX2 were over-represented, whereas at day 45, FOXO4, CREB, and CREBP1 were enriched, showing a switch toward more restricted neuronal commitment as these TFs specifically activate genes involved in neuronal differentiation pathways. Moreover, pathway analysis was assessed to uncover the molecular signaling specifically associated with cell fate commitment. Each differentiation step was compared with the pluripotent stage (day 0) and the highest statistically ranked pathways were associated with hESC pluripotency, axonal guidance, or other stem cell-associated pathways, such as TGFβ and Wnt signaling (Fig. 3E). These pathways also displayed a decrease of the P value, which is associated with a greater statistical significance in the progressive switching off in the stemness-associated genes whose promoters were hypermethylated compared with day 0. Instead, cell cycle and proliferation appeared only at later stages of differentiation.
Pluripotent and in vitro differentiated H9-hESCs showed no methylation-based oncogenic features
The translational use of in vitro differentiated hESCs necessitates stringent quality controls and, in particular, it is extremely important to verify that these cells do not acquire tumorigenic features in the DNA methylation profiles due to in vitro manipulation and continuous culturing. Pathway analysis revealed the involvement of genes related to the molecular mechanism of cancer: this was not surprising since almost a complete overlap between cancer- and pluripotency-associated genes occurred (82%–86%, Fig. 3F). Nonetheless, we checked the possible role of each putative cancer-associated gene that did not overlap with stemness functions (Supplementary Table S4).
Among the 21 genes, 19 were possible oncogenes (on the basis of their function); however, they were identified in committed cells as hypermethylated, therefore, no increased tumorigenic potential could be attributed to the methylation changes of these genes. The remaining two genes are possible oncosuppressors: BCL2L11 and RBL2. We compared their expression with the GBM stem cell line (G144, Supplementary Fig. S5). While RBL2 revealed no correlation between the expression and its promoter methylation state, BCL2L11 promoter hypermethylation resulted in downmodulation of its expression in committed cells (day 45) and BCL2L11 mRNA levels decreased to a level comparable with that of G144 cancer cells. Therefore, BCL2L11 methylation changes may likely contribute to tumorigenicity in committed cells.
We also compared possible methylation-associated tumorigenic features of the H9-hESCs at days 0 and 45 with that of the G144 GBM stem cell line [23]. DMRs between G144 and H9, both at pluripotent and differentiated stages, were analyzed using a pairwise comparison applying the same threshold method. This approach identified 2,498 and 6,099 DMRs for days 0 and 45, respectively (23% and 56.3%), compared with G144 (Fig. 4A). These results enabled us to classify the H9-hESC time points into a separate arm when performing a hierarchical cluster analysis (Fig. 4B). Cancer cells are characterized by hypermethylation of Polycomb repressive complex 2 (PRC2)-associated genes.
