
Abstract
Induced pluripotent stem cells (iPSCs) have brought great promises for disease modeling and cell-based therapies. One concern related to the use of reprogrammed somatic cells is the loss of genomic integrity and chromosome stability, a hallmark for cancer and many other human disorders. We investigated 16 human iPSC lines reprogrammed by nonintegrative Sendai virus (SeV) and another 16 iPSC lines generated by integrative lentivirus for genetic changes. At early passages we detected cytogenetic rearrangements in 44% (7/16) of iPSC lines generated by lentiviral integration whereas the corresponding figure was 6% (1/16) using SeV-based delivery. The rearrangements were numerical and/or structural with chromosomes 5 and 12 as the most frequently involved chromosomes. Three iPSC lines with chromosome 5 aberrations were derived from one and the same donor. We present in this study the aberrant karyotypes including a duplication of chromosome 5q13q33 that restricts a candidate region for growth advantage. Our results suggest that the use of integrative lentivirus confers a higher risk for cytogenetic abnormalities at early passages when compared to SeV-based reprogramming. In combination, our findings expand the knowledge on acquired cytogenetic aberrations in iPSC after reprogramming and during culture.
Introduction
I
The dynamics of genetic integrity after reprogramming to iPSC has been studied using high-resolution comparative genomic hybridization (arrayCGH) and this demonstrated that cells at early passages carry high levels of acquired copy number variations (CNVs) [11,12]. However, many of these alterations disappear with early clonal expansion suggesting that cell reprogramming has a transient effect on genome integrity with a growth advantage for cells that have retained a balanced diploid genome. This has been confirmed by whole-genome sequencing of mouse iPSC lines [13] showing that the genome-wide mutation rate does not exceed that of their parental cells. However, with prolonged culture and high number of passages, the risk for cytogenetic rearrangements and aneuploidies increases in stem cells similar to other types of cells [2,12,14].
The overall frequency of acquired genetic variants after expansion of iPSC clones and ESC appears comparable although with some differences in the types of abnormalities [4,12]. The reason for differences in the types of karyotypic abnormalities in iPSC versus ESC is yet unclear and it may be hypothesized that it is related to the methods used for reprogramming [15]. Furthermore, only a few studies have compared the frequency of genomic rearrangements in iPSC with respect to different methods of reprogramming [16]. The techniques for derivation of iPSCs can be broadly categorized into integrative and nonintegrative delivery of reprogramming factors. The latter has been developed to reduce DNA damage after cell reprogramming and to avoid insertional mutagenesis. The most frequently used methods until now are based on integrating adenovirus or lentivirus [17,18] and nonintegrating vectors such as oriP/EBNA1 episomal vectors [19], nonintegrating lentiviral vectors [20], and temperature-sensitive Sendai virus (SeV) vectors [21]. Alternative methods have been developed that are free of both virus and DNA such as synthetic microRNAs [22]. However, it is still unclear how several of these different reprogramming methods are associated with frequency and type of cytogenetic rearrangements.
To address these questions we have analyzed and compared the chromosome abnormalities in iPSC reprogrammed by two different and frequently used viral delivery methods. In this study, we present the spectrum of karyotypic aberrations in iPSC lines generated by integrating lentivirus that belongs to the Retroviridae family, and SeV a member of the Paramyxoviridae family of RNA viruses that replicates in the cytoplasm without integration into the host genome, respectively. Both methods used the OKSM set of reprogramming factors. In this study, we show that iPSC lines generated by lentivirus show a higher propensity for chromosomal aberrations at early passages when compared to SeV-derived iPSC. Furthermore, we present a duplicated chromosome 5q region that restricts a candidate region for growth advantage after reprogramming.
