
Abstract
Germ line development is crucial in organisms with sexual reproduction to complete their life cycle. In mammals, knowledge about germ line development is based mainly on the mouse model, in which genetic and epigenetic events are well described. However, little is known about how germ line development is orchestrated in humans, especially in the earliest stages. New findings derived from human in vitro models to obtain germ cells can shed light on these questions. This comprehensive review summarizes the current knowledge about mammalian germ line development, emphasizing the state of the art obtained from in vitro models for germ cell-like cell derivation. Current knowledge of the pluripotency cycle and germ cell specification has allowed different in vitro strategies to obtain germ cells with proven functionality in mouse models. Several reports during the last 10 years show that in vitro germ cell derivation with proven functionality to generate a healthy offspring is possible in mice. However, differences in the embryo development and pluripotency potential between human and mouse make it difficult to extrapolate these results. Further efforts on both human and mouse in vitro models to obtain germ cells from pluripotent stem cells may help to elucidate how human physiological events take place; therefore, therapeutic strategies can also be considered.
Introduction
E
Infertility causes have been widely studied. However, there are infertility situations that cannot be overcome with current techniques. Thus, a better knowledge of the events that take place during human germ line development would help in the design of new strategies to treat infertility. In this report, we will review the most important events during germ line development in mammals, with special focus on humans. Moreover, we will pay attention to how regenerative medicine can be a tool to study human germ line development and help us to understand its insights, deepening the current knowledge of how the pluripotency cycle is closed along development in vivo and demonstrating its plasticity in vitro.
Overview of Germ Line Development in Mammals
The differentiation between germ and somatic cells occurs very early in development with the escape of a group of mesodermic cells from their somatic fate during gastrulation and is characterized by two fundamental facts: reacquisition of the pluripotency and extensive epigenetic remodeling. Next, after sexual determination, each one of the respective sexual features of future eggs and spermatozoa is acquired during gametogenesis sensu stricto, as gonads are synchronically formed.
In the mouse model, during gastrulation, the founder population of primordial germ cells (PGCs) is specified by extrinsic signals driven by bone morphogenetic proteins (BMP4 and BMP8b), members of the TGFβ superfamily, from the adjacent extraembryonic ectoderm (ExE) at embryonic day 6.25 (E6.25) [4]. BMPs secreted by the ExE induce Prdm1 and Prdm14 expression in PGCs which, together with Tcfap2c, will act as master regulators over a very reduced group of epiblastic cells located close to the base of Allantois [5]. Thus, PGCs are specified from somatic precursors in a process called epigenesis, which has many implications. First, at a signaling level, PGCs need to be specified by external signals, which will turn on the germ cell program and turn off the somatic program. Opposite to this situation, in other model animals (eg, Drosophila melanogaster, Caenorhabditis elegans), germ line is specified thanks to inherited maternal determinants, called germ plasm, already present in the oocyte [3,6] in a process known as preformation. Second, mammals and other animals following epigenesis induction endure a cellular reorganization locating PGC precursors outside the embryo to prevent their exposure to somatizing signals [6]. This reorganization is not needed in the preformation model since the somatizing signals are directly suppressed by the inherited maternal determinants. Therefore, PGC specification in response to extrinsic signals is revealed as the first crucial step in germ line development, which determines future events, such as the need for pluripotency reacquisition and epigenetic reprogramming [7,8]. Thus, just after their specification, PGCs are characterized by the expression of several pluripotency markers such as Oct4 (also known as Pou5f1), Nanog, and Sox2 [9], as well as other markers such as Fragilis and Stella [10,11].
Epigenetics comprises all the modifications of the DNA that do not imply changes in its sequence and includes DNA methylation [12] and histone modifications [13], both controlling the gene expression pattern. At the moment of their specification, PGCs are epigenetically indistinguishable from surrounding epiblastic cells and show inherited epigenetic modifications such as DNA methylation and X chromosome inactivation that represent epigenetic barriers to totipotency acquisition in somatic tissues. After their specification, PGCs start their migration at E8.5–9.5 through the posterior hindgut to the forming gonad [7,14], and during their migration, PGCs begin the first wave of a profound epigenetic remodeling to acquire the epigenetic state necessary to form functional gametes [15]. The first epigenetic changes in PGC reprogramming happen at the histone modification level, leading to a large chromatin remodeling. Only in the X chromosome, the inactive X chromosome of female PGCs show decreased levels of histone 3 lysine 27 trimethylation (H3K27me3), a repressive histone modification, as an indication of its reactivation, whereas in the somatic line, H3K27me3 levels remain unchanged [15,16]. These changes in histone modifications occur in parallel to an overall reduction in DNA methylation during PGC migration [14,15]. Apparently, methylation levels do not change dramatically from PGC specification until their arrival to the genital ridges at E10.5, when a deeper reset of methylated DNA occurs in a second wave of epigenetic remodeling events [17]. This process involves a global erasure of DNA methylation patterns, including imprinted genes, and PGCs reach a basal epigenetic state by approximately E13.5, even though there are some sequences in the genome that are resistant to demethylation such as the intracisternal A particle (IAP) retrotransposon family and adjacent CpG islands (CGIs) [15,18], suggesting a possible epigenetic inheritance between generations.
