
Abstract
Wolfram syndrome 2 (WFS2) is a premature aging syndrome caused by an irreversible mitochondria-mediated disorder. Cisd2, which regulates mitochondrial electron transport, has been recently identified as the causative gene of WFS2. The mouse Cisd2 knockout (KO) (Cisd2−/− ) recapitulates most of the clinical manifestations of WFS2, including growth retardation, osteopenia, and lordokyphosis. However, the precise mechanisms underlying osteopenia in WFS2 and Cisd2 KO mice remain unknown. In this study, we collected embryonic fibroblasts from Cisd2-deficient embryos and reprogrammed them into induced pluripotent stem cells (iPSCs) via retroviral transduction with Oct4/Sox2/Klf4/c-Myc. Cisd2-deficient mouse iPSCs (miPSCs) exhibited structural abnormalities in their mitochondria and an impaired proliferative capability. The global gene expression profiles of Cisd2+/+ , Cisd2+/− , and Cisd2−/− miPSCs revealed that Cisd2 functions as a regulator of both mitochondrial electron transport and Wnt/β-catenin signaling, which is critical for cell proliferation and osteogenic differentiation. Notably, Cisd2−/− miPSCs exhibited impaired Wnt/β-catenin signaling, with the downregulation of downstream genes, such as Tcf1, Fosl1, and Jun and the osteogenic regulator Runx2. Several differentiation markers for tridermal lineages were globally impaired in Cisd2−/− miPSCs. Alizarin red S staining and flow cytometry analysis further revealed that Cisd2−/− miPSCs failed to undergo osteogenic differentiation. Taken together, our results, as determined using an miPSC-based platform, have demonstrated that Cisd2 regulates mitochondrial function, proliferation, intracellular Ca2+ homeostasis, and Wnt pathway signaling. Cisd2 deficiency impairs the activation of Wnt/β-catenin signaling and thereby contributes to the pathogeneses of osteopenia and lordokyphosis in WFS2 patients.
Introduction
I
However, the functions of the novel protein Cisd3, which contains two CDGSH domains and no transmembrane domain, remain unclear. Patients with a Cisd2 homozygous mutation are diagnosed with Wolfram syndrome 2 (WFS2), an autosomal recessive inherited disease characterized by juvenile-onset neurodegeneration of the central and peripheral nervous systems [6]. Chen et al. generated Cisd2 knockout (KO) mice that exhibited WFS2-like clinical symptoms, including early senescence, protruding ears, corneal opacities, thin bones, and low muscle mass [7]. Mitochondrial biogenesis and dynamic homeostasis are important for supplying a sufficient amount of energy for development and differentiation [8]. Notably, Chen et al. have demonstrated that Cisd2 deficiency leads to structural damage of the outer mitochondrial membrane in mice, resulting in mitochondrial dysfunction with reduced electron transport activity and oxygen consumption. However, whether Cisd2 affects mitochondrial function to further modulate stem cell biology and cellular differentiation during early development remains unclear.
Mitochondria depend on the activity of the mitochondrial electron transport chain, as mediated by respiratory complexes I, III, and IV, which drive ATP synthesis through complex V (ATP synthase) [9]. Mitochondrial electron transport generates not only ATP but also by-products, including ROS and other metabolites [10]. In mitochondrial oxidative phosphorylation, mitochondria generate more ATP and ROS than is produced by glycolysis. Mitochondria are necessary for supporting active proliferation; therefore, they are essential for cell reprogramming and maintaining human embryonic stem cell identity [11]. Mitochondria regulate cell proliferation and differentiation, particularly in osteoblasts and adipocytes [12 –14]. Consistent with these functions, the inhibition of mitochondrial respiration via chemical treatments or overexpression of transcription factors increases pluripotency, whereas activation of mitochondrial activity impairs reprogramming [10].
The intracellular distribution of mitochondria has been associated with the degree of stemness in adult monkey stromal stem cells [15], suggesting that their differential distribution affects the maturation of developing embryonic stem cells [16]. Gene KOs of critical factors (Tfam, Polg, and Polg2) in mitochondrial biogenesis cause embryonic lethality at a late stage due to an insufficient supply of energy and metabolites required for cell differentiation [17]. Mitochondrial inhibitors retard osteogenic differentiation [18]. Similarly, Cisd2 modulates the differentiation of adipocytes by regulating intracellular Ca2+ homeostasis [19]. However, how the loss of Cisd2 delays differentiation remains unclear. A recently developed promising technique for recapitulating developmental and pathological processes involves the reprogramming of differentiated human/mouse somatic cells into induced pluripotent stem cells (iPSCs) [20,21].
iPSCs are generated by transducing 4 transcription factor-encoding genes, known as the OSKM genes, including Oct4 (Pou5F1), Sox2, Klf4 (Kruppel-like factor 4), and c-Myc, and these cells have been confirmed to exhibit embryonic stem cell-like characteristics in studies of the mechanisms and progression of diseases and in medical applications [21]. In this study, we established Cisd2 KO mouse iPSCs (miPSCs), representing naïve precursors to multiple lineages present in Wolfram syndrome. We sought to elucidate the transcript profile of these Cisd2-deficient miPSCs and mitochondria-associated parameters to further evaluate the specific role of Cisd2 in transcriptional regulation. The capacity of Cisd2-deficient miPSCs for differentiation into multiple lineages, particularly osteogenic lineages, was also investigated. The results of this study allow elucidation of the role of Cisd2 in mitochondria and suggest that this protein maintains the expression of developmental genes by affecting Wnt signaling.
Materials and Methods
Generation of iPSC lines and cell culture
Cisd2 deficiency (Cisd2+/− , Cisd2−/− ) and wild-type (Cisd2+/+ ) C57BL/6 mice have been described [7]. The mice were bred and treated according to the National Research Council's Guide for the Care and Use of Laboratory Animals. Fibroblasts were collected from postnatal mice, and cultured in DMEM medium supplemented 15% fetal bovine serum (FBS). For reprogramming into iPSCs, fibroblasts were plated at 1 × 105 cells per six-well dish for infection with OSKM retrovirus. After infection, cells were cultured and medium changed every 2 days. After 16 days, embryonic stem cells like colonies were transferred onto feeder layer and cultured in DMEM supplemented with 15% FBS, 0.1 mM nonessential amino acids (11140-050; Invitrogen), 0.55 mM 2-mercaptoethanol (M7154-100 ML; Sigma), 50 μg/mL penicillin-streptomycin (15140-122; Invitrogen), and 0.3% recombinant leukemia inhibitory factor (LIF, PMC9484; Invitrogen) at 37°C, 5% CO2. Cisd2+/+ , Cisd2+/− , and Cisd2−/− miPSCs were cultured in the ES medium supplemented with LIF.
Embryoid body-mediated osteogenic differentiation
For embryoid body (EB) formation, miPSCs were dissociated into a single cell suspension using 0.25% trypsin-EDTA and plated onto nonadherent bacterial culture dishes at a density of 2 × 106 cells/100 mm plate, where they were allowed to aggregate. After 4 days in floating culture, EBs were transferred onto gelatin-coated plates and maintained in the same medium for 24 h. EBs were then assigned to different groups for in vitro differentiation into multiple lineages as previously described [22], including hepatic, adipogenic, and neural differentiation lineages. Next, EBs were transferred to gelatin-coated plates and cultured with osteogenic induction medium (DMEM-HG containing 10% FBS, 10−8 M dexamethasone, 50 μg/mL ascorbic acid, 10 mM β-glycerolphosphate, and 1% penicillin-streptomycin). For osteogenic differentiation, cells were cultured at 37°C and 5% CO2 for 21 days, and the medium was changed every other day.