Materials and Methods
Primary fibroblast lines and culture
Primary fibroblast lines were established from punch biopsies obtained from the thigh of healthy individuals (n=6) and patients (n=6) (Fig. 1A). The donors were between 5 and 56 years of age, with a male/female ratio of 5/7. Patient-derived fibroblasts were from two patients with Lissencephaly (Liss1 and Liss2) with DCX gene mutations; two patients with Incontinentia pigmenti (IP5 and IP7), both carrying deletion of exons 4–10 in the NEMO gene, and two patients with Mowat–Wilson syndrome (MWS26 and MWS27) with mutations in ZEB2. None of the listed mutations are known to affect pathways critical for chromosome stability and the disorders are not associated with cytogenetic rearrangements [23
–25]. Primary fibroblast lines were established following standard protocols [26]. Fibroblasts were maintained in human foreskin fibroblast (HFF) medium [Dulbecco's modified Eagle's medium (DMEM, high glucose; Sigma) supplemented with 10% fetal bovine serum (FBS; Sigma), 2 mM

Reprogramming methods, cell lines used, and frequency of cytogenetic aberrations.
Cell reprogramming and stem cell culture
Lentivirus particles carrying a polycistronic Oct3/4, Klf4, c-Myc, and Sox2 construct [17] were prepared in HEK293T cells. Briefly, 7×105 HEK293T cells were seeded onto a 6-cm culture dish in 5 mL HFF medium, and transfected next day with 2 μg pEF1α-StemCCA/LoxP, 1.5 μg psPAX2 (Plasmid No. 12260; Addgene), and 0.5 μg pMD2.G (Plasmid No. 12259; Addgene), respectively, using FuGENE HD Transfection Reagent and associated protocol (Promega). The medium was replaced the next day and virus particles were harvested after 24 and 48 h, respectively.
For lentiviral transduction, we seeded 150,000 primary fibroblasts onto a 10-cm culture dish in HFF medium. Next day, the medium was changed to HFF medium supplemented with 1 μg/mL polybrene, and 2 mL of lentivirus supernatant was added. The medium was replaced with fresh medium every 24 h. On day 7, cells were harvested with TrypLE Express and 50,000 cells were seeded onto a 10-cm dish preseeded with Mitomycin C-treated human foreskin fibroblasts (HFFC, CRL2429; ATCC). After overnight recovery, medium was replaced with human embryonic stem cell (hES) medium (Knockout® DMEM supplemented with 20% serum replacement, nonessential amino acids,
Transduction by SeV was performed using the CytoTune®-iPS Sendai Reprogramming Kit (Life Technologies), following the manufacturer's protocol with minor modifications. Briefly, 25,000 skin fibroblasts [passage (P1-3) from healthy donors] were plated per 1-well of a 24-well plate (Corning) with 200 μL mixture of the four CytoTune Sendai factors (each 3×106 CIU) diluted in prewarmed HFF medium [DMEM (Sigma), 15% FBS (Sigma), 2% penicillin–streptomycin (Gibco), 1% nonessential amino acids (Gibco)] per well. Twenty-four hours post-transduction, the medium was replaced with fresh HFF medium, and cells were cultured under standard conditions for another 6 days with medium changed every second day. On day 7 after transduction, we seeded 1.5×105 cells from every cell line onto 100-mm dishes preplated with 3.5×106 Mitomycin C-inactivated feeders (HFFC) in HFF medium. Medium was then replaced every day with hES medium [Knockout DMEM, Knockout Serum Replacement, nonessential-amino acids, Glutamax, penicillin–streptomycin (Gibco), and 10 ng/mL bFGF (R&D systems)] until days 28–35 after transduction when passage 0 (P0) colonies were picked. The iPSCs were picked clonally and separate clones were cultured on feeders with manual passaging.
iPSC or human ESCs were cultured on dishes coated with HFFC in hES medium supplemented with 10 ng/mL bFGF. iPSC colonies were passaged manually or enzymatically using collagenase IV (Gibco). Alternatively, cells were transitioned to feeder-free culturing on Matrigel™ (BD bioscience) in E8 medium (Stem Cell Technologies) with Dispase passaging following the manufacturer's recommendations.