Recent studies suggest that hydroxymethylcytosine (hmC), a derivative of methylcytosine (mC) [19], is involved in PGC reprogramming. DNA methylation can be passively erased by rounds of replication not followed by the DNA methylation maintenance system [20], but also actively removed by ten-eleven translocation (TET) enzymes [19,20]. TET enzymes are 5mC-dioxygenases that catalyze the oxidation of mC into hmC, as well as into its other derivatives such as formylcytosine (fC) and carboxylcytosine (caC). The role that all these oxidated forms of mC play in the epigenome remains unclear, but some studies suggest that they not only act as intermediates preceding DNA methylation erasure but also have a structural function controlling gene expression [21]. Thus, in the PGC reprogramming context, the conversion of 5mC to 5hmC by TET enzymes provides a substrate for base excision, repair-mediated active demethylation that could give rise to unmodified cytosine, hence actively erasing DNA methylation marks [22 –24]. However, not all methylated regions are prone to be demethylated in the first wave of demethylation, since imprinted loci or specific single copy genes maintain their methylation in CGIs [17]. It is during the second wave of demethylation, which starts at E10.5, when these genomic regions with epigenetic memory are completely erased by E13.5 [17]. There is only one other epigenetic reprogramming event during the life cycle comparable to PGC reprogramming, and it takes place in the zygote just after fertilization and during preimplantation development. However, the PGC reprogramming event is much deeper and more extensive than that which occurs in the zygote (in which parental imprints remain) and represents the most basal epigenetic state of the mammalian life cycle [15].
When PGCs colonize the genital ridges, they start a process of proliferation and sex determination called gametogenesis that ends with the formation of sperm and eggs. Sex determination depends on the gonadal microenvironment, regardless of the sex chromosome [25,26] being synchronous with genital ridge formation and maturation. In the mouse, the bipotential genital ridges are formed on the ventral surface of the mesonephros, accompanied by proliferation of the coelomic epithelium around E9.5 [27]. During sex determination, PGCs must establish their sex-specific epigenetic patterns, which include paternal or maternal imprinted marks. The imprinting genes involve genomic sequences that exhibit differences in CpG methylation according to the parental origin, being one allele completely methylated and the other fully demethylated [28]. The establishment of these epigenetic patterns occurs before meiosis in the sperm at E14.5–E16.5 [17] and after the first meiotic division in eggs after birth [29 –33]. As a result of sex determination, spermatozoa are highly methylated (around 80%), while oocytes are about 30% methylated [17].
Thus, according to the somatic sex of the embryo, germ cells follow a different maturation pattern to finally give rise to mature and functional gametes. However, events that take place in the early phases of germ cell development have been demonstrated to be essential for future gametes to be able to restore totipotency after fertilization in the newly formed embryo. It is in this moment when the zygote endures epigenetic reprogramming to acquire totipotency, and in which some genomic regions escape demethylation, including centromeric repeats, IAP retrotransposons, and differentially methylated regions that are present in parentally imprinted genes [34]. Therefore, soon after fertilization, the paternal genome undergoes rapid and active DNA demethylation, while the maternal one is passively and slowly demethylated during segmentation. Subsequently, in the blastocyst stage, the methylation profile characteristic of the new individual starts to be acquired as pluripotency potential is lost and first cellular fate decisions are taken, moving forward to a cycle that will be completed with the specification of PGCs of the new generation [35 –37].
Germ Line and Pluripotency
Fertilization of the oocyte gives rise to a totipotent zygote, which is the only totipotent cell that can give rise to all cell types in an organism. Hence, this potential is hidden in a pluripotency cycle, which is only completed and closed between generations with germ line development [1,7,38]. In the case of mammals, due to the epigenesis specification mechanism of PGCs, this pluripotency cycle presents a gap between generations; postsegmentation embryos are composed entirely of somatic cells, and germ line is specified by extrinsic signals that return early germ cells to a state of pluripotency. Opposite to epigenesis, in preformation mechanisms, the pluripotency cycle results are more obvious, as it is closed due to the fact that germ cells inherit the germ plasm that maintains their plasticity [7,39].
Consequently, germ cells harbor a latent pluripotent potential (Fig. 1), although they have been traditionally considered unipotent. Therefore, germ cell specification through epigenesis requires a tight orchestration of signaling pathways that confer germ cell pluripotency re-establishment [7,40,41].