Teratomas assay
miPSCs were maintained and propagated on the feeder cell. Three days later, miPSCs were trypsinized, washed twice with phosphate buffered saline (PBS), and then subcutaneously injected into the bilateral inguens of male NOD-SCID mice (3 × 107 cells per mL in saline, 100 μL per injection). After 3–4 weeks, teratoma were collected and fixed in 4% paraformaldehyde, before embedding in paraffin. This study was approved by the Institutional Review Board (VGHIRB 2014-05-002C) of Taipei Veterans General Hospital. Paraffin-embedded tissue was sectioned and stained with H&E.
Chimera mouse production by blastocyst injection
The introduction of Cisd2 +/+ and Cisd2 −/− miPSCs (derived from C57BL/6J strain, black coat color) into mouse blastocysts-derived from C57BL/6J-Tyrc2J strain (albino) was followed as previously described with some modifications [23]. The adult chimeras were confirmed by coat color, demonstrating that miPSCs were competent to produce adult chimeric mice. This study was assisted by the Transgenic Mouse Model Core Facility, Academic Sinica, Taiwan.
Alkaline phosphatase staining
For detecting the alkaline phosphatase (AP) activity of cells on original plates, cells were fixed with 80% alcohol, and then fixed cells stained using the Blue Alkaline Phosphatase Substrate kit III (Vector Laboratories) according to the manufacturer's instructions.
Western blot analysis, immunofluorescence assay, and flow cytometry
Western blots were performed as previously described [22]. Details of antibodies can be found in Supplementary Table S1 (Supplementary Data are available online at
Cells were fixed with 4% paraformaldehyde, permeabilized in 1% triton X-100, and blocked with 5% FBS. Cells were stained with primary antibodies, and then incubated with Alexa Fluor 568-conjugated anti rabbit (1:300; Invitrogen) or FITC-conjugated anti rabbit (1:300; Invitrogen). Details of antibodies can be found in Supplementary Table S1. Cells were then washed with PBS and photographed under a fluorescence microscope (Olympus).
Membrane potential (Δψm) and mitochondrial mass
Δψm was assessed in the iPSC lines using a MitoProbe JC-1 Assay Kit by flow cytometry (Life Technologies), according to the manufacturer's protocol. Briefly, miPSCs were seeded on six-well plates at a density of 5 × 105 cells/well and cultured for 1 day. The cells were then resuspended in 500 μL of warm media and incubated with JC-1 dye (2 mM final concentration) at 37°C and 5% CO2 for 30 min and washed twice with PBS before flow cytometry analysis. At high concentrations, JC-1 monomers form aggregates. The monomers emit green fluorescence, and the aggregates emit red fluorescence. A decrease in delta psi is determined according to a decrease in red fluorescence emission [24,25], and a decrease in monomeric fluorescence (JC-I green) indicates a loss of mitochondrial mass [26]. Cells were collected via centrifugation and resuspended in PBS. The relative membrane potential was calculated by normalizing the intensity of red JC-1 fluorescence to that of green JC-1 fluorescence, which represents the mitochondrial mass. The samples were analyzed by flow cytometry (BD LSRFortessa cell analyzer; Becton Dickenson) and BD FACS Diva software (Becton Dickenson).
To determine mitochondrial mass, miPSCs were seeded on six-well plates at a density of 5 × 105 cells/well and cultured for 1 day. The cells were then trypsinized, suspended in PBS, and transferred into flow tubes. One millimolar 10-N-nonyl acridine orange (NAO) dye was added to the tubes, which were protected from light, for 10 min at room temperature. The fluorescence intensity of 10,000 cells, as determined by flow cytometry, was recorded at an excitation wavelength of 488 nm and an emission wavelength of 535 nm.
Quantitative real-time polymerase chain reaction
Total mRNA was obtained using RNeasy kit (Qiagen) reverse-transcribed using the superscript III first strand synthesis system (Invitrogen). Quantitative polymerase chain reaction was carried out with ABI 7900 Fast System and SYBR Green Master Mix. Primer sequences are shown in Supplementary Tables S2 and S3. The expression values are normalized against the corresponding Gapdh value.
Transmission electron microscopy
miPSCs were fixed in the mixture of glutaraldehyde and paraformadehyde in cacodylate buffer (pH 7.4). They were postfixed in 1% OsO4 and 1.5% potassium hexanoferrate (pH 6.0), then rinsed in cacodylate and 0.2 M sodium meleate buffers (pH 6.0), and blocked with 1% uranyl acetate. Following dehydration, the cells were embedded in Epon and sectioned for TEM analysis.
Cell growth rate and MTT assay
For evaluation of cell growth rate, miPSCs were plated on 24-well plates at a density of 2 × 104 cells/well. Cells were trypsinized and determined by cell counting at day 0, 1, and 3. For determination of viable cell number at different passage, cells were seeded on 24-well plates at a density of 2 × 104 cells/well. After aspirating the culture medium, cells were incubated with DMEM medium with the addition of methyl thiazol tetrazolium (3-(4,5-dimethylthiazoyl-2)-2,5-dipheyl tetrazolium bromide at 0.5 mg/mL; SIMGA) at 37°C for 1 h. After incubation, we aspirated the medium and added dimethyl sulfoxide to dissolve the MTT formazan product. The amount of MTT formazan product was determined using a microplate reader at an absorbance of 560 nm (SpectraMax 250; Molecular Devices).
Microarray, gene set enrichment analysis, and ingenuity pathway analysis
Total RNA was extracted from mouse Cisd2+/+
, Cisd2+/−
, and Cisd2−/−
miPSCs using an RNeasy Kit (Qiagen). Reverse transcription and hybridization were performed at the gene expression analysis core facility of the National Yang-Ming University Genome Research Center. Arrays (Affymetrix Mouse 430A 2.0) processed according to the manufacturer's instructions were scanned using an Affymetrix microarray platform. Raw microarray data were preprocessed and normalized using the RMA algorithm implemented in Affymetrix Power Tools. The expression levels of individual genes were calculated by averaging the signal intensities of all of the corresponding probesets. Heatmap and hierarchical clustering was performed for selected RNAs based on city-block distances. The distances were computed based on the average mRNA expression levels in the mouse Cisd2+/+
, Cisd2+/−
, Cisd2−/−
miPSC, and mESC groups. The genes used for functional classification in Fig. 2B are listed in Supplementary Table S4. To gain functional insights from these gene expression data, gene set enrichment analysis (GSEA) was conducted using predefined gene sets (ectoderm, mesoderm, and endoderm) [27
–29] and a customized R script for GSEA. The datasets were submitted to Gene Expression Omnibus database (
Alizarin red S staining
Following previous described osteogenic differentiation method, the ability of calcium deposition was determined at day 21. After aspirating the culture medium, cells were washed with PBS. Ice-cold 70% alcohol was added to fix the cells. After fixing for 1 h, cells were washed thrice with ddH2O. Twenty millimolars alizarin red S (ARS) solution was added into each well and incubated for 20 min. After aspirating the solution, cells were washed thrice with ddH2O. After aspirating the solution, CPC buffer [10 g cetylpyridinium chloride (CPC; Sigma) and 0.1469 g Na2HPO4 (Sigma) dissolved in 100 mL ddH2O] was added and then incubated for 1 h. The amount of ARS was determined at an absorbance of 405 nm (SpectraMax 250; Molecular Devices).
Flow cytometry analysis for HCC cell lines
To detect the expression profiles of alkaline phosphatase (ALP) in each miPSC lines, cells were incubated with rabbit anti-ALP antibody (AF2910; R&D Systems) and visualized by using Alexa 568-conjugated goat anti-mouse (Invitrogen). Samples were analyzed using an LSR II flow cytometry and CellQuest Software (BD Biosciences). Live cells were gated, and dead cells and cell debris were excluded using FCS and SSC biparametric plots.