Karyotyping
Cells were treated with 200 μL of colcemid per 1 mL of culture medium [KaryoMax Colcemid Solution (10 μg/mL); Gibco] for 4 h. TrypLE and Trypsin Inhibitor (Invitrogen) were used for cell harvesting. Harvested cells were treated with freshly prepared hypotonic solution (75 mM KCl: 0.6% Na citrate in proportion 1:2, respectively, both by Sigma) for 30 min at 37°C followed by triple fixator treatment (3:1 methanol:acetic acid, both by Sigma) for 20 min each. Slides with metaphases were prepared by dropping fixed cell suspension on cold wet microscope slides (VWR). Metaphases were trypsinized with Trypsin (Sigma) and stained with Giemsa staining solution for further G-banding analysis. Metaphase analysis was performed using the Metafer slide scanning platform and Ikaros software (MetaSystems). Description and specification of chromosomal aberrations were done according to ISCN 2013.
Immunofluorescent staining
iPSC colonies were passaged onto glass coverslips coated by Geltrex (Life Technologies), either preplated with feeders or feeder-free, and cultured for 4 days before immunofluorescent (IF) labeling. IF staining was performed using standard techniques [27] on colonies fixed with freshly prepared ice-cold 4% paraformaldehyde and subsequently permeabilized in the blocking solution (1×phosphate-buffered saline pH 7.4, 1% bovine serum albumin, 0.1% Triton X-100). Primary antibodies against Oct4 (1:100; Abnova), Tra-1-60 (1:500; Invitrogen), Nanog (1:100; Millipore), and SSEA4 (1:500; Invitrogen), respectively, were allowed to bind overnight separately or in appropriate combinations at 4°C. After washing in 1×TBS, 0.05% Tween, the secondary antibodies anti-mouse IgG AlexaFluor 488, anti-mouse IgG AlexaFluor 555, anti-mouse IgM AlexaFluor 555, anti-rabbit IgG AlexaFluor 555, or anti-rabbit IgG AlexaFluor 633 (1:1,000; Invitrogen) were applied alone or in appropriate combinations for 1.5 h at room temperature in the dark. Visualization was performed on a Zeiss 510 confocal microscope (Carl Zeiss Microscopy) using Zen 2009 imaging software.
In vitro embryoid body differentiation
Embryoid bodies (EBs) were obtained from 20 to 30 large iPSC colonies that were manually cut and transferred to ultra-low attachment six-well plates (Sigma) and maintained in hES medium without bFGF for 3–4 weeks. Medium was changed every 24 h. Alternatively, EBs were formed using an AggreWell® 400 plate (Stem Cell Technologies) following the manufacturer's protocols. The EBs were harvested and centrifuged at 1,500 rpm for 5 min. The pellet was used for RNA extraction with the PureLink® RNA Mini Kit (Life Technologies) according to the manufacturer's instructions.
Analysis of stem cell marker expression
iPSC colonies were plated feeder-free on a 35-mm plate for one passage until confluent and harvested by lysis in TRIzol ®. RNA extraction was performed with the PureLink RNA Mini Kit (Life Technologies) according to the manufacturer's instructions. RNA quality was checked using an RNA nano6000 chip on a Bioanalyser2100 (Agilent). Only samples with an RNA integrity number (RIN) of ≥8 were used for further analysis. One microgram of total RNA was converted into cDNA using the SuperScript VILO cDNA Synthesis Kit (Life Technologies), mixed with TaqMan® Gene Expression Master Mix and loaded to a TaqMan Human Stem Cell Pluripotency Low-Density Array (TLDA; Life technologies) following the manufacturer's protocols. The TLDA was run on a 7900HT Real-Time PCR System (Applied Biosystems) and data were processed with RQ Manager® 1.2.1 software. The CT values obtained were used to calculate the expression relative to β-actin (dCT) and compared across all lines using GraphPad Prism 6. Hierarchical cluster analysis was performed by Genesis [28].
Results
Generation of iPSC lines from fibroblasts
Twelve different parental fibroblast cell lines (K1, K2, K4, K5, Liss1, Liss2, K7, K8, IP5, IP7, MWS26, and MWS27) with normal karyotypes were established from healthy individuals and patients. Six fibroblast lines (K1, K2, K4, K5, Liss1, and Liss2) were reprogrammed with integrative lentivirus resulting in 16 iPSCs lines (ie, with the prefix “L-”; Fig. 1A) and eight fibroblast lines (K1, K2, K7, K8, IP5, IP7, MWS26, and MWS27) were used as parental cells for reprogramming by SeV, resulting in another 16 iPSC lines (ie, prefix “S-”; Fig. 1A). The two fibroblast lines K1 and K2 were reprogrammed by both methods.