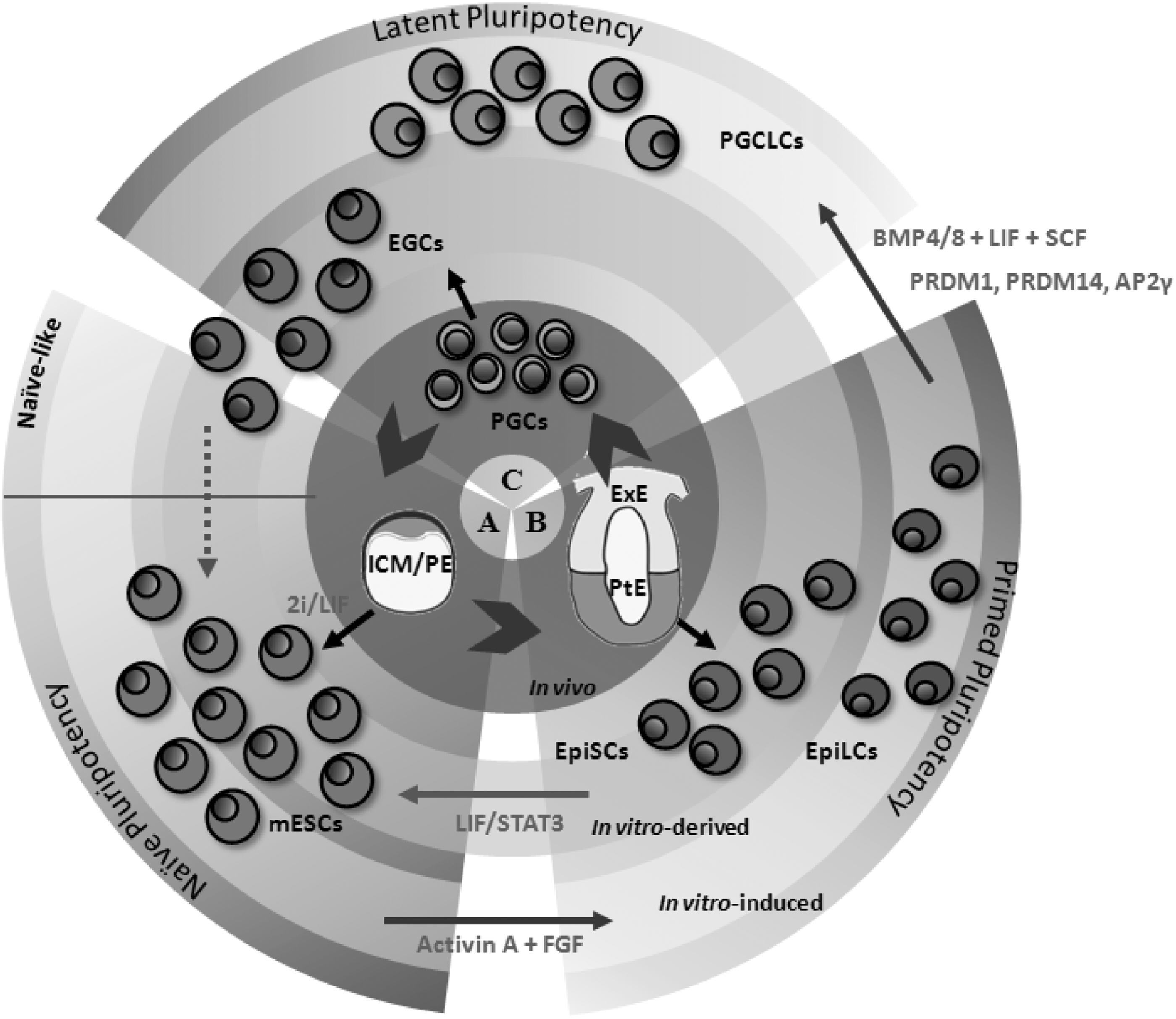
Pluripotency cycle. The inner circle represents pluripotent states in vivo; the gray circle represents in vitro-derived pluripotent states; and the external circle represents the in vitro-induced pluripotent states.
Specification of PGCs in the mouse: the golden tripartite
As previously discussed, in the mouse model, PGCs originate from the most proximal region of the postimplantation epiblast in response to external signals coming from the ExE [2]. These cells are developed from the inner cell mass (ICM), where cells expressing Sox2, Oct4, and Nanog become the precursors from which PGCs will arise [42,43]. At E5.5, epiblast cells are able to respond to BMP4 which, in turn, activates the key PGC transcriptional regulator Prdm1 through SMAD1 and SMAD5 [44]. Thus, Prdm1 (also known as Blimp1) expression at E6.25 is the first component of a tripartite of transcription factors involved in mouse germ line specification and marks the onset of PGC specification, demarcating germ cells from the neighboring somatic cells through the repression of the incipient mesodermal program [45 –47]. Also involved in this specification network is LIN28 [48], considered to be essential for PGC specification, thanks to its role as a negative regulator of Let-7 micro-RNA (miRNA) [49]. Since Let-7 is known to inhibit Blimp1 mRNA translation by binding to its 3′ UTR [50], it is proposed that LIN28 contributes to the activation of Blimp1 by blocking the maturation of functional Let-7 miRNA [49].