Free intracellular Ca2+ measurement
Intracellular Ca2+ levels were determined using a Fluo3-AM probe (Molecular Probes), which binds Ca2+ with high specific affinity. After trypsinization, cells were washed once with Ca2+-free PBS and resuspended in fresh buffer. The cells were then incubated in the dark with 5 μM Fluo-3/AM at 37°C for 30 min, and fluorescence was measured at FL-1 (530 nm) by flow cytometry (BD LSRFortessa cell analyzer; Becton Dickinson), with excitation at 488 nm. The fluorescence intensities of the Fluo-3/AM probes were measured via flow cytometry and analyzed using BD FACS Diva software (Becton Dickenson). At least 10,000 events per sample were acquired. Samples without Fluo-3AM fluorescence were considered blank controls, for which fluorescence was represented as F0. The Geo Mean of fluorescence (F) of each sample was subtracted by F0 to eliminate background fluorescence.
Immunoprecipitation assay
miPSCs were lysed in Buffer G, incubated overnight with 6 μg antibody, and captured with Protein G beads according to manufacturer's instructions. Two milligrams cell lysate was incubated with 20 μg Protein G beads. Protein complexes were eluted by boiling in loading buffer. Ten microliters was used for each immunoblot with 2% input.
Statistical analysis
Data were expressed as mean ± SD. Statistically significant differences among groups were detected by one-way analysis of variance, and when significance was observed, post hoc Tukey tests were performed using SPSS (SPSS 12.0; SPSS). Data were plotted using GraphPad Prism program (GraphPad Prism 5.0; GraphPad). The criterion for significance was set as P < 0.05.
Results
Characterization of Cisd2-deficient miPSCs
The somatic fibroblasts used to generate iPSCs were collected from Cisd2-deficient embryos, which carried mitochondria-associated phenotypic defects. However, whether cells containing the mutation for the genetic disease that causes premature aging can be reprogrammed into miPSCs is unknown. Cisd2 is an important regulator of life span [30]. Cisd2 KO mice exhibit features of severe premature aging and early onset degeneration of multiple tissues. Most of these degenerated tissues were originally considered as mitochondria-enriched or dependent tissues. Thus, we attempted to determine whether Cisd2 deficiency affects miPSC generation. Cisd2 deficiency causes mitochondria-associated phenotypic defects in mice [7]. Cisd2 KO mice have been demonstrated with many manifestations of premature aging, such as osteopenia, lordokyphosis, early onset degeneration of central nerves, and impaired glucose tolerance [7]. Systemic defects have been attributed to the differential global distribution and dysregulation of mitochondria. However, little is known about the potential role of the Cisd2-mediated developmental process in developing blastocysts. Indeed, Cisd2 deficiency has been reported to delay adipocyte differentiation. Considering the relative difficulty in obtaining sufficient embryos from Cisd2−/− mice and in culturing embryos in vitro for long periods of time, we established an iPSC-based model to develop an efficient platform for investigating Cisd2. To establish this model, we reprogrammed Cisd2-deficient fibroblasts using the OSKM genes. We collected individual fibroblasts from aging C57BL/6 (B6) mice with the Cisd2+/+ , Cisd2 +/−, and Cisd2 −/− genotypes and then plated 4 × 104 cells/well in six-well plates 1 day before viral infection. We subsequently transduced fibroblasts with the OSKM genes on day 0. We observed an ES-like morphology in the reprogrammed fibroblasts by day 7 and detected ALP activity by day 18 (Fig. 1A). Finally, we obtained miPSCs with Cisd2+/+ , Cisd2+/− , and Cisd2−/− genotypes via retroviral infection.
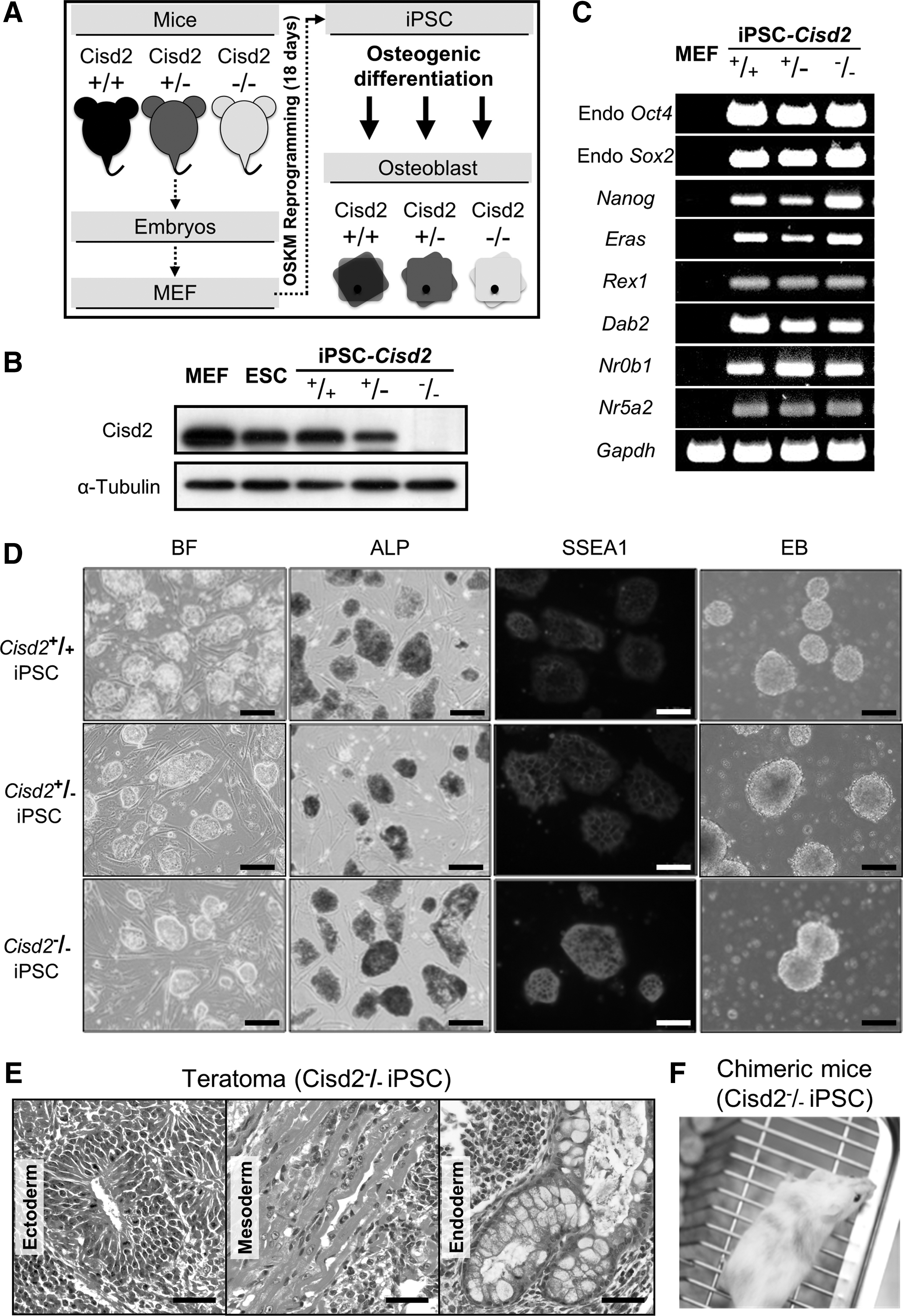
Generation of miPSCs derived from wild-type (Cisd2+/+
) or Cisd2-deficient (Cisd2+/−
and Cisd2−/−
) fibroblasts using the four OSKM factors.