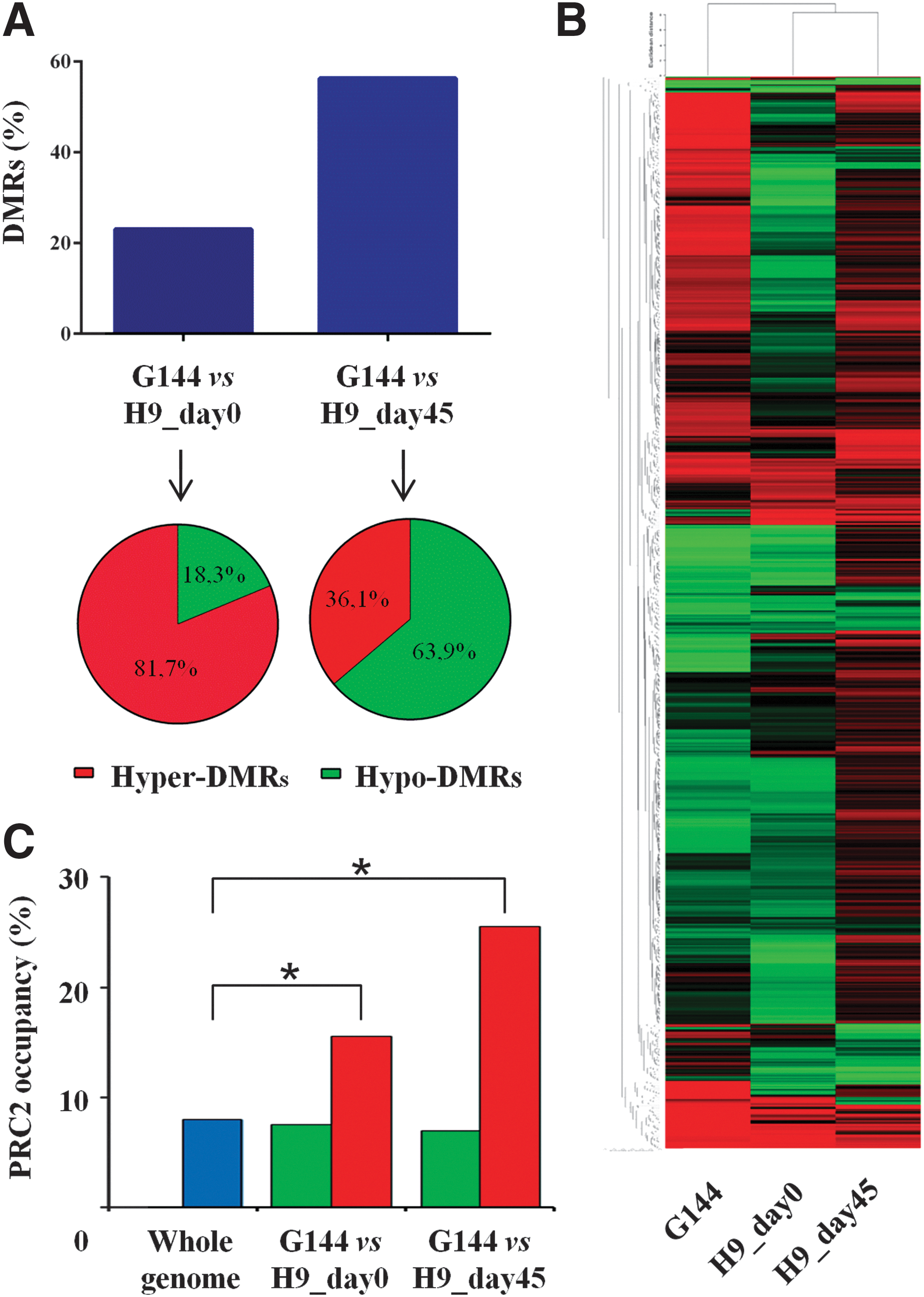
H9-hESCs show no epigenetic tumorigenic properties when compared with the G144 glioma stem cell line.
We generated two lists of hypermethylated DMRs in G144 when compared with undifferentiated and committed H9-hESCs (days 0 and 45, respectively). Thus, we compared these lists with the genes associated with Polycomb occupancy in hESCs [41]. These hypermethylated DMRs in G144 showed a statistically significant increase of genes associated with PRC2 occupancy compared with whole-genome general occupancy [41]. On the other hand, hypomethylated DMRs in G144 revealed the same frequency as the global genomic occupancy.
This analysis pointed out that H9-hESCs, both at pluripotent and differentiated stages, displayed significant differences in the methylation profiles when compared with glioma stem cells and that the differences between embryonic and cancer cells were associated with hypermethylated DMRs linked to PRC2 occupancy, a peculiar feature of malignant cells. These results indicate that H9-hESCs showed no or very little DNA methylation pattern associated with cellular transformation.