Together with Prdm1, Prdm14 expression is also enhanced in response to the activation signaling of BMP4. The onset of Prdm14 expression is not Prdm1 dependent, but Prdm1 is essential for Prdm14 expression maintenance in a dose-dependent manner, reinforcing its activity [51,52]. Therefore, while Prdm1 represses the somatic program, Prdm14 acts to induce the epigenetic reprogramming in early germ cells. In fact, mutations in Prdm14 led to the formation of aberrant PGCs with a defective epigenetic programming by failure of global erasure of H3K9me2 histone modification at E8.5, losing PGCs by around E11.5 [20].
Tcfap2c, which encodes AP2γ, has also been described in the mouse as a critical element that acts downstream of Prdm1, being the third component of the tripartite of transcription factors crucial for PGC specification. AP2γ might also be involved in the repression of somatic genes, including early mesodermal markers, such as Hoxb1 [11]. The induction of AP2γ by Prdm1 is crucial for the combinatorial role of Prdm14 and AP2γ in the induction of PGC-specific genes such as Dnd1 and Nanos. They also bind to the distal enhancer of Oct4 to maintain its expression in PGCs.
Thus, Prdm1, Prdm14, and AP2γ together constitute the tripartite transcriptional network that is mutually interdependent for PGC specification in mice [53]. Hence, this tripartite transcriptional network enables several programs to initiate in early PGCs, including the repression of somatic genes. For example, the migratory program, as well as the expression of PGC-specific genes, is executed mainly by Prdm14 and AP2γ, whereas Prdm14 and Prdm1 collaborate in both the induction and repression of epigenetic modifiers [54]. At an epigenetic level, the tripartite transcriptional network induces the global DNA demethylation characteristic of PGC reprogramming [17,19]. Prdm1 and Prdm14 promote DNA replication-coupled DNA demethylation by repressing the de novo DNA-methyltransferase Dnmt3b, as well as Uhrf1, a cofactor for the DNA methylation maintenance system. Hydroxylation of 5-methyl-cytosine is mediated by TET1 and TET2, which are recruited to DNA target loci by Prdm14 [19,55]. The expression of Prdm1 prevents the somatic fate at the onset of PGC specification, initiating a reversion of some of these properties, including X-reactivation and re-expression of key pluripotency genes, such as Sox2 and Nanog [1]. Finally, the reactivation of the inactive X-chromosome in PGCs might also be facilitated by Prdm14 binding to intron 1 of Xist [54,56,57].
Comparison between human and mouse models of specification: unsolved questions
Due to the limited access to the early stages of human development, current knowledge about very early germ line development in vivo is based on the mouse model [58]. Interestingly, the timing and formation of the extraembryonic tissues where early germ cells are specified are significantly different in mice and humans. This fact, hence, might have some implications for PGC specification and determination [59].
More specifically, before gastrulation, the mouse epiblast can be visualized as a thick-walled cup of tissue, which forms the entire embryo and some of the placental membranes. At this moment, the ExE arises from the polar trophectoderm underlying the epiblast. In humans, by contrast, the embryo consists of a bilaminar disc, the epiblast, and the primitive endoderm or hypoblast. The epiblast cells will form the amnioblast and the amniotic cavity [59], whereas the polar trophectoderm over the epiblast differentiates toward the syncytiotrophoblast and the cytotrophoblast, which remain in contact with the epiblast (Fig. 2). In other words, no equivalent structure to the mouse ExE, from which PGC specification is triggered, is apparently formed in the human embryo [59].