Before investigating whether the developmental process is affected by Cisd2 deficiency, we confirmed that Cisd2 expression in miPSCs was consistent with that in fibroblasts. Western blot analysis revealed that OSKM reprogramming had limited effects on the expression of this protein (Fig. 1B). We determined that Cisd2 expression was similar in Cisd2+/+ mouse embryonic fibroblasts (MEFs) from primary culture from fetal mice, Cisd2+/+ aging fibroblasts and miPSCs, suggesting that its expression was maintained at similar levels in these different cells. In contrast, Cisd2 expression was significantly lower in Cisd2+/− and Cisd2−/− aging miPSCs than in Cisd2+/+ miPSCs. These results indicated that retroviral infection of cells with the OSKM genes did not affect the expression of this Cisd2. Next, we examined the expression of pluripotency markers by reverse transcription–polymerase chain reaction (RT-PCR). RT-PCR analysis revealed that all of the stemness genes were expressed (Fig. 1C), including Endo-Oct4, Endo-Sox2, Nanog, Eras, Rex1, Dab2, Nr0b1, and Nr5a2 [21]. Despite the absence of Cisd2, these iPSC colonies exhibited all of the characteristics of embryonic stem cells, including the characteristic morphology, ALP activity, expression of the stemness marker SSEA1 and formation of EBs (Fig. 1D). Collectively, our data demonstrated that the miPSCs with the genotypes Cisd2+/+ , Cisd2+/− , and Cisd2−/− were embryonic stem cell-like. To further investigate the role of Cisd2 in regulating development in vivo, we transplanted Cisd2−/− miPSC cells (3 × 106 cells) subcutaneously into immune-compromised NOD-SCID mice. Teratoma-containing cells from the three germ layers, namely the ectoderm (neural epithelium, Fig. 1E), mesoderm (muscle, Fig. 1E), and endoderm (respiratory epithelium, Fig. 1E), were observed upon histological analysis, indicating that the cells had a considerable level of pluripotency. To further investigate the pluripotency of Cisd2-deficient cells, we performed a chimeric experiment to evaluate the developmental potential of Cisd2-deficient miPSCs. Notably, we obtained surviving Cisd2−/− chimeric mice by injecting Cisd2-deficient miPSCs into C57BL/6J-Tyrc2J blastocysts. Compared with the chimeric mice generated using Cisd2+/+ miPSCs, live Cisd2−/− chimeric mice were distinguished based on their coat color (Fig. 1F), indicating that Cisd2-deficient miPSCs are still able to differentiate in vivo. These data indicate that the loss of Cisd2 does not inhibit iPSC generation and that miPSCs with this disease-causing mutation exhibit the typical capacity for simple differentiation in vitro.
Cisd2 regulates the membrane potential of iPSCs
Cisd2 is located in the ER and mitochondrial membrane and contains two redox-active 2Fe-2S clusters [7,31]. It may play a critical role in maintaining the integrity and function of mitochondria [7]. Interestingly, it is also a causative gene of a premature aging phenotype [7]. However, the consequences of Cisd2 deficiency in both somatic cells and stem cells are not yet fully understood. First, we evaluated the mitochondrial structure of Cisd2-deficient miPSCs. TEM analysis revealed normal mitochondrial structures in Cisd2+/+ miPSCs but not in Cisd2−/− miPSCs (Fig. 2A). Only a few mitochondria were detected in Cisd2−/− miPSCs, in which cristae were rarely visible compared with those in Cisd2+/+ miPSCs, suggesting an altered mitochondrial structure resulting from Cisd2 deficiency. A pie plot based on the microarray analysis results revealed six major classifications of predominantly downregulated transcripts in Cisd2−/− miPSCs, including mitochondrial localization, fission and fusion, inner membrane translocation, outer membrane translocation, mitochondrial protein import, and membrane polarization and potential (Fig. 2B). Among these classifications, Cisd2 predominantly affected the transcripts that function to maintain mitochondrial membrane polarization and potential.

Deficiency of Cisd2 in miPSCs results in the inhibition of the mitochondrial potential.
Based on the findings of TEM and microarray analyses (Fig. 2A, B) and because Cisd2 has been demonstrated to be associated with mitochondrial dysfunction and degenerative disorders in aging mammals [7], we hypothesized that senescent somatic cells (MEFs) or senescent stem cells (iPSCs) would also exhibit notable abnormalities in mitochondrial function. Serial passages have been shown to induce cellular senescence [32]; therefore, we assessed and compared mitochondrial potentials (ΔΨm) by JC-1 analysis [24] among cells with various Cisd2 genotypes (Cisd2+/+, Cisd2+/−, and Cisd2−/−) at passages 1 to 5, and we investigated whether Cisd2 deficiency impairs the mitochondrial membrane potential in somatic cells or stem cells. In general, somatic cells (MEFs) with any given genotype exhibited a higher mitochondrial membrane potential. Remarkably, both MEFs and miPSCs showed signs of Cisd2 deficiency at higher passages (ie, they consistently exhibited impaired mitochondrial potentials; Fig. 2C). NAO assay also revealed decreased mitochondrial masses in Cisd2-deficient cells (MEFs and miPSCs) compared with their Cisd2+/+ counterpart cells (Supplementary Fig. S1A). These results indicated that Cisd2 deficiency impaired regular mitochondrial functioning in both somatic and stem cells.
Several proteins have been closely associated with mitochondrial function. HS-1-associated protein X-1 (Hax-1), which is a mitochondrial protein with antiapoptotic functions, is a regulator of Ca2+ signaling. Hax-1 has been implicated in severe congenital neutropenia and neurological disorders [33]. BCL2/adenovirus E1B 19-kDa interacting protein 3 (Bnip3) encodes a mitochondrial outer membrane protein that is associated with cell death, membrane permeability, and the proton electrochemical gradient in normal tissues [34]. Superoxide dismutase 1 (Sod1) encodes a mitochondrial protein that converts harmful superoxide radicals into less toxic metabolites [35]. Tumor protein P53 (Trp53) encodes the most frequently mutated tumor suppressor in human cancers that is directly involved in mitochondrial outer membrane permeability [36]. To explore the mechanism associated with the mitochondrial dysfunction mediated by Cisd2 deficiency, we performed western blotting and quantitative real-time polymerase chain reaction (qRT-PCR) to examine the expression of mitochondria-associated factors in miPSC lines with various Cisd2 genotypes (Cisd2+/+, Cisd2+/−, and Cisd2−/−). Western blot analysis revealed downregulation of the Hax1, Bnip3, and Sod1 proteins in Cisd2−/− cells but not in Cisd2+/+ cells (Fig. 2D). qRT-PCR further revealed downregulation of several mitochondria-associated genes, including Hax1, Brinp3, Sod1, and Trp53 (Fig. 2E). Taken together, our findings indicate that Cisd2 modulates regular mitochondrial function and the gene expression and integrity of several mitochondria-associated factors.