All 32 iPSC lines showed stable growth over passaging and fulfilled established morphological criteria, including colony formation with tightly packed roundish cells and a high nucleus-to-cytoplasm ratio. The iPSC lines were positive for pluripotency markers Nanog, Oct4, Tra-1-60, and SSEA4 as demonstrated by immunostaining.
Cytogenetic and molecular analysis of iPSC lines
All 32 iPSC lines were first karyotyped between passages 10 and 21 (early passages) using standard G-banding procedures. We observed that the cell lines reprogrammed by lentivirus had a higher number of karyotypic abnormalities (7/16 or 43.75%) compared to SeV-reprogrammed cell lines (1/16 or 6.25%) at these early passages (Fig. 1B). This difference is statistically significant (χ2 =4.17, P<0.05). The aberrant karyotypes are listed in Table 1.
iPSCs, induced pluripotent stem cells; LeV, lentivirus; SeV, Sendai virus.
We identified both numerical and structural karyotypic aberrations (Table 1 and Fig. 1C). Three lines (L-K1B, L-K4A, and L-Liss2A) showed a combination of structural and numerical chromosomal abnormalities, three cell lines (L-K2B, L-K5A, and S-IP5D) carried only structural aberrations and two lines (L-K2A and L-Liss1E) carried only numerical aberrations (Table 1 and Fig. 2A). Two cell lines (L-K4A and L-Liss2A) carried small chromatin fragments containing centromeres of unknown origin classified as marker chromosomes. Notably, three out of seven abnormalities in iPSC generated by lentiviruses were mosaic for chromosome 12 trisomy (Table 1). The single abnormal karyotype among the SeV-generated lines at early passages revealed a balanced translocation between chromosomes 10 and 18 [46,XX,t(10;18)(p15;q11.2)] (Fig. 2A). Aberrations involving chromosome 5 were identified in three lines. Two of these clones (L-K2A and S-K2A), generated by either reprogramming method, showed a chromosome 5 trisomy. Interestingly, a third line (L-K2B) showed a duplication of chromosome 5q13q33 in 9/20 cells (46,XY,dup(5)(q13q33)[9]/46,XY[11]). The duplication was detected already at P15 and after induction with lentivirus. Furthermore, the duplicated region spans approximately half of the long arm of chromosome 5 corresponding to about 80 Mb of DNA. We then picked colonies from the mosaic L-K2B line by picking colonies based on morphological criteria and we derived four different sublines (L-K2B2, L-K2B3, L-K2B4, and L-K2B5). The sublines were cultured individually for multiple passages and they all showed normal karyotypes confirming genetic heterogeneity at P15 with a proportion of cytogenetically normal clones.