Integrative analysis revealed epigenetically regulated genes during striatal lineage commitment
It is well known that DNA methylation regulates gene expression; hence, we performed an integrative analysis between promoter DMRs and previously published cross-referenced expression data [22], which were obtained using exactly the same cell line, differentiation protocol, and time points. The gene list was generated by comparing the fold change in expression with delta methylation values of mature neurons (day 45) versus the pluripotency stage (day 0).
A panel of 97 DMRs, corresponding to 95 genes, showed matching between methylation changes and differential expression (Fig. 5). Interestingly, the DMR panel contained epigenetically regulated genes during striatal lineage differentiation. Moreover, an enrichment of functional annotations related to development and differentiation, cell proliferation, and metabolic process was identified. Finally, among these differentially expressed and methylated genes, a subcluster of 18 genes was marked out as stage-specific genes at day 45 of striatal differentiation in the H9-hESC line, as identified in the cross-referenced expression analysis. Interestingly, the DMR panel identified epigenetically regulated genes during striatal lineage differentiation.
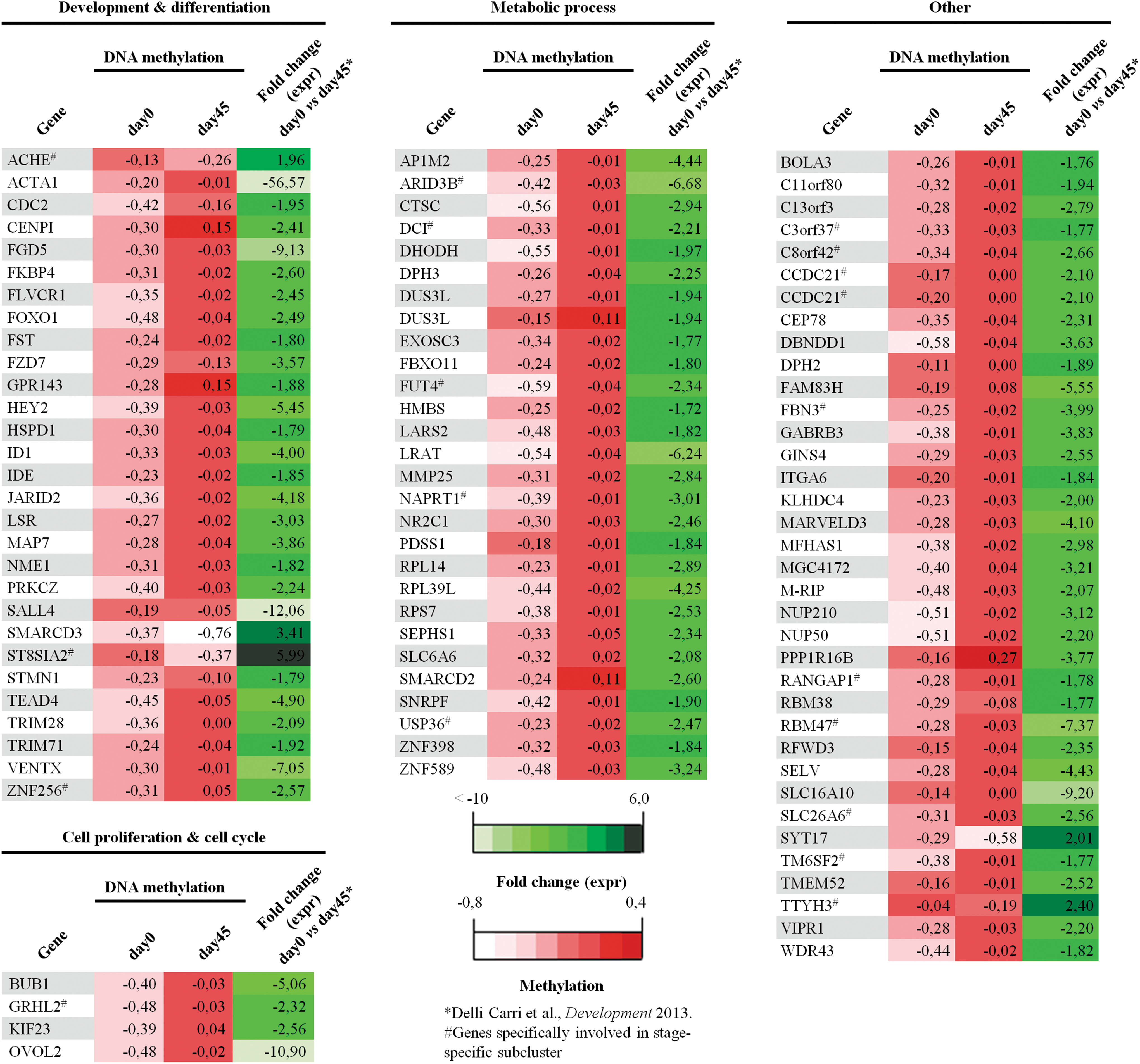
Comparison between DMRs at gene promoters and differentially expressed genes by cross-references of gene expression profiles. Ninety-seven loci corresponding to 95 genes display a concordance between methylation difference and expression during striatal differentiation at day 45 when compared with the pluripotency stage (day 0). These genes can be divided into different functional categories, such as development and differentiation, cell cycle, and metabolic process. A subcluster of 19 stage-specific genes (#) associated with striatal differentiation is marked out. Color images available online at
Functional validation of promoter methylation changes
We selected five genes (BCL2L11, DLL1, HEY2, ID1, and SMARCD3) to validate the functional implications of DNA methylation changes on gene expression. qRT-PCR on RNA extracted from day 0 to 45 differentiated cells showed that promoter CpG methylation and mRNA expression were inversely correlated: hypermethylation resulted in downregulation of gene transcripts, whereas hypomethylation revealed an increase of gene expression (Supplementary Fig. S6). Although only five genes were used to validate the methylome platform, the complete concordance assures the use of such approach in further qualifying stem cells for down-the-line applications.