Cisd2 sustains activation of canonical Wnt signaling
Because a recent study has reported that Cisd2-deficient mice exhibit osteopenia and lordokyphosis at ∼8 to 12 weeks of age [7], we investigated the extent to which the known transcriptional network regulates early development and the Wnt pathway. The formation of the mesoderm from embryonic stem cells depends on canonical Wnt/β-catenin signaling, and inhibition of endogenous Wnt signaling abolishes the differentiation potential toward the mesoderm and endoderm [37,38]. Early development genes exhibit Wnt-dependent expression, indicating that Wnt/β-catenin signaling plays an essential role in development. We performed microarray (Affymetrix mouse 430 2.0)-based analysis to compare the global transcript profiles of Cisd2+/+
, Cisd2+/−
, and Cisd2−/−
miPSCs. We identified 2,267 (944 + 1,323) and 2,406 (1,083 + 1,323) genes that were downregulated in Cisd2+/−
and Cisd2−/−
miPSCs, respectively, compared with Cisd2+/+
miPSCs. A comparison of the expression in these two groups revealed that 1,323 genes were significantly downregulated under Cisd2 deficiency (Fig. 3A). IPA (
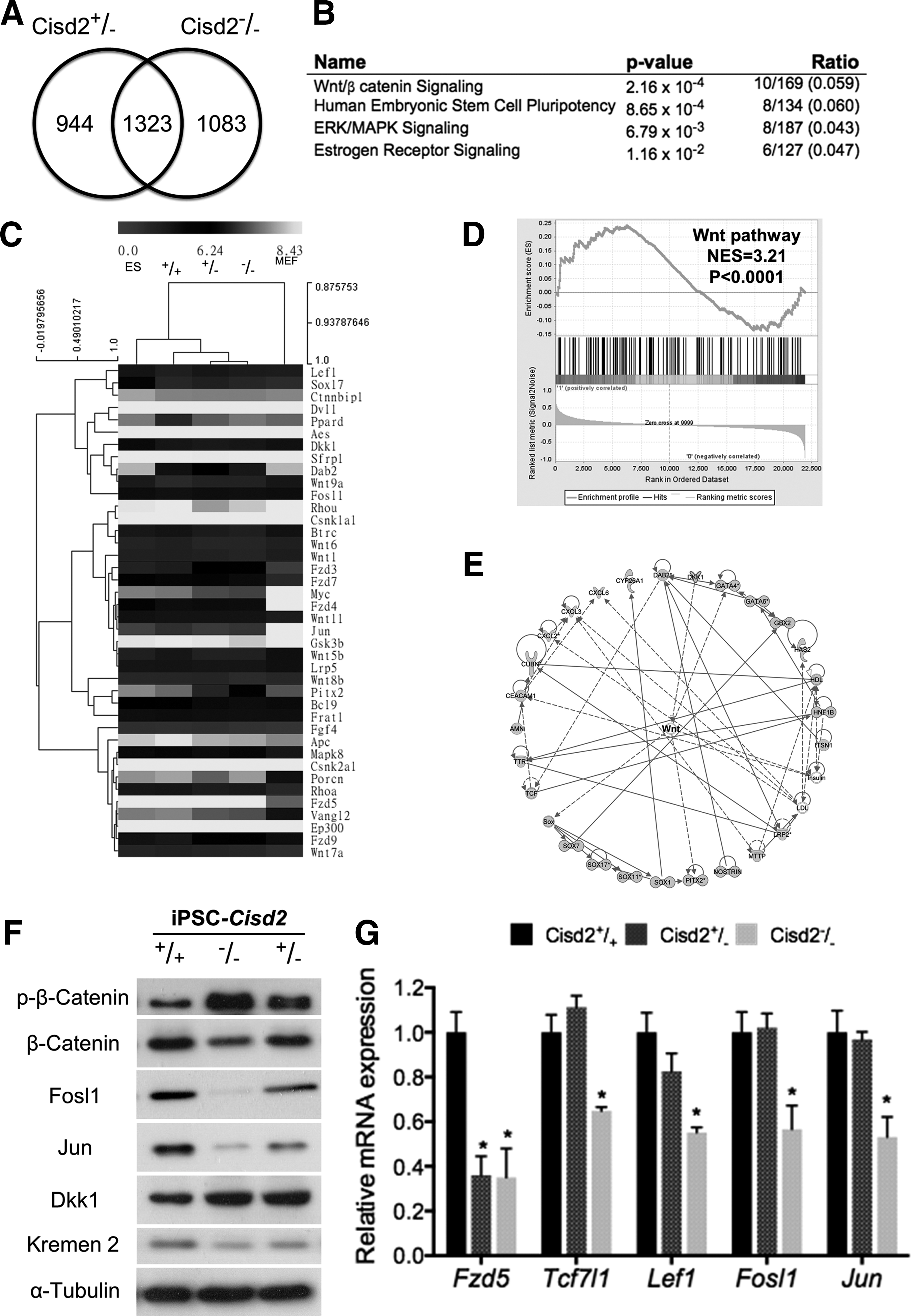
Cisd2 deficiency impairs Wnt/β-catenin signaling. Microarray analysis is used to examine perturbation of endogenous gene expression by Cisd2 deficiency.
Cisd2 deficiency impairs the proliferative ability of miPSCs
To investigate the role of Cisd2 in the Wnt pathway, we investigated whether proliferation was affected by Cisd2 deficiency. Although mitochondria are important for cell proliferation, pluripotent stem cells rarely rely on mitochondria-mediated oxidative phosphorylation [8]. We performed GSEA, and the results further confirmed that most of the genes regulated by Cisd2 were closely associated with cell proliferation (Fig. 4A). To further quantitatively investigate the genes that participate in regulating cell proliferation, we performed qRT-PCR to determine the expression of the genes that were examined in GSEA. Apc, EP300, Dab2, and c-Myc are important transcriptional regulators that promote cell proliferation [42]. In addition, PCNA and MKi67 are highly expressed in the cells with high proliferation rates [42]. The expression levels of Apc, EP300, Dab2, c-Myc, PCNA, and Ki67 were downregulated in Cisd2−/− miPSCs compared with Cisd2+/+ miPSCs (Fig. 4B). Western blot analysis also indicated that Cisd2 deficiency decreased the EP300, Dab2, and PCNA levels, whereas the Apc level was unchanged (Fig. 4C).
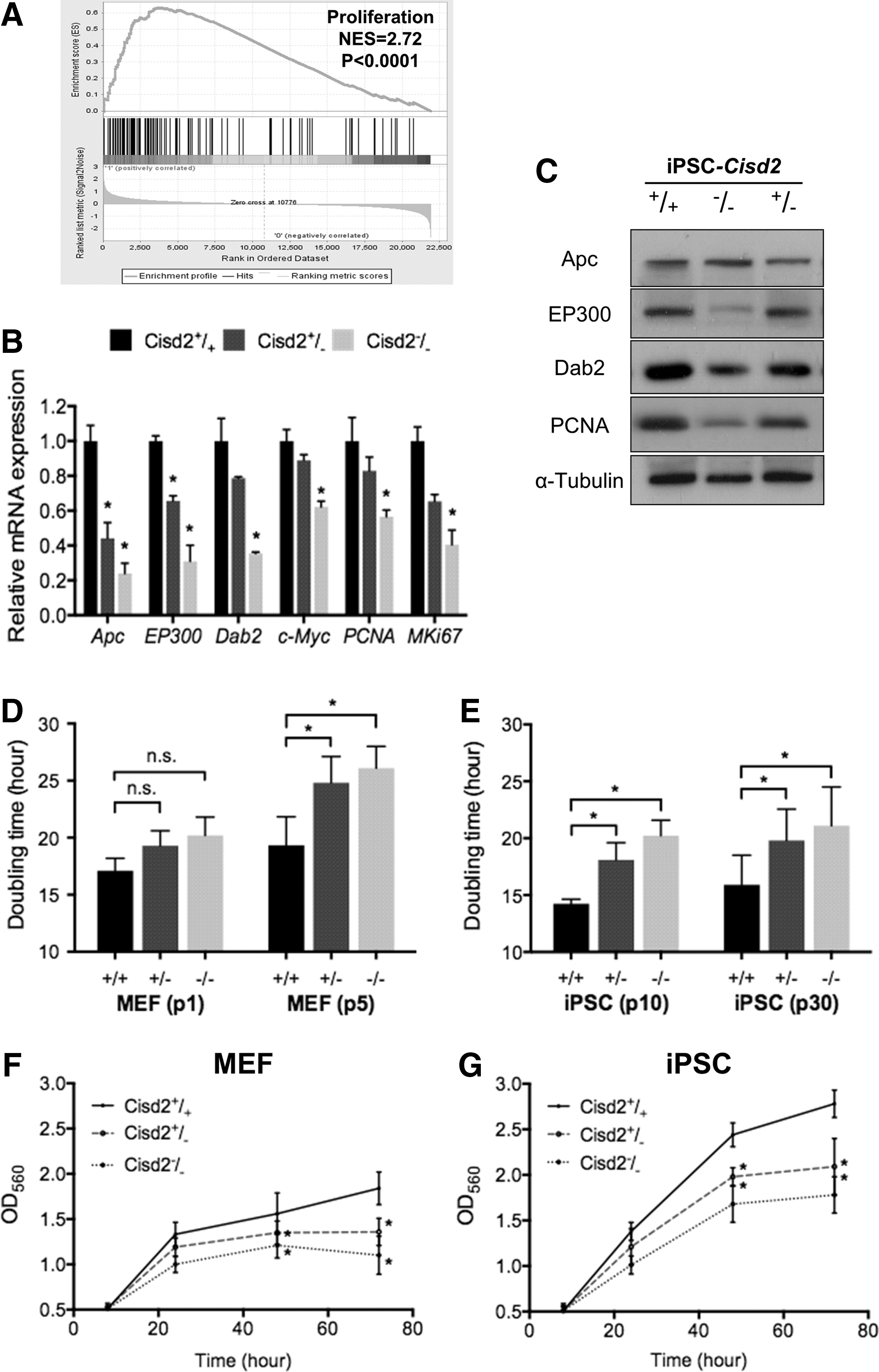
Cisd2 deficiency impairs cell proliferation.