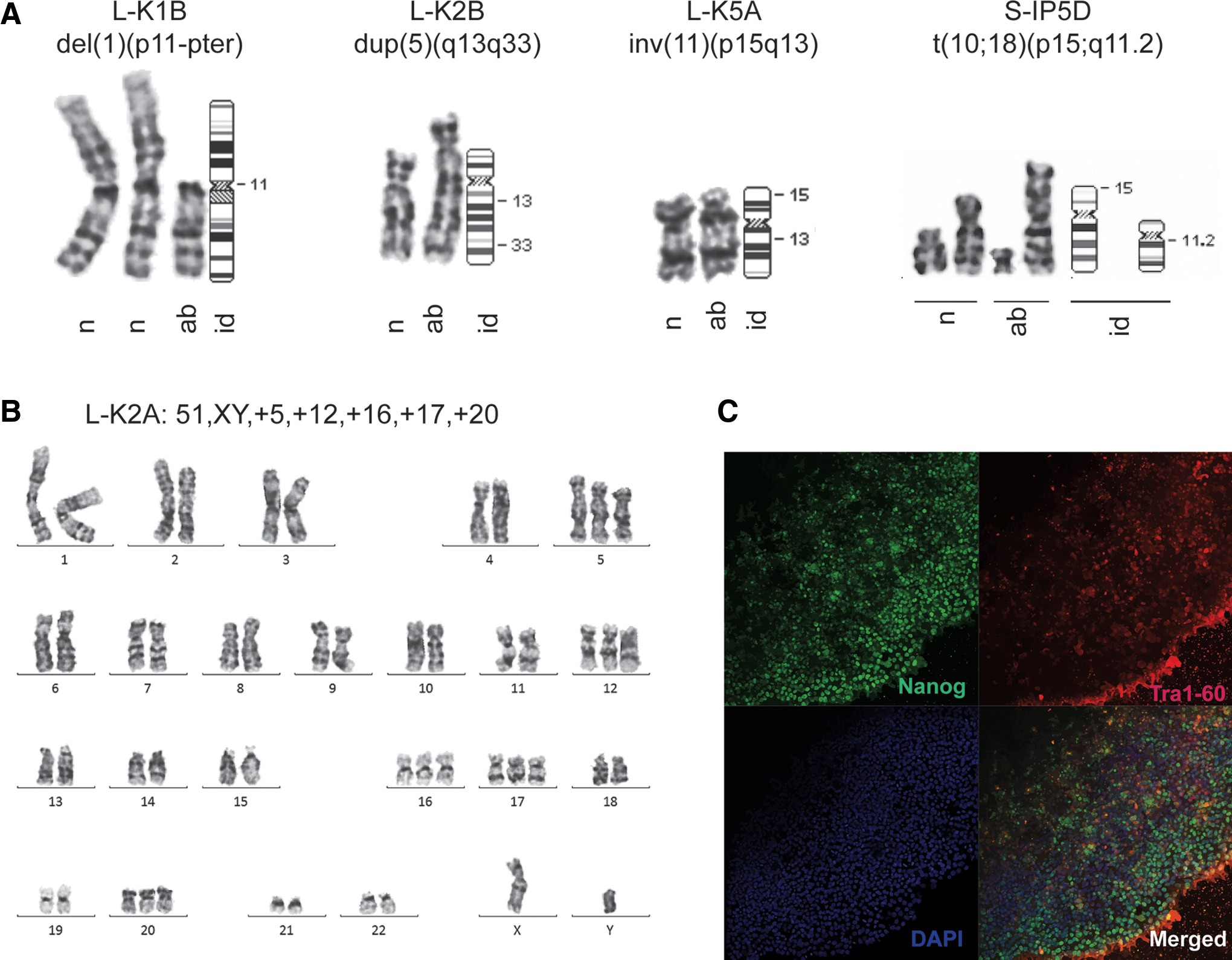
Aberrant karyotypes and stem cell characteristics of iPSCs.
We next asked at what stage the karyotypic abnormalities occurred in the iPSC lines L-K2A, L-K2B, L-Liss2A (Table 1). Cells were thawed from P12 of the L-Liss2A line with a mosaic trisomy of chromosome 12 (47,XY,+12[4]/47,XY, +mar[1]/46,XY[25]) identified at P17. These clones showed a normal karyotype after additional 16 passages (P28) suggesting that trisomy 12 was acquired after P12. Another karyotypic difference between early and late passages was observed for the iPSC line L-K2A. This line showed a nonmosaic aberrant karyotype (51,XY,+5,+12,+16,+17,+20) at P17 (Fig. 2B, C). An early passage (P13) was thawed and analysis showed the mosaic karyotype (50,XY,+5,+12,+16,+20[1]/50,XY,+9,+12,+16,+20[1]/49,XY,+12,+16,+20[10]) after expansion to P22. This suggests a primary rearrangement before P13 resulting in secondary changes and clonal heterogeneity. In combination, these findings demonstrate that the vast majority of the structural and numerical aberrations observed herein are acquired at early passages and after reprogramming with lentiviruses.