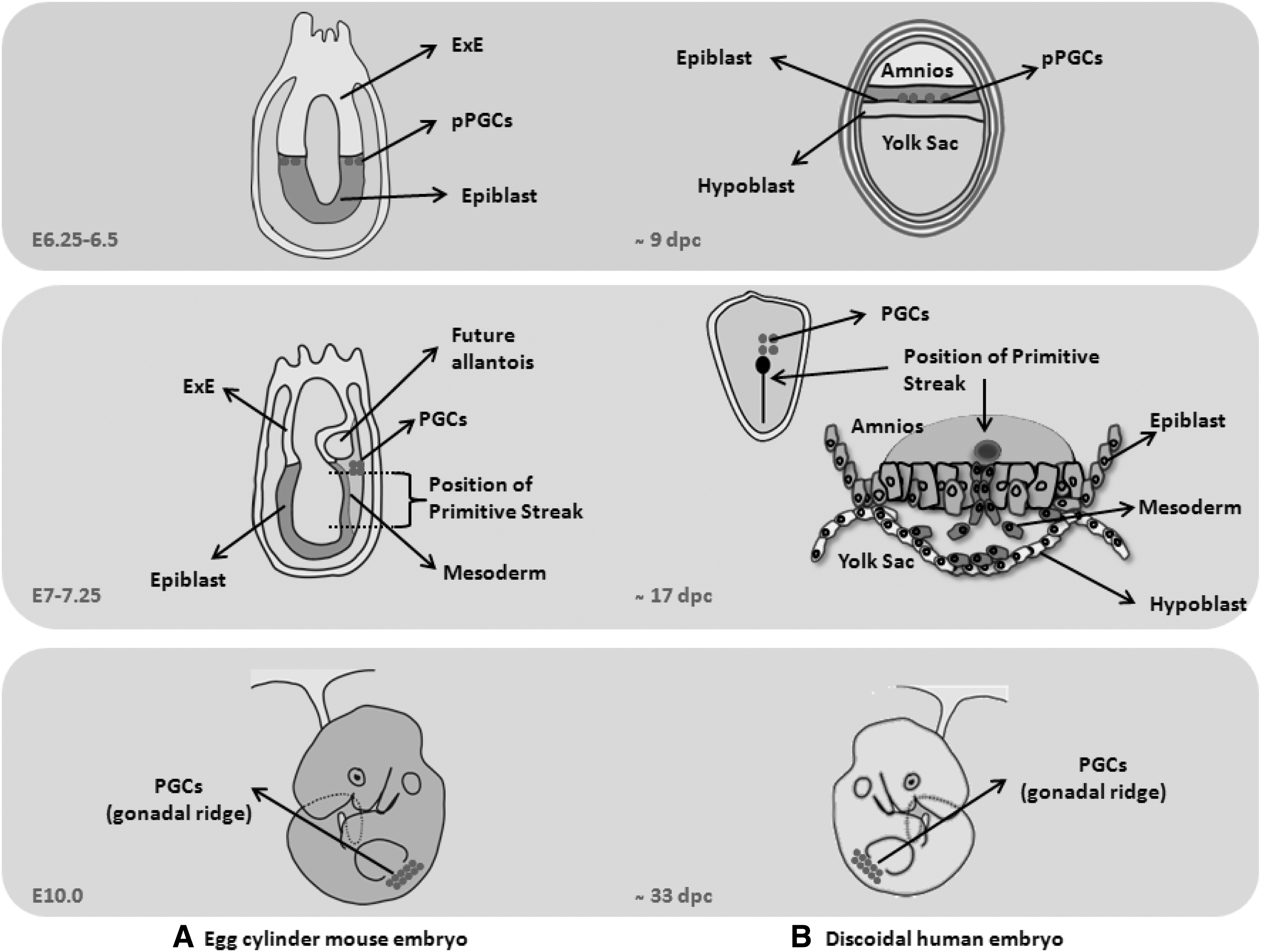
Schematic anatomy models of mouse and human embryo.
Thus, although mice have been widely used as the mammalian model to define, at a molecular level, several signaling pathways in vivo, it does not seem to be the best model to compare PGC determination in humans. On the other hand, the use of other animal models with a discoidal epiblast may help to clarify some questions. Interestingly, in rabbits, PGCs arise in the primitive streak as well as in mice, but BMP2 is expressed from the hypoblast and yolk sac epithelium at the boundary of the embryonic disc, which is equivalent to the proximal and extraembryonic visceral endoderm in mice. Hence, BMP2 seems to play a more essential role in rabbit PGC specification than BMP4, since BMP4 is expressed after BMP2, and Blimp1-positive cells are identified close to BMP2 expression, following its same pattern [41,60]. In pigs, also with discoidal embryos, PGCs are not specified at an equivalent time to mouse embryos. In the proximal epiblast of pregastrulating pig embryos, few cells start to express OCT4 as the unique germ line marker. After that, and similar to other nonrodent mammals, including humans, OCT4-positive cells move from the embryonic disc to the extraembryonic mesoderm of the yolk sac wall and become restricted to the ventral wall of the hindgut [61].
To date, there is no well-defined nonrodent animal model that enables us to identify where PGC specification signal(s) come from. In fact, precursors of human PGCs have been suggested to emerge from the epiblast around 10–11 days postcoitum (dpc) by the action of BMP signals from the hypoblast and cytotrophoblast, and probably from amnioblast or both, but this last point remains unclear. Two to 4 days after, these precursors migrate to the extraembryonic mesoderm and reach the region of the yolk sac where the allantois is formed at around 16 dpc [59]. Then, during the fourth week of gestation, PGCs are passively incorporated into the embryo with the yolk sac wall by folding the embryonic disc, and they start to migrate toward the gonadal ridges (Fig. 2).
