
Abstract
Stem cells have attracted great interest for their therapeutic capacity in tissue regeneration. Cyclic adenosine 3′,5′-monophosphate (cAMP), existing in high concentration at wound sites, mediated various signaling pathways such as cytoskeleton dynamics, cell adhesion, and cell migration in stem cells, which suggest the critical roles of cAMP in the wound healing process through functional regulation of stem cells. However, the mechanisms behind the effect of cAMP on mouse embryonic stem cell (mESC) motility and its roles on skin wound healing remain to be fully elucidated. In the present study, 8-Bromo cAMP-treated mESCs showed significant wound closure and improved neovascularization. Moreover, 8-Bromo cAMP stimulated mESC migration into the wound bed. 8-Bromo cAMP also increased ESC motility in in vitro migration assay. 8-Bromo cAMP induced myosin light chain phosphorylation through Rac1 and Cdc42 signaling, which were involved in 8-Bromo cAMP-induced decrease in expression of junction proteins (connexin 43, E-cadherin, and occludin) at the plasma membrane. Subsequently, 8-Bromo cAMP induced the disruption of cell junctions (including gap junctions, adherens junctions, and tight junctions), which reduced the function of the gap junctions and cell adhesion. In addition, 8-Bromo cAMP-induced Rac1 and Cdc42 activation increased Arp3, TOCA, PAK, and N-WASP expression, but decreased cofilin phosphorylation level, which elicited actin cytoskeleton remodeling. In contrast to the control, 8-Bromo cAMP evoked a substantial migration of cells into the denuded area, which was blocked by the small interfering RNAs of the signaling pathway-related molecules or by inhibitors. In conclusion, cAMP enhanced the migration of mESCs through effective coordination of junctional disruption and actin cytoskeleton remodeling, which increased the wound healing capacity of ESCs.
Introduction
S
Cyclic adenosine 3′,5′-monophosphate (cAMP) is an example of a cellular regulator that mediates diverse effects in cytoskeletal dynamics, cell adhesion, and cell migration [7,8]. Indeed, various studies have proposed that cAMP is one of the important small molecules that acts as a novel biofactor in tissue repair and regeneration [9]. Therefore, elucidating the role of cAMP in the functional regulation of stem cells is both a challenge and an opportunity in stem cell biology, as well as stem cell therapy. Paulucci-Holthauzen et al. showed the existence of protein kinase A (PKA) activity gradients in single migrating cells and that these gradients are properly localized to influence key regulators of actin cytoskeletal reorganization within the lamellae [10]. In contrast, elevation of intracellular cAMP promotes collagen-dependent dissociation of cell junctions, but inhibits the migration of carcinoma cells [11,12]. Based on those studies, the effects of cAMP on cell migration can be both stimulatory and inhibitory, depending on the cell type and ligand used [13,14]. The primary downstream signaling effectors of cAMP are protein kinase A, exchange protein activated by cAMP (Epac), and cyclic nucleotide-gated channels [15]. PKA and Epac are involved in cell migration through small GTPases and which modulate cell–cell junctions mainly by controlling the assembly and contractility of the actin cytoskeleton, an essential structural scaffold of junctions [16,17]. Although it is clear that small GTPases dynamically regulate cell migration, the mechanisms through which PKA and Epac activates small GTPases during these processes remain quite unclear.
Embryonic stem cells (ESCs), which possess the features for infinite self-renewal and the capacity to differentiate into the cellular derivatives of three lineages, attracted great interest in cell-based regenerative medicine for their differentiation ability and paracrine effect, although the exact mechanisms remain poorly understood [18 –20]. The mechanisms underlying the regulation of ESC therapeutic capacity have gained significant attention in the utilization of exogenously administered ESCs in tissue repair. Despite limitations in ESCs in skin wound healing, previous reports that application of mESCs simulates reconstitution into fully differentiated skin and accelerates wound healing in diabetic rats show the potential of ESCs in regenerative medicine [21,22]. In this respect, chemical approaches have been applied in vitro and in vivo to manipulate cell fate toward the desired therapeutic applications, which include cell activation and expansion. Thus, dissecting the regulatory pathways and deciphering the underlying molecular mechanisms of stem cell fate through cAMP are necessary to advance stem cell therapy in the management of wound healing. This is a knowledge-based field in which we have come a long way in the use of mESCs in transplantation for regenerative medicine. Therefore, in the present study, we investigated how cAMP affects mESC migration, focusing in particular on the cell junctions and the cytoskeleton, as well as skin wound healing.