Discussion
hESCs hold an enormous potential for regenerative medicine, including neurodegenerative disorders such as Huntington disease (HD). DNA methylation is extensively remodeled/reorganized during mammalian development, a process that influences gene regulation and expression. However, little is known about the DNA methylation dynamics and the possible epigenetic signatures that occur during cellular differentiation. Indeed, the investigation of the epigenetic safety is a crucial aspect for clinical trial scale-up of hESCs. Thus, in this study, we took advantage of an array-based technology to explore the DNA methylation changes that occur during in vitro development, using a fine-tuned ontogeny-recapitulating differentiation protocol that is able to generate a high proportion of MAP2ab+ neuronal cells, 20% of which are functional DARPP-32+ medium-sized spiny neurons. We investigated the developmental statuses that represent pluripotent, neural committed cells, neural rosettes, and mature neurons to obtain a chronological profile of the genome-wide DNA methylation changes of H9-hESCs during striatal differentiation.
Our findings show that H9-hESCs are mostly characterized by low levels of methylation at CpG sites. DNA hypomethylation at the pluripotent stage constitutes the ground level for a highly dynamic and transcriptionally active chromatin and reflects the ability of hESCs to initiate a wide range of lineage commitment programs [42,43]. In our striatal protocol, the process of differentiation is characterized by two waves of methylation: the first wave during neural commitment and the second in mature neurons, divided by a global demethylation at the rosette stage.
First of all, this dynamic reveals that differentiation is a combination of de novo methylation and hypomethylation where cells acquire appropriate cell type-specific marks that reflect their developmental stages and functional identities [44,45]. The early hypomethylation in H9-hESCs likely mirrors the initial decondensed chromatin and transcriptional competence of the pluripotent inner cell mass cells. These events are necessary to trigger the expression of those genes needed to start the process of specialization and subsequently, as differentiation proceeds, hypermethylation locks the cell fate decision [46,47].