In conclusion, anatomical differences between rodents and other mammals during early development are relevant. On the other hand, many legal and ethical limits imply that very little is known about early human embryo development and, particularly, germ line specification. Moreover, this first step in germ cell development reveals as crucial for proper reprogramming and pluripotency reversal in order for future germ cells to be able to generate a new organism. The role that the starting signaling pathway plays and the putative function the specification tripartite has in humans, however, are not yet determined. In this sense, the pluripotent background that epiblastic cells possess might be different between mouse and human, conditioning the way that the first steps in PGC determination take place.
Are the anatomical differences between human and mouse embryos relevant to obtain gametes in vitro?
The anatomical differences between mouse and human embryos might have an effect on the pluripotency reacquisition mechanism. In fact, the consequences of this differential pattern should also be reflected in vitro (Fig. 1).
In mice, the epiblast derives from the ICM, as embryonic stem cells (ESCs) do in vitro [62 –67]. Mouse ESC (mESC) pluripotency is defined by their ability to differentiate into representative cell types from the three germinal layers (ectoderm, mesoderm, and endoderm) in vitro and in vivo by the formation of teratomas after injection in immunosuppressed mice. Moreover, both chimera formation [68] and tetraploid complementation are the last and most consistent features of pluripotency (Table 1) [69]. In addition, pluripotent stem cells (PSCs) can also be derived from the epiblast of postimplantation mouse embryos. These cells are called mouse epiblastic stem cells (mEpiSCs) as they express pluripotency markers such as Oct4, Nanog, and Sox2 and can be differentiated in vitro into the three germ layers and, also, into the germ line [70,71]. Furthermore, mEpiSCs are able to form teratomas in vivo, although they show a very low potential to contribute to chimera formation (Table 1) [72]. Interestingly, mEpiSC can be converted into ESC-like cells when cultured under conditions similar to mESCs with BMP4 and leukemia inhibitory factor (LIF)/STAT3; likewise, the ICM-derived mESC can convert to EpiSC-like cells when cultured under EpiSC conditions, that is, under FGF and Activin signaling (Fig. 1) [73,74].
hESC, human embryonic stem cells; ICM, inner cell mass; mEGCs, mouse embryonic germ cells; mEpiSCs, mouse epiblastic stem cells; mESC, mouse embryonic stem cells; mPGCs, mouse primordial germ cells; Xa, X chromosome activated; Xi, X chromosome inactivated.
The slightly different features that PSCs show reveal the existence of several pluripotency states, distinguishing between naive and primed pluripotent states [42]. Naive pluripotency is defined as the initial ground state of pluripotency, in which the cells have not yet lost their ability to form every cell type in the organism, including germ cells. However, as cells become specified into a defined fate, they restrict their potential to a primed pluripotency state [7]. The mEpiSCs derived from postimplantation blastocysts exhibit primed characteristics since they lose the expression of some pluripotency factors, such as Rex1 or Klf4, and are unable to form chimeras (Table 1) [75]. Interestingly, mouse PGCs from the postimplantation epiblast can be driven to become embryonic germ cells (EGCs) in vitro. EGCs are able to reverse this primed state toward a naive pluripotent state as shown by their ability to form chimeras and differentiate into all cell types, including germ line (Table 1) [76]. Consequently, PGCs from the postimplantation embryo are considered to have a latent pluripotent potential [7].
First, reports on germ line differentiation demonstrated that mESCs spontaneously differentiated in vitro toward germ cell lineages, forming oocyte-like cells [77] and sperm-like cells able to participate in spermatogenesis in vivo [78] and to form blastocysts after injection into oocytes by intracytoplasmic sperm injection (ICSI) [79]. These attempts, however, failed in overcoming meiotic checkpoints to give rise to a healthy and fertile offspring, probably because earlier events such as establishment of primed pluripotency were not properly reached (Table 2).
5hmC, 5-hydroxymethylcytosine; 5mC, 5-methylcytosine; bFGF, basic fibroblast growth; BMP, bone morphogenetic protein; EGF, epidermal growth factor; EpiLCs, epiblast-like cells; hiPSC, human induced pluripotent stem cell; ICR, imprinted control region; ICSI, intracytoplasmic sperm injection; LIF, leukemia inhibitory factor; miPSC, mouse induced pluripotent stem cell; RA, retinoic acid; ROCK, Rho-kinase; SCF, stem cell factor; Xa, X chromosome activation.