In search for aberrations that are acquired with prolonged culture, we performed a second karyotyping between P22 and P37 on 10 cell lines that were cytogenetically normal at earlier passages. The majority of the lines retained a normal karyotype (Table 1) with the SeV-generated S-K2A line as a single exception. This line showed trisomy for chromosome 5 (47,XY,+5) acquired between passages 11 and 31.
Analysis of iPSC and EBs
Expression of stem cell markers and markers for pluripotency was confirmed in total RNA from all iPSC at a stage corresponding to the karyotypes made at early passages. The analysis was performed by TaqMan Low-Density Human Pluripotency array (TLDA). The expression profile of pluripotency marker genes among all iPSC lines was similar and comparable to that of the three ESC lines used as references (Fig. 3A) [3]. Furthermore, a heat map analysis of the 47 core markers for undifferentiated cell state and pluripotency showed that all iPSC lines expressed markers for undifferentiated and pluripotent cells at levels similar to that in the ESC lines despite cytogenetic abnormalities in several iPSCs. We further performed hierarchical cluster analysis to compare marker expression in iPSCs with both the ESCs and the parental fibroblast lines (Fig. 3B). The analysis revealed that all stem cell lines (iPSC and ESC) cluster together and separate from the parental human dermal fibroblasts (HDF). The ESC lines are interspersed with the iPSC lines, suggesting that a distinction between iPSC and ESC is not possible using TLDA. We further investigated the possibility of differences in expression pattern of markers between lentivirus or SeV-derived iPSC using two-way analysis of variance (ANOVA). However, no significant difference could be detected between the two groups.
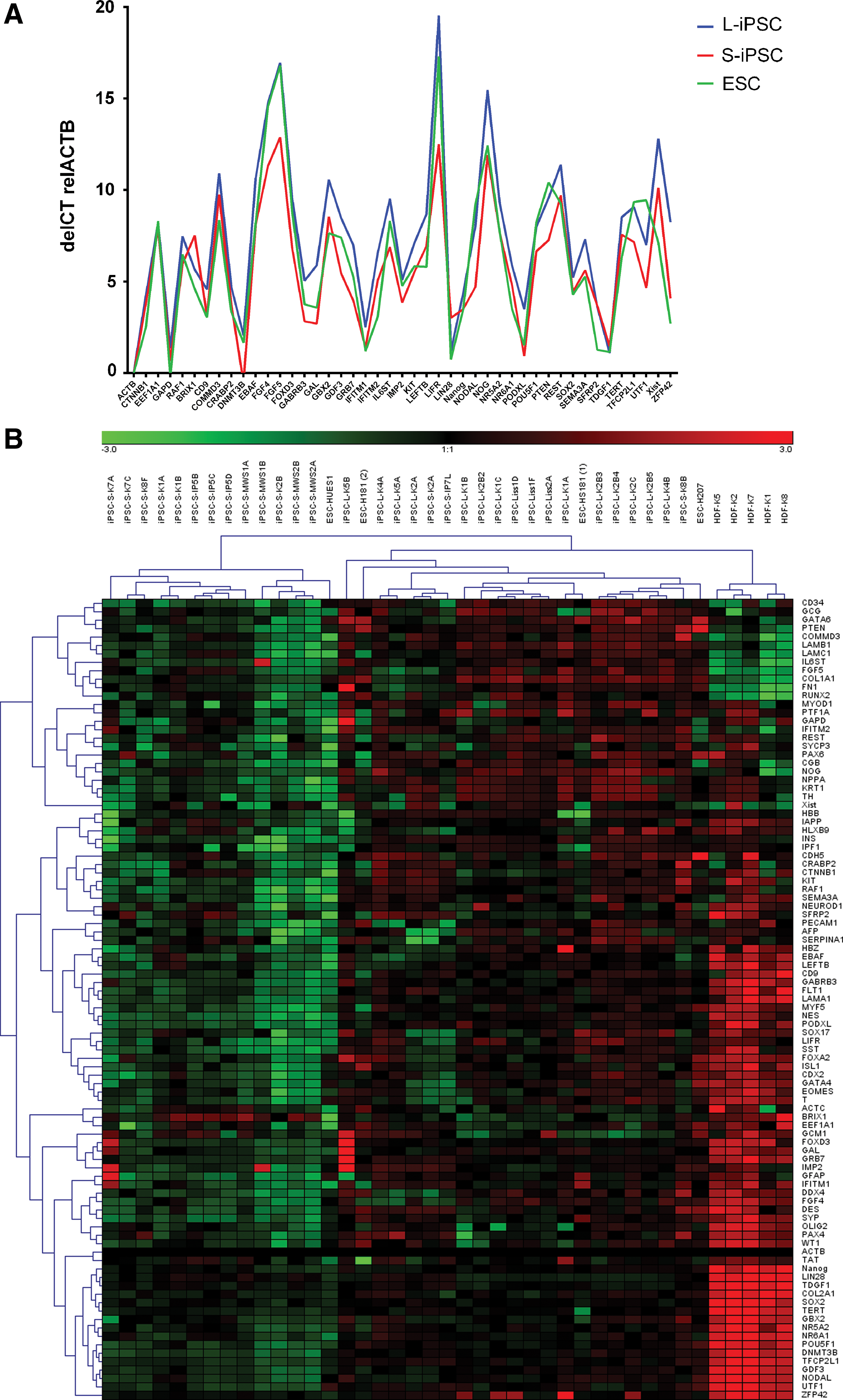
Expression analysis of iPSCs, embryonic stem cells (ESCs), and parental fibroblasts.