DNA demethylation is another important player in the epigenetic scenario during cell commitment as it implies a sort of reshuffling of pluripotent cells into lineage-specific progenitors. This step represents the end of pluripotency and the beginning of the lineage-determining process [1]. Progenitor cells, such as the rosette stage, definitely constitute a subtle equilibrium between self-renewal and multilineage differentiation and entail the transition into lineage-restricted paths and also the CpG methylation profile mirrors this equilibrium. Subsequently, the last phase of commitment is marked by gain of DNA methylation more than loss [48], which consequently abolishes the risk of lineage unrelated gene expression and locks cell fate decision [26].
We identified a percentage of stage-specific DMRs at promoter regions comparable with published literature [17,26,49,50]. In this study, we report on the identification of differentiation-specific DMRs, which turn out to be really significant at day 15, because even if the global CpG methylation is very similar to that of the pluripotent stage (day 0), the statistical analysis revealed a considerable percentage of DMRs, showing that these cells represent two specific and distinct entities that could be recognized at the epigenetic level. Bioinformatics confirmed the dynamics of the methylome in the different cellular subtypes as specific functional categories were involved at each differentiation stage.
Silencing of pluripotency was first observed through hypermethylation of genes involved in embryonic pattern specification and progressive gain of hypermethylated DMRs in signals involved in pluripotency and self-renewal, such as TGF, Wnt, and Nanog pathways. Furthermore, mature neurons were characterized by an enrichment of methylated regions associated with cell cycle and mitosis, consistent with the reduced proliferative potential of these cells, whereas hypomethylated genes were related to nervous system differentiation.
Hypomethylation of a specific set of genes during differentiation also suggested the prevalence of peculiar TF binding sites at different stages of commitment. Indeed, chromatin remodeling through hypomethylation may involve TFs that activate the initiation of a transcriptional cascade of key tissue-specific genes and thus cell lineage choices [51]. Hence, genes with reduced DNA methylation could be targeted and occupied by specific TFs and could activate cell fate options with an appropriate timing during commitment [52,53].
Particularly, we identified PITX2 that is actually expressed in neuroepithelial and nestin-positive neural progenitor cells derived from ESCs and important for neural development. These progenitor cells may overlap with the developmental stage of our cells after neural induction and patterning at day 15 [54 –56]. The switch toward a more specific neuronal and striatal fate is also accompanied by the involvement of more definite TFs, such as CREB and CREBP1, which are known to be involved in the expression of striatal genes containing CRE-responsive elements and thus influence the expression of a specific phenotype of the developing striatum [57 –59].
Moreover, the consistency of orchestrated DNA methylation changes during striatal commitment is proved by the identification of a subcluster of 18 differentially methylated and expressed genes that could be directly associated with a specific stage and striatal-enriched gene signature [22]. All these findings support the hypothesis that DNA methylation is an epigenetic mechanism that governs hESC fate, where different methylation patterns underpin specific developmental stages and thus the methylome could be considered an appropriate biomarker for the evaluation of the cellular commitment [31]. Despite the abundance of neuronal cells (∼70%) in our cultures, only a fraction carried markers of authentic MSNs. This analysis was performed on the bulk cell population and future studies on the purified cellular population are needed to confirm the specificity of our findings.
DNA methylation not only plays a role in hESC pluripotency and differentiation but also influences the safety and efficacy of the derived cell product to be used in stem cell-based therapies. Both culture conditions and differentiation protocols may encourage the development of dangerous DNA methylation abnormalities that may promote and sustain cell proliferation. Therefore, understanding of the methylation signatures that go with striatal differentiation of hESCs is a critical step to promote an effective and safe cell-based treatment for neurodegenerative diseases, including HD [60 –62]. We addressed this issue by comparing the methylation patterns of pluripotent hESCs and their mature neuronal derivatives to glioblastoma stem cells.