Similar results from human embryonic stem cells (hESCs) were later obtained by spontaneous differentiation, suggesting that hESCs consist of a heterogeneous population, in which some cells are predisposed toward germ cell fate differentiation [80]. Moreover, hESCs show low potential to contribute to chimera formation, although they are able to give rise to the three germ layers and form teratomas in vitro, suggesting that they are more similar to mEpiSCs than to mESCs (73). Hence, hESCs represent a pluripotency state closer to primed pluripotency (72) from where they could directly respond to meiotic entry induction (82–85). In fact, acquisition of a primed pluripotent state has revealed as essential for functional mouse gamete generation in vitro [53,81,82], as discussed below (Fig. 1).
In Vitro Models to Explore Human Germ Line Development
Recent works highlighted the relevance of the specification signaling on PGC specification in vitro, and hence, germ line development into functional gametes (Table 2) [53,81,82]. In these reports, mESCs cultured in 2i conditions to induce their naive pluripotency state were first differentiated into epiblastic-like cells (EpiLCs), evolving toward a primed stated by treating them with Activin A, basic fibroblastic growth factor, and 1% knockout serum replacement. Subsequently, EpiLCs were differentiated into PGC-like cells (PGCLCs) by two methods: adding BMP4/8b to the culture media, mimicking the BMP signal specification from the ExE in vivo, together with stem cell factor (SCF), LIF, and epidermal growth factor (EGF) [81,82]; or alternatively, with the overexpression of Prdm1, Prdm14, and Tfap2C genes, which act downstream of the BMP signaling pathway [81]. The obtained PGCLCs showed imprinting patterns corresponding to their embryonic somatic sex and when transplanted into host mice, they went through both complete spermatogenesis [53,82], oogenesis, and produced a fertile offspring following ICSI [81]. With these experiments, the exclusive capability of epiblastic cells to respond to BMP signaling, as well as the important role of the BMP4/Prdm1 signaling pathway for PGC specification, was demonstrated for the first time in vitro. This has been, indeed, the only strategy able to generate viable gametes in vitro with a proven, healthy, and fertile offspring.
In the same way that these works contributed to a better understanding of the germ line specification process in mice, in vitro models can also be useful for similar purposes on human germ cell development. Following this perspective, a recent publication highlighted some important differences in human germ line specification [83]. In this study, hESCs were cultured under 4i conditions to induce a distinct pluripotent state closer to the naive state before preinducing them into an EpiLC state. In a similar way that was previously reported in the mouse model [81,82], preinduced hESCs differentiated into PGCLCs in suspension under the induction with BMPs, LIF SCF, and EGF. Interestingly, resulting human germ-like cells displayed several differences compared to the ones obtained in mice, such as null activation of SOX2, a delayed expression of PRDM14, and an upregulation of SOX17 and GATA4. Due to the unexpected upregulation of SOX17, traditionally considered an endodermic transcription factor, its role in human PGC specification was investigated. In contrast with mouse PGC specification, SOX17 might play an essential role in human germ line specification by acting upstream and synergistically with BLIMP1 as the master regulators of PGC specification, at the same time that BLIMP1 acts as a repressor of the endodermic differentiation of PGCs [83].
In addition, the first suggestions of germ line formation from hESCs in vitro were reported by spontaneous differentiation [80,84]. Undifferentiated hESCs expressed some early germ cell markers, such as c-KIT and DAZL, but not late markers such as VASA or SYCP3 [85], a structural protein of the synaptonemal complex, suggesting that hESCs could be a heterogeneous pluripotent population, in which some cells had a predisposition toward a germ cell fate. However, meiotic progression remained as one of the main obstacles. In fact, complete meiotic progression of in vitro-derived human germ cells was achieved with the ectopic expression of the DAZ gene family members DAZ2, DAZL, and BOULE not only in hESC but also in human induced PSC (hiPSC) lines subjected to spontaneous differentiation [86,87]. The expression of these highly conserved RNA-binding proteins leads the correct meiotic progression of human germ cells in vitro in the absence of a gonadal niche (Table 2). Along the same line, another report highlighted the possible role that RNA-binding proteins may have in meiotic entry control, showing how the ectopic expression of VASA also leads to meiotic induction of in vitro-derived human germ cells [88]. In fact, recent reports demonstrate that DAZL limits pluripotency as well as differentiation to control apoptosis in PGCs through a robust interactive network. If this network is broken or DAZL expression is lost, the consequence is teratoma formation [89].