Pluripotency of iPSC lines was further demonstrated by EB formation and subsequent differentiation for 3–4 weeks. The EBs were harvested, total RNA was isolated and analyzed on a TLDA for expression of markers from all three germ layers. Notably, EBs from the iPSC lines L-K2A with the nonmosaic karyotype 51,XY,+5,+12,+16,+17,+20 and S-IP5D with the karyotype 46,XX,t(10;18)(p15;q11.2) showed normal abilities to differentiate into all three germ layers. Furthermore, both lines showed TLDA expression profiles comparable to EBs from karyotypically normal iPSC lines (not shown). This observation illustrates the need for analysis of genetic integrity of pluripotent cell lines before using these for further experiments.
Discussion
Identification of compromised genomic integrity of human iPSC is crucial with respect to their use for disease modeling and cell therapy. In this study, we examined the cytogenetic integrity in iPSC reprogrammed by two different viral-based methods. The use of integrative vectors has been questioned as it is a potential source of mutations and for reactivation of oncogenes. Indeed, the use of episomal or protein-based delivery methods have shown a lower frequency of cytogenetic aberrations when compared to procedures based on integrative viruses [18,19]. Furthermore, complementary high-resolution methods have indicated a high rate of de novo subchromosomal aberrations and CNVs in early passages of iPSC using integrative methods [2,11,12]. Interestingly, analysis of CNVs showed a different distribution in iPSC when compared to ESCs [12]. In combination, these studies suggest that the reprogramming procedure and/or culture of iPSC influence the rate of acquired genomic rearrangements. However, no systematic investigation has been performed that compares cytogenetic aberrations after reprogramming by lentiviruses and SeV. To this end, we used karyotyping to assess genomic integrity of iPSC as it is the most frequently used method for the assessment of genomic integrity in iPSC [15]. It may be argued that minor rearrangements escape detection using karyotyping as the resolution is limited compared to, for example, arrayCGH analysis. However, it has been shown that the vast majority of acquired duplications in iPSC are >10 Mb in size and thus detectable by karyotyping [4,5].