Methods
Materials
Mouse ESCs were obtained (product code: ES E14TG2a) from the American Type Culture Collection (ATCC). Fetal bovine serum (FBS) was purchased from Bio Whittaker. cAMP agonist (8-Bromoadenosine-3′,5′-cyclic monophosphate [8-Bromo cAMP]), ML-7, chloroquine, lactacystin, and fluorescence isothiocyanate-conjugated (FITC-conjugated) goat-anti mouse IgM were acquired from Sigma Chemical Company. PKA inhibitor 14–22 amide (PKI) was acquired from MERCK. Anti-ZO-1, E-cadherin, occludin, Cx43, Rac1, Cdc42, RhoA, β-actin, MLC, p-MLC, MLCK, PAK, Arp3, TOCA, N-WASP, cofilin, and p-cofilin antibodies were obtained from Santa Cruz Biotechnology. Goat anti-rabbit IgG was acquired from Jackson Immunoresearch. Liquiscint was purchased from National Diagnostics. All reagents were of the highest commercial purity available.
Mouse skin wound healing model
Seven-week-old male ICR mice were purchased from Han Lim Experimental Animal (Suwon, Korea). Experiments with animals were carried out under approval by the Institutional Animal Care and Use Committee of Seoul National University (SNU-150508-4) and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Methods for the mouse skin excision wound splinting model and stem cell transplantation were performed according to a previous study [23]. Mice were randomly divided into groups (n = 5, each group). Mice were anesthetized by intraperitoneal injection using a combination of Zoletil50®, Rompun®, and saline (a ratio of 2:1:2, respectively) and placed on a heating table kept at 37°C to maintain constant body temperature. Their back was shaved, sterilized with povidone–iodine, followed by a 70% ethanol, and the wound was created by using a 6-mm diameter sterile biopsy punch surgically. In the cell-treated group, 0.7 × 106 mESCs in 60 μL Matrigel (BD Biosciences) were injected into the dermis at four sites around the wound. We also topically apply 0.3 × 106 mESCs in 20 μL Matrigel onto the wound site. In mESCs with the 8-Bromo cAMP group, cells were preactivated with 8-Bromo cAMP as well as bromo-2′-deoxyuridine (BrdU, 2 μM) for 24 h, and then mESCs were applied with 8-Bromo cAMP. The wound was placed with the splint around the wound (with several stitches and/or glue) and dressed with Tegaderm (3M) sterile transparent dressing. Mice were placed in individual cages under a warm and humid incubator. Images of the wound were acquired on days 0, 1, 5, 7, 8, and 9 with a digital camera system (40D, Canon) at the same distance. At day 9, the wound tissues were embedded in O.C.T. compound (Sakura Finetek), stored at −70°C, cut into 6-μm frozen sections using a cryosection machine, and mounted on SuperFrost Plus slides (Thermo Fisher Scientific) for hematoxylin and eosin (H&E) staining.
ESC culture
The mouse ESCs were cultured in DMEM (Gibco-BRL) supplemented with 3.7 g/L sodium bicarbonate, 1% penicillin and streptomycin, 1.7 mM
SL/DT assay
The gap junctional intercellular communication level was measured using a slight modification of the scrape loading/dye transfer (SL/DT) method [24,25]. The mouse ESCs on 60-mm dishes were rinsed twice with 2 mL of PBS, and 0.05% Lucifer Yellow was then applied to the center of the dish. The scrapes were performed to ensure that the scrape traversed a large group of confluent cells. The cells were incubated in the dye mix for precisely 10 min, rinsed quickly thrice with PBS, and finally examined by confocal microscopy (Fluoview 300; Olympus). Lucifer Yellow is a small molecule that can move freely through the gap junctions from the loaded cells to the neighboring ones [26].