To investigate whether Cisd2 regulates cell proliferation, we evaluated and compared the proliferation rates of MEFs and miPSCs with various Cisd2 genotypes (Cisd2+/+ , Cisd2+/− , or Cisd2−/− ) within 72 h. In MEFs at higher passages (fifth passage), the doubling time of Cisd2−/− MEFs was significantly higher than that of Cisd2+/+ MEFs (Fig. 4D). At the 10th passage of miPSCs, Cisd2−/− miPSCs also exhibited a longer doubling time than their counterpart Cisd2+/+ miPSCs, and the doubling time of miPSCs slightly increased at the 30th passage for all Cisd2 genotypes (Fig. 4E). MTT assay further revealed that the proliferative capacities of Cisd2-deficient MEFs (5th passage) and Cisd2-deficient miPSCs (30th passage) were significantly decreased compared with those of their Cisd2 +/+ counterpart cells (Fig. 4F, G, respectively). Collectively, our data demonstrate that Cisd2-deficient MEFs and miPSCs exhibit reduced proliferation rates, demonstrating a crucial role of Cisd2 in maintaining cell proliferation in both somatic and pluripotent stem cells.
Cisd2 deficiency impairs osteocyte differentiation
Because the Wnt pathway is essential for the development of embryonic stem cells, we hypothesized that Cisd2 deficiency may impair the development and differentiation of stem cells into various dermal lineages. To evaluate whether the differentiation capacity of Cisd2-deficient miPSCs was impaired, we assessed three germ layer markers after employing various differentiation protocols in miPSCs with various Cisd2 genotypes (Cisd2+/+ , Cisd2+/− , or Cisd2−/− ). Immunofluorescence staining showed that miPSCs with each Cisd2 genotype were capable of tri-dermal differentiation in vitro and expressed the typical markers corresponding to each lineage, including alpha-fetoprotein, smooth muscle actin, and Nestin, after differentiation (Fig. 5A). In general, immunofluorescence staining did not reveal obvious defects in endodermal, mesodermal, or ectodermal differentiation of miPSCs with each Cisd2 genotype. Nevertheless, qRT-PCR indicated that several markers specific for the differentiation of the three dermal lineages were substantially decreased in Cisd2−/− miPSCs (Ncam1, Olig2, Fgf5, and En2 as ectodermal markers; Bmp4, T, Tbx3, and Bmp2 as mesodermal markers; and Hnf4a, Gata6, Sox7, and Sox17 as endodermal markers; Fig. 5B). These data indicate that Cisd2 deficiency blunts the capacity of miPSCs for differentiation into multiple dermal lineages.
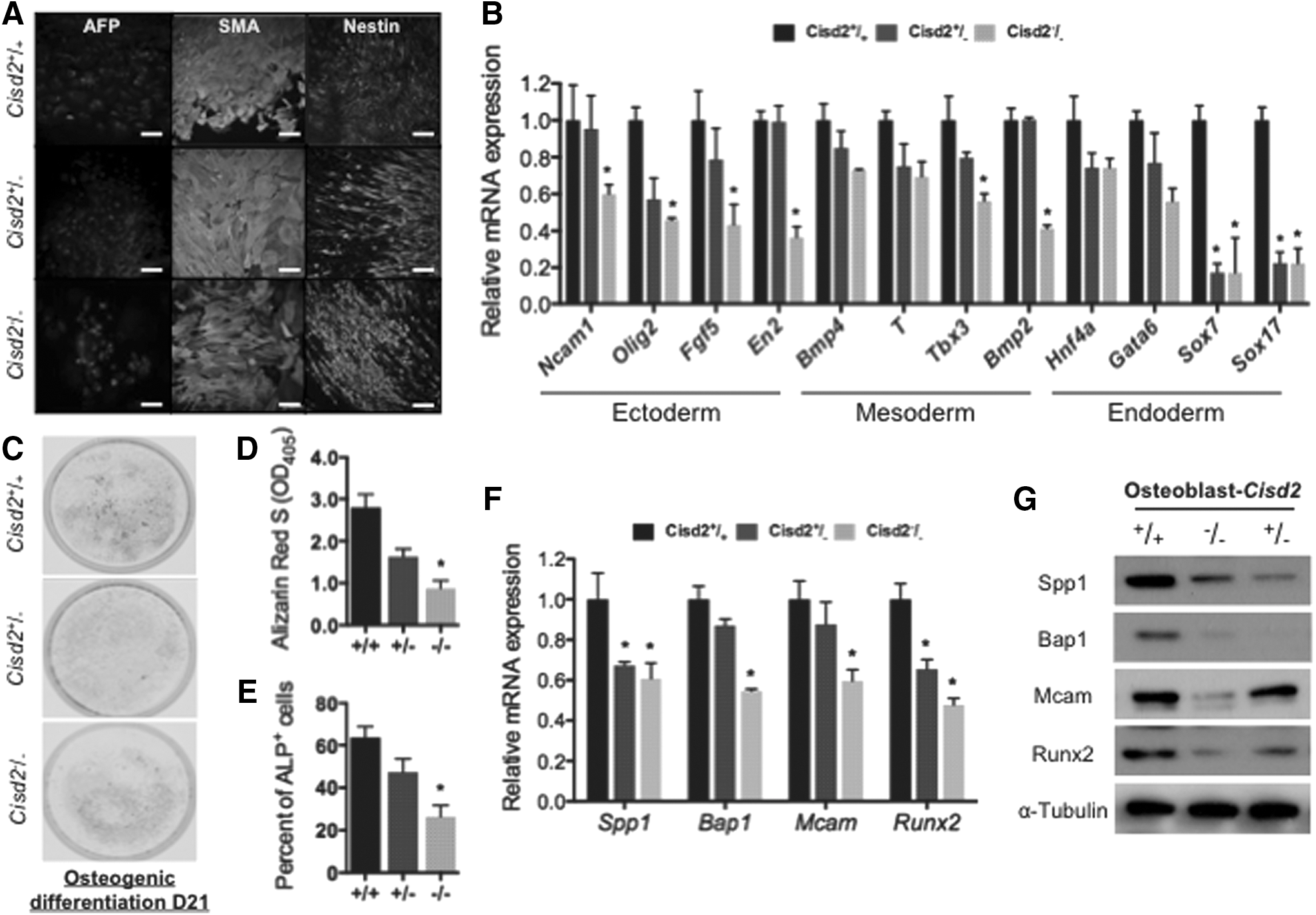
Cisd2 deficiency impairs osteogenesis in miPSCs.