However, the current state of the art of germ line derivation from human pluripotent cells presents some limitations. Differences in the pluripotential status of mESCs (naive-like state) and hESCs (primed state) make it difficult to compare their potential to fully develop into functional germ cells. Moreover ethical restrictions make it difficult to carry out functional assays with the germ cells obtained from human pluripotent cells. Thus, xenotransplantation of human germ cell-like cells into seminiferous tubules of immunosuppressed mice to test their potential to colonize the seminiferous epithelium, like human spermatogonial stem cells (SSCs) do, could be an important tool to assay the functionality of in vitro-derived human germ cells. In this context, the role of RNA-binding proteins has also been tested, demonstrating that hiPSCs improved their ability to colonize the lumen of the seminiferous tubules of sterilized immunodeficient upon VASA overexpression in combination with pluripotency factors. These results highlight the role that VASA plays controlling the pluripotency state in a combined in vitro/in vivo model [90]. This model also represents a very interesting analysis method from a clinical point of view and has already been used to analyze the capability of hiPSCs derived from azoospermic men with different deletions in the Y chromosome, demonstrating how the genetic background affects their capability to differentiate into germ cells and colonize the seminiferous niche in vivo [91]. Finally, this same approach has been used recently to demonstrate that germ cell-like cells can be formed in vivo independently of X chromosome's genetic profile [92]. In this case, hiPSCs from Turner syndrome patients were derived and xenotransplanted into seminiferous tubules, demonstrating that, regarding infertility in women with X chromosome aneuploidies, two X chromosomes are not required for human germ cell formation but for their maintenance until adulthood [92].
Discussion and Concluding Remarks
The mammalian germ line development can be considered as a noncontinuous pluripotency cycle between generations, due to the need of external signals for PGC specification during the embryonic development of each individual.
Knowledge derived from germ line development is increasing since interest in this research field is getting bigger, in part, due to its possible clinical applications to treat infertility. Moreover, in vitro models give researchers the opportunity to improve the knowledge of human germ line development, a process that involves key milestones, mainly divided in two steps: pluripotency reacquisition (including epigenetic reprogramming) and gamete differentiation (including meiosis).
According to the mouse model, the tripartite of transcription factors composed of Prdm1, Prdm14, and Tfap2C allows the postimplantation epiblast, a primed cell subpopulation, to be prone to reacquire the naive pluripotency state. However, differences between rodents and other mammalian species are evident [83]. Rodents show an unusual method of early formation of egg cylinder epiblast and also form unique characteristic PSCs with a different pluripotency potential (ie, mESCs and EpiSCs), probably due to the ability of rodent blastocysts to temporarily arrest their development depending on the niche (ie, endometrium) conditions, known as diapause [93]. Mouse embryos arrested in diapause resume their development in response to LIF signaling, which is in concordance with the need of the LIF/STAT3 pathway to derive mESCs [93]. In addition, diapause capacity could also have a role in the explanation of why only mESCs and mEGCs, but not hESCs, respond to 2i conditions to acquire the true naive pluripotent state [93]. Instead, other mammals, such as primates, show epiblast delamination, forming a simple flattened embryonic disc [60,94 –98]. Both mESC and miPSCs show the capability to give rise to all cell types, including germ cells, and to form teratomas and chimeras, demonstrating a ground naive pluripotent state. Otherwise, hESCs share characteristics with mEpiSCs, which are derived from the postimplantation epiblast, both showing limited capability to form chimeras corresponding to a primed pluripotency status. It is not known whether naive pluripotency exists in the human embryo or, if it does exist, our inability to recapitulate in vitro might be due to this status being only displayed in a very short window of time in vivo [93]. This important issue requires resolution.
A more challenging approach is the study of human in vitro gamete generation. According to the complexity of the meiotic process, this seems to be the biggest obstacle in the in vitro gametogenesis reconstitution [99]. First, since meiosis is a reductional division, the DNA content must be haploid (1 N) for all chromosomes and must show an appropriate organization that could be analyzed by karyotyping and fluorescence in situ hybridization. Second, the recombination process must be assessed by immunolabeling of proteins involved in homologous recombination synapsis to demonstrate an appropriate localization [99]. Finally, the in vitro gametes must be able to form a euploid and viable offspring and not only fertilization or early embryos. However, this last characteristic is the main handicap for human models due to its ethical controversy.
Differences between mice and human embryology prevent the direct translation of knowledge obtained from mouse early germ line development to humans. Thus, in vitro models for germ line differentiation may be an important tool to fully understand this process in humans (Table 2) [83,90 –92]. Herein, further efforts must be addressed in the future to improve these models to complete our current knowledge on human germ line development, overcoming the main handicaps and, eventually, to allow improving human infertility treatments.
Footnotes
Acknowledgments
This work was supported by a research project grant PI13/00546 from ISCIII, cofounded by the European Regional Development Fund “A way to make Europe,” the KY Cha Award in Stem Cell Technology 2014 conceded by the American Society for Reproductive Medicine (ASRM), a VAL I+D grant conceded to JMM (ACIF/2014/077), a Sara Borrell grant conceded to J.V.M. (CD12/00568) by the ISCIII, and an FPU grant conceded to AMMA (AP-2009-4522) by the Spanish Ministry of Education.
Author Disclosure Statement
No competing financial interests exist.