Cell adhesion assay
A 96-well plate was coated with Laminin-1 at 37°C for 1 h and washed twice with PBS. The plate was blocked with 0.5% BSA in DMEM at 37°C for 1 h and then washed with PBS. Cells were seeded at 2 × 104 cells in each well, and then incubated in a CO2 incubator at 37°C for 120 min. The plate was shaken at 1,500 rpm for 15 s and washed with PBS. Cells were fixed with 3.5% paraformaldehyde for 10 min and washed twice with PBS. The cells were stained with crystal violet for 10 min and then washed twice with PBS. The plates were completely dried and 2% SDS was added. The plates were incubated in the RT for 30 min and quantified using a microplate reader at 550 nm.
Cell migration assay
Oris™ assay
Cells were seeded at 3 × 102 cells/100 μL in each well (Platypus Technologies; cat# CMA 1. 101). Inserts were carefully removed when cells had reached 70% confluence. Then, migrated cells were stained with 5 μM calcein-AM and quantified through measurement of fluorescence signals using a microplate reader at excitation and emission wavelengths of 485 nm and 515 nm, respectively.
Wound healing assay
Cells were seeded at 4 × 104 cells/200 μL in culture dishes with inserts (Ibidi). After 70% confluence, inserts were removed to create a wound field of approximately 500 μm. After acquiring 0 h images using a phase-contrast microscope, the cells were treated with 8-Bromo cAMP for 24 h and the image was acquired. The migrated areas were analyzed with MetaMorph v. 7.01 software (Molecular Devices) and represented as % of control.
Live cell imaging microscopy
Cells were placed in temperature/CO2 control chambers (Tokai) with or without 8-Bromo cAMP installed with an Olympus IX81-ZDC zero-drift microscope. A series of images were acquired for 0–24 h at 5-min intervals, using a Cascade 512B camera (Roper Scientific) operated by the multidimensional acquisition package of MetaMorph v. 7.01 software.
Immunofluorescence staining
Cells were fixed with 3.5% paraformaldehyde in PBS, permeabilized for 5 min with 0.1% Triton X-100, and washed thrice for 10 min each with PBS. Cells were preincubated with 10% BSA in PBS for 30 min to decrease nonspecific antibody binding. Cells were incubated with primary antibody against target protein (1:50; Santa Cruz Biotechnology, Inc.) for 1 h, and then incubated for 1 h with a 1:100 dilution of Alexa Fluor 488 or 555-conjugated secondary antibody. Fluorescent images were visualized using a FluoView 300 fluorescence microscope (Olympus).
Preparation of cytosolic and total membrane fractions
The cytosolic and total membrane fractions were prepared by slight modification of the method reported by Mackman et al. [27]. The medium was removed and replaced with serum-free DMEM, including all supplements and LIF, for 24 h before the experiments. After removing the medium, the cells were washed twice with ice-cold PBS, scraped, harvested by microcentrifugation, and resuspended in buffer A (137 mM NaCl, 8.1 mM Na2HPO4, 2.7 mM KCl, 1.5 mM KH2PO4, 2.5 mM EDTA, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, 0.5 mM sodium orthovanadate, pH 7.5). The resuspended cells were then mechanically lysed on ice by trituration with a 19-gauge needle. The cell lysates were initially centrifuged at 1,000 g for 10 min at 4°C. The supernatants were collected and centrifuged at 100,000 g for 1 h at 4°C to prepare the cytosolic and total particulate fractions. The supernatant (cytosolic fraction) was precipitated with 5 volumes of acetone, incubated on ice for 5 min, and centrifuged at 20,000 g for 20 min at 4°C. The resulting pellet was resuspended in buffer A containing 1% (v/v) Triton X-100. The particulate fractions, which contain the membrane fraction, were washed twice and resuspended in buffer A containing 1% (v/v) Triton X-100.
Western blot analysis
The cells were harvested, washed twice with PBS, and lysed with buffer [20 mM Tris (pH 7.5), 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mg/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride, and 0.5 mM sodium orthovanadate] for 30 min on ice. The lysates were then cleared by centrifugation (10 min at 15,000 rpm, 4°C), and protein concentration was determined by the Bradford method [28]. Equal amounts of protein (40 μg) were resolved by electrophoresis on 6–10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membranes. After washing with TBST [10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.05% Tween 20], the blots were blocked with 5% skim milk for 1 h, and then incubated with the appropriate primary antibody at the dilution recommended by the supplier. The membranes were then washed, and the primary antibodies were detected with goat anti-rabbit IgG or goat anti-mouse IgG conjugated to horseradish peroxidase. The bands were visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech).