We selected day 45 as the time point of analysis in addition to early stages of differentiation, such as days 5 and 15. At these time points, cells are too immature (day 5) and the striatal differentiation protocol is still in the patterning phase, with many progenitors in active cell division (day 15). Cells at day 45 of differentiation constitute a possible cell product in the transplant and are thus important to be controlled [22]. Our results suggest that the methylation profiles of either pluripotent or striatal committed cells strongly differ from the DNA methylation signature of GBM stem cells, indicating that these cell populations are distinct epigenetic entities [23].
During cell commitment, even if we identified pathways related to the molecular mechanism of cancer and glioma signaling among the promoter DMRs, a close overlap of cancer-associated and stem cell genes was identified and constitutes an acceptable hypothesis. This phenomenon is consistent with loss of pluripotency and gain of lineage-specific choices [63] and is a crucial point as hESCs turn into fate-restricted progeny by dropping their self-renewal ability before being used for cell replacement [64,65]. The striatal differentiation protocol used here revealed a further difference in the methylation profiles of our cells from cancer cells. Actually, a significantly lower hypermethylation level of PRC2-targeted genes in our cellular model was identified when compared with GBM stem cells.
Polycomb occupancy can trigger de novo DNA methylation at a subset of promoters and restrict the potential of neuronal progenitors during differentiation [26]. Misregulation of this process can result in cancer-specific aberrant promoter methylation. A Polycomb connection was identified in several works on human cancer cells, where hypermethylation of gene promoters targeted by PRC2 in hESC cultures was evidenced [66,67]. This methylation difference has a pivotal role as one hallmark of GBM stem cells is the hypermethylation of PRC2 target genes that can arrest arresting cancer cells in a sort of continuous self-renewal loop [23,68].
H9-hESCs and their derivatives possess peculiar methylation profiles that distance themselves from the unique Polycomb signature characteristic of cancer cells. Whereas on analyzing the methylation data more deeply, we identified BCL2L11 as a potential oncogenic methylation maker during exposure to our striatal protocol because it was found to be hypermethylated and its transcript downregulated during neuronal commitment. BCL2L11 is a proapoptotic gene and thus an oncosuppressor: its downregulation may support prosurvival pathways and stimulate cell proliferation [69]. Moreover, BCL2L11 was recently identified as hypermethylated in human cancers, such as chronic myeloid leukemia and breast cancer [70,71].
Therefore, the evaluation of the relative risk–benefit ratio has a crucial importance when translating hESCs in clinical applications, and the study of the methylation profiles contributes to unveil and harness the power of hESCs to offer safe and effective cell-based therapies [65,72].
All these findings provide several evidences of the importance of the DNA methylome in hESCs and striatal differentiation. Our well-defined ontogeny-recapitulating protocol helped us to identify unique signatures that could distinguish differentiation-specific stages, provide a blueprint of the methylome changes during striatal commitment, and offer a new repository of cell derivatives for regenerative medicine [73]. Subsequently, our results revealed a complete evaluation of the methylation safety of hESCs and their striatal derivatives; anyway, the tolerance level in the type and number of methylation changes to consider these cells safe for regenerative medicine should be further investigated, emphasizing the challenge to produce a reliable and complete screening to predict the biosafety of hESCs.
Footnotes
Acknowledgments
The authors thank Dr. Stefano Camnasio, Dr. Barbara Motta, and Dr. Alessia Delli Carri for the assistance with neural differentiation protocol of hESCs. This work has been supported by grants from NeuroStemcell (EU Seventh Framework Programme, grant agreement no. 222943), NeuroStemcellRepair (European Union Seventh Framework Programme, grant agreement no. 602278), Ministero della Salute (RF-MUL-2008-1248034), MIUR Regione Lombardia Network Lombardo iPS (NetLiPS, Project ID 30190629-2011), Progetto Quadro Regione Lombardia-CNR (RSPPTECH 2013–2015), and InterOmics Flagship Project (no. 0092174-16/12/2014).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.