Recent studies have reported that the Wnt pathway is highly associated with osteogenesis in mouse embryonic stem cells [43]; therefore, we examined whether Cisd2 also influences the osteogenic potential of miPSCs. After employing the protocol for the generation of osteoblasts from miPSCs with various Cisd2 genotypes, ARS staining was used to examine Ca2+-rich deposits in miPSCs that had differentiated into osteoblasts (Fig. 5C). A cetylpyridinium chloride solution was then used for extraction of ARS from cells and subsequent quantification of Ca2+-rich deposits in these extracts via colorimetric detection at 405 nm [44]. Among all miPSCs with the various Cisd2 genotypes, Cisd2−/− miPSCs exhibited the least amount of Ca2+ deposits (Fig. 5D). In addition, flow cytometry analysis revealed that the level of ALP, a well-accepted osteogenic marker, was also substantially decreased in Cisd2−/− miPSCs (Fig. 5E and Supplementary Fig. S1B). Further, qRT-PCR indicated that the levels of osteogenic markers, such as Spp1, Bap1, Mcam, and runt-related transcription factor 2 (Runx2), were decreased in these cells (Fig. 5F). Western blot analysis confirmed the downregulation of these markers at the protein level (Fig. 5G). Collectively, our findings indicate that Cisd2 deficiency resulted in impaired differentiation of various dermal lineages, in which prominent abnormalities were observed in osteogenic differentiation.
Cisd2 and Gimap5 co-regulate Ca2+ homeostasis in both MEFs and miPSCs
A recent study has shown that Cisd2 deficiency inhibits adipogenesis by affecting intracellular Ca2+ levels. In addition, Cisd2 and endogenous Gimap5 have been demonstrated to interact with each other [19]. Notably, cells lacking both Cisd2 and Gimap5 exhibit additive increases in intracellular Ca2+, suggesting that these proteins are essential for controlling intracellular Ca2+ levels [19]. To examine whether the interrelationship between Cisd2 and Gimap5 is also essential for regulation of Ca2+ homeostasis, several experiments were conducted to examine miPSCs with the genotype Cisd2+/+ , Cisd2+/− , or Cisd2−/− . The immunoprecipitation assay results showed that Cisd2 and Gimap5 interacted with each other in miPSCs (Fig. 6A). In addition, Cisd2−/− fibroblasts had higher intracellular Ca2+ levels in both MEFs and miPSCs compared with the corresponding cells with the other two Cisd2 genotypes, as determined using a Fluo-3-AM fluorescence probe (Fig. 6B). After continuous passaging, the intracellular Ca2+ levels remained the highest in Cisd−/− cells compared with Cisd2+/+ and Cisd2+/− cells (Fig. 6C, D), and these high Ca2+ levels under Cisd2 deficiency were observed in both MEFs (Fig. 6C) and miPSCs (Fig. 6D). These results suggest that the association of Cisd2 with Gimap5 contributes to the maintenance of intracellular Ca2+ homeostasis in both somatic and stem cells and that Cisd2 deficiency contributes to abnormal elevations in intracellular Ca2+ concentrations and impaired mitochondrial function, which may negatively affect regular Wnt pathway signaling (Fig. 6E).

Cisd2-mediated regulation of osteogenesis and Ca2+ homeostasis in miPSCs.
Discussion
Cisd2 is a member of the CDGSH iron-sulfur cluster family, which maintains mitochondrial integrity and regulates the life span and development of multiple cell lineages [30]. A mutation in Cisd2 is a major cause of Wolfram syndrome, an autosomal recessive inherited disease that is characterized by multiple degenerative anomalies [7]. Cisd2 KO mice exhibit significant clinical features, including neurodegeneration of the central and peripheral nervous systems, decreased muscle mass, osteopenia, and lordokyphosis [7]. In addition, Cisd2 deficiency inhibits adipocyte differentiation in mice [19]. These findings indicate a critical but unclear role of Cisd2 during development. Cisd2 forms a homodimer that harbors two redox-active 2Fe-2S clusters and localizes to the ER or mitochondrial membrane [7,31]. Mitochondrial functions may play critical roles in controlling the early differentiation of embryonic stem cells [16]. To understand the importance of Cisd2 in development, we established miPSCs from Cisd2−/− mice for the first time to investigate the progression of reprogramming, pluripotency maintenance, and differentiation. Cisd2 deficiency induced mitochondrial dysfunction, which was associated with reductions in membrane potential, mitochondrial mass, and expression of genes encoding electron transport chain proteins. Interestingly, we observed that Cisd2 deficiency enhanced the efficacy of reprograming but decreased the growth rate and efficiency of osteogenesis. Microarray analysis revealed that the major influence on osteogenic differentiation was the commitment of Cisd2−/− miPSCs toward an osteoclast fate. Collectively, our data suggest that Cisd2-dependent mitochondrial integrity may be fundamental for maintaining the growth rate of miPSCs and the differentiation efficacy of osteogenesis.
Cisd2 is involved in mitochondrial membrane potential maintenance
Classification of mitochondrial proteins into several groups according to their specific functions revealed that the expression levels of the proteins associated with membrane potential were severely decreased in Cisd2−/− miPSCs. This finding is remarkable because Cisd2 KO mice have been previously shown to exhibit obvious impairments in the functioning of mitochondrial complexes I and III [7], whereas our bioinformatics analysis indicated that mitochondrial membrane potential-associated proteins were primarily destroyed, particularly during the embryonic stages [7]. The mitochondrial membrane potential controls the release of cytochrome c to mediate cellular apoptosis [45]. Reducing the membrane potential has been shown to stimulate G0/G1 arrest to diminish cell growth without inducing apoptosis in colonic epithelial cells and promyelocytic leukemia cells [46,47]. These findings are consistent with our results, indicating that Cisd2 regulates the membrane potential, thereby promoting cellular growth.
Cisd2 determines mesodermal differentiation
Mitochondria-enriched organs, such as those of the nervous system, exhibit significant abnormalities in Cisd2 KO mice [7]. Most of these organs develop from the ectodermal lineage, and the mesoderm is not considered the primary impaired germ layer. Lordokyphosis and osteoporosis have been previously noted as significant phenotypes in Cisd2 KO mice after 8 weeks of age, emphasizing the importance of Cisd2 in mesodermal development [7]. The easily manipulable miPSC platform provided insights into the influence of Cisd2 during the embryonic stage. Cisd2−/− miPSCs exhibited lower expression levels of several mesodermal markers compared with wild-type miPSCs, as determined by qRT-PCR. Interestingly, some of these mesodermal markers are also endodermal markers, such as Sox7 and Sox17, which were significantly downregulated [48]. Sox17 deficiency suppresses the mesodermal transcription factors Mesp1 and Mesp2, inhibiting cardiomyocyte genesis. Sox7 functions in Flk1 mesodermal precursors, and Sox7 shRNA prevents formation of hematopoietic progenitors [48 –51]. Two other notable mesodermal markers are Bmp2 and Bmp4. The Bmp family regulates critical determining factors involved in formation of the mesoderm, heart, cartilage, and bone. Bmp2 stimulates the expression of bone-associated markers, such as osteocalcin, by activating downstream Smad 1/5 cross-linked to Runx2. Bmp4 is a crucial factor for transmitting signals from the extracellular matrix (ECM) to intracellular signaling pathways [52 –54]. Heteromeric complexes formed by β3-integrin and Bmp4 receptor BMPRIA upon Bmp4 stimulation promote the activity of Erk2 and Smad1/5 to arrest the cell cycle at G0/G1 via upregulation of p21 and p27, which initiates differentiation, particularly osteogenesis [55,56]. The significant inhibition of expression of the early to late mesodermal markers Sox7/Sox17 and Bmp2/Bmp4 in Cisd2−/− miPSCs indicates that Cisd2 has an essential role in cell fate determination.