Following widespread practice for iPSC generation, we derived 32 iPSCs from fibroblasts with normal karyotypes using either integrative lentivirus (n=16) or nonintegrating SeV (n=16), for transgene expression of the four reprogramming factors OKSM. Our overall comparison of karyotypes revealed that, at early passages (<P21), 7 out of 16 iPSC lines reprogrammed by lentivirus carried a cytogenetic aberration. This is in the same range as previously reported for both ESC and iPSC in other studies [4,5]. The corresponding figure for cytogenetic aberrations in our lines reprogrammed by SeV was 1 out of 16 and clearly below the reported average for iPSC using integrative methods. Furthermore, the rate of cytogenetic aberrations at early passages and after reprogramming with SeV in our study is in line with studies using episomal-based reprogramming [16]. After prolonged culture, we detected one acquired aberration in a SeV-reprogrammed line. This occurrence of aneuploidies with time in culture is consistent with previous reports on both ESCs and iPSCs with preponderance for chromosomes 8, 12, 17, 20, and X [4]. The explanation for differences in cytogenetic aberrations with the two reprogramming methods used herein is unclear. However, one plausible mechanism is that the stable integration using lentivirus may lead to the reactivation or transcriptional leakage of transgenes, for example, c-Myc. Another possible mechanism is direct effects on the host genome at the sites of integration leading to altered or silenced expression of targeted genes.
Some of the individual aberrant karyotypes in our study exhibit similarities. Three lines showed aneuploidy for chromosome 12 previously described as a predominant abnormality during ESC and iPSC culture adaptation. The abnormality was observed after lentiviral reprogramming and before P21. Trisomy 12 was associated with trisomy of both chromosomes 5 and 20 in the line L-K2A. This is consistent with earlier reports showing recurrent aneuploidy of these chromosomes in stem cell lines [5,29]. Furthermore, mapping of overlapping duplications in independent ESC clones has identified a critical region on chromosome 20q11 [5] and it has been suggested that overexpression of the BCL2L1 gene, located within this part of the genome, is responsible for growth advantage. Interestingly, we identified three cell lines with aberrant karyotypes involving chromosome 5 including an isolated trisomy 5 in the S-K2A line acquired at a late passage. Moreover, in the L-K2B line we identified a duplication spanning the chromosome 5q region [dup(5)(q13q33)] as the only abnormality. In combination with previous reports on isolated gains of chromosome 5 and chromosome 5q, our observation supports a genetic mechanism on chromosome 5 that promotes cell survival in iPSC. Furthermore, the candidate region is restricted by the dup(5)(q13q33). Noteworthy, all three iPSC lines with cytogenetic aberrations involving chromosome 5 are derived from one and the same parental fibroblast line. Thus, it may be argued that they originate from a very low degree of mosaicism that escaped detection in the parental cells. However, this is not supported by the normal karyotype at P11 in S-K2A with an acquired chromosome 5 trisomy detected at P31. Furthermore, the aberrations involving chromosome 5 are different in each of the three lines.
Finally, the expression pattern of markers for pluripotency was very similar in iPSC lines generated by either a lentivirus or SeV-based method. Furthermore, the expression profiles of the iPSCs were almost identical to that of ESCs used as reference. Importantly, among the iPSCs, we did not observe any correlation between the cytogenetic aberrant lines and the expression of 96 stem cell markers. This illustrates that analysis of these markers may not reveal major cytogenetic aberrations and stresses the importance of genetic analysis of iPSC lines performed at different passages before further applications in biomedicine.
In summary, we have demonstrated that reprogramming of human iPSC using SeV significantly reduces cytogenetic aberrations in early culture passages when compared to integrative lentivirus. Thus, SeV-based generation of iPSC maintains genomic integrity better than integrative lentivirus. Because we compared these two viral-based methods, we do not know how the SeV-based method relates to other nonintegrating methods, for example, mRNA or protein-based methods in our setting. We further identified three iPSC lines with aberrations involving chromosome 5. One of these lines showed a duplication of chromosome 5q13q33 as the single abnormality that restricts a candidate locus for iPSC survival. Our results have improved understanding on cytogenetic aberrations associated with reprogramming methods and further investigations are now required to clarify the significance of chromosome 5q for growth advantage in iPSC.
Footnotes
Acknowledgments
The authors thank C. Annerén for data on the ESC lines. This work was supported by grants from Astra-Zeneca, Uppsala University Hospital, the Swedish Research Council (K2013-66X-10829-20-3), Uppsala University and Science for Life Laboratory. M.S. was supported by grant from the Sävstaholm Society.
Author Disclosure Statement
No competing financial interests exist.