Cisd2 is involved in osteogenic differentiation
Consistent with the analytical results, ARS staining revealed osteogenic variations between Cisd2+/+ and Cisd2−/− miPSCs. RNA expression analysis indicated that the expression levels of most of the precursor and mature markers of osteoblasts, such as Spp1, Bap1, Mcam, and Runx2, were reduced, further emphasizing the importance of Cisd2 in osteogenesis. Runx2 is a key transcription factor that serves as a scaffold for nucleic acids and regulates several essential skeletal-related genes [57]. It acts bidirectionally to target the promoter of Bmp2/Bmp4 and recruit R-Smad to trigger Bmp signaling pathways [43,58 –61]. Interestingly, nearly all of the genes with the most drastically altered expression function downstream of the Wnt pathway, indicating that Cisd2 is involved in this signaling pathway through an unknown mechanism during the osteogenic process.
Wnt and Bmp4 are two well-known signals involved in osteophysiology [43,58,59]. Canonical Wnt signaling is initiated when Wnt3a binds to Lrp5/6 and Fzd, consequently dephosphorylating β-catenin and enabling its interaction with Tcf/Lef to promote the activation of downstream targets, including Runx2, Fosl, and Jun [58,62]. Wnt is involved in many pivotal cellular physiological processes, including proliferation, differentiation, and the ECM mineralization of osteoblasts [62,63]. It also blocks the conversion of mesenchymal cells into adipocytes [64] and the differentiation of chondrocytes [65]. Bmp2 is induced downstream to promote Runx2 expression, which is upregulated during osteogenesis. The Wnt pathway reportedly interacts with Bmp signaling and vice versa. For example, Bmp2 inhibits Wnt7a and β-catenin via p38 to enhance chondrogenesis, whereas their co-regulation reduces osteoblast differentiation [66]. However, studies of the involvement of Cisd2 in the crosstalk between these two signaling pathways to direct osteogenesis are limited. Overexpression of mitochondrial transcription factor A (Tfam) activates the canonical Wnt pathway through Lrp5 to promote β-catenin/Tcf transcriptional activity, further contributing to the induction of osteogenesis-related gene expression [67]. Bmp2 also functions as an apoptotic regulator of osteoblasts by inducing the mitochondrial release of cytochrome c and Bax/Bcl-2 [68,69]. Collectively, these recent studies have demonstrated the importance of the mitochondrion in the Wnt/Bmp signaling pathway, but the detailed mechanism remains to be further elucidated. Bone remodeling is a balanced state between bone formation and bone resorption involving interactions between osteoblasts and osteoclasts [70]. Clinical findings have indicated that imbalances between bone formation and resorption lead to mineral over-accumulation and the subsequent stimulation of abnormal growth, resulting in lordokyphosis and osteoporosis [70,71]. As previously mentioned, the Wnt/β-catenin and Bmp pathways are the two most critical signaling pathways that mediate bone formation [72]. Bmps act synergistically with Wnt to inactivate GSK3β, thereby stabilizing β-catenin, which upregulates Runx2 expression [72]. The Wnt and Bmp pathways collaborate in several biogenic processes, particularly during the early stages of differentiation [73]. In addition to the molecular mechanism driving osteodifferentiation, bone remodeling is regarded as a reversible process. Osteoblasts are the major regulators of osteoclastogenesis through the binding of the surface marker M-CSF to c-FMS, activating NF-κB/c-Fos to induce the expression of osteoclast-associated proteins or the secretion of osteoprotegerin, which interferes with the RANKL pathway in osteoclast progenitors [74,75]. Osteoblasts and osteoclasts vary slightly in structure. Osteoblasts have an abundance of ERs, whereas osteoclasts are enriched in mitochondria [76]. Because Cisd2 is important in the mitochondria, we speculate that osteoclastogenesis is likely disrupted, consistent with our findings, but the underlying mechanism is unclear. Cisd2 was defined as the pivotal gene in WFS2 due to its role in the ER, acting together with Bcl-2, to modulate Ca2+ homeostasis [4,5]. This role has also been described by Wang et al., who demonstrated that Cisd2 is localized to mitochondria-associated ER membranes [19], indicating that this protein maintains not only the function of mitochondria but also that of the ER [77]. Therefore, osteoporosis induced by the loss of Cisd2 function may be caused by abnormal Ca2+ deposition in the ER.
Dkk1 specifically inhibits canonical Wnt signaling by binding to the Wnt-receptor complex [41]. In addition, transmembrane protein Kremen2 are high-affinity Dkk1 receptors that functionally cooperate with Dkk1 to block Wnt/beta-catenin signaling. Kremen2 forms a ternary complex with Dkk1 and Lrp6, and induces rapid endocytosis and removal of the Wnt-receptor complex from the plasma membrane. Our results indicated that Cisd2−/− iPSCs contained higher amount of phosphorylated β-catenin than the other two Cisd2 counterpart lines. Remarkably, Cisd2 deficiency substantially upregulated Dkk1 but did not affect Kremen2 protein amount (Fig. 3F). These data indicated that regulation of Wnt pathway by Cisd2, at least, is partially exerted through modulating Dkk1 to antagonizing the signaling of Wnt pathway.
Taken together, these findings suggest that the expression of Cisd2 and the Cisd2-related pathway may affect osteogenic differentiation and that osteogenesis-related signaling candidates may respond to the activities of Cisd2. Future studies should determine whether the two above-mentioned signaling pathways participate in crosstalk through Cisd2 to regulate dynamic bone remodeling and should further clarify the role of Cisd2 in relation to the mitochondrion and ER in osteophysiology (Fig. 6E).
Our study reports the promising generation of miPSCs from Cisd2 KO mice that recapitulate developmental and differentiation processes. We have demonstrated that the loss of Cisd2 decreases the efficiency of osteogenic differentiation and affects certain aspects of mitochondrial function via undetermined mechanisms in Cisd2-deficient miPSCs. Notably, the role of Cisd2 in the differentiation and development of specific lineages, particularly those related to the aging process and senescence, should be further explored. Moreover, through the application of cellular reprogramming and ex vivo transplantation, we hope to better understand the Cisd2-associated biomolecular mechanisms and Cisd2-dependent premature aging process, and this information will potentially be of benefit to researchers of related clinical syndromes, such as WFS2.
Footnotes
Acknowledgments
This study was funded by the Ministry of Health and Welfare [Excellent Clinical Trial Center & Cancer Center Research of Excellence (MOHW104-TDU-B-211-124-001/TD-B-111-02 & MOHW104-TDU-B-211-113-003)], National Health Research Institutes (NHRI-EX102-10258SI), MOST (103-2321-B-010-025, 104-2627-M-010-004,104-2325-B-010-006), MOST Ingenious Plan (NYMU-NHRI-2015), Yen-Tjing-Ling Medical Foundation (CI-102/103/104), TVGH-NTU Joint Project (VN104-09), Taipei Veterans General Hospital (103- V-B-027), National Health Research Institutes (NHRI-EX104-10258SI), and the Genomic Center Project and Cancer Center Project of National Yang-Ming University (Ministry of Education, Aim for the Top University Plan), Taiwan.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.