Affinity precipitation of cellular GTP-Rac1 and -Cdc42
Rac1 and Cdc42 activation was determined using an affinity precipitation assay incorporating the glutathione S-transferase (GST)-tagged fusion protein corresponding to the p21-binding domain (PBD, residues 67–150; Millipore) of human PAK-1 for Rac1 and Cdc42, which bind only to the active GTP-bound form [29]. Cells that had reached 70% confluence were incubated in the presence of 8-bromo cAMP for 30 min at 37°C before the addition of lysis buffer (25 mM HEPES, [pH 7.5], 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 1 μg/mL aprotinin, 10 μg/mL leupeptin, and 1 mM Na3VO4) at 4°C. Whole cell lysates were then incubated with agarose-conjugated GST-PBD, 20 μg, for 45 min at 4°C, and were then washed thrice with lysis buffer. Agarose beads were boiled in sample buffer to release active Rac1and Cdc42. Sample proteins were resolved by 15% SDS-PAGE, followed by immunoblotting.
Immunoprecipitation
Cells were lysed with lysis buffer (1% Triton X-100 in 50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 5 mM EDTA, 2 mM Na3VO4, 2.5 mM Na4PO7, 100 mM NaF, 200 nM microcystin lysine–arginine, and protease inhibitors). Cell lysates (300 μg) were mixed with 10 μg of mouse anti-ZO-1 antibody. The samples were incubated for 4 h, mixed with protein A/G PLUS-agarose immunoprecipitation reagent (Pierce), and then incubated for an additional 12 h. The beads were washed four times, and the bound proteins were released from the beads by boiling in SDS-PAGE sample buffer for 5 min. Samples were analyzed by western blotting with target antibodies.
Small interfering RNA transfection
Cells grown until 50% confluence in each dish were transfected for 24 h with either a SMARTpool of small interfering RNA (siRNA) specific for target protein (100 nM) or a nontargeting siRNA as a negative control (100 nM; Dharmacon) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 24 h, the transfection mixtures were replaced with regular medium and cells were maintained in normal culture conditions (DMEM supplemented with LIF and FBS) as they were before the experiments. The efficacies of siRNAs used in the present study were confirmed in our experimental condition (Supplementary Fig. S1; Supplementary Data are available online at
Statistical analyses
The results are expressed as the mean ± the standard error (SE). All experiments were analyzed by ANOVA, and some experiments were compared with the control using a Bonferroni–Dunn test. A P value < 0.05 was considered significant.
Results
Effects of 8-Bromo cAMP-treated mESCs on the wound healing model
To investigate the effect of 8-Bromo cAMP-treated mESCs on wound closure, we created a full-thickness skin wound on the skin of ICR mice. Four groups of mice were prepared: vehicle, mESCs, 8-Bromo cAMP, and 8-Bromo cAMP-treated mESCs. We applied the mESCs alone, 8-Bromo cAMP alone, or 8-Bromo cAMP-treated mESCs onto the skin wound and measured the wound area on days 0, 1, 5, 7, 8, and 9 until complete epithelialization in the 8-Bromo cAMP-treated mESCs. Macroscopically, the recovery rate apparently was faster in the 8-Bromo cAMP-treated mESCs than other groups at 9 days after wounding (Fig. 1A, B). Figure 1C, with the histological analysis using H&E staining, shows the representative micrographs of the wounded skin at day 9 postwounding. The wound sites were covered by epithelium, except in the vehicle group, and granulation tissue formations were observed in all groups, but not completely filled. The 8-Bromo cAMP-treated mESC group showed rapid reformation of granulation and reepithelialization of the wounds compared with the 8-Bromo cAMP and mESC groups. In the wounds of the vehicle, 8-Bromo cAMP, and mESC groups, the blood vessel was thin or had a few branches in the skin surrounding the wounds. In contrast, the wounds of the 8-Bromo cAMP-treated mESC group showed thick vessels with many fine branches extended into the wound center, forming complex networks (Fig. 1D), suggesting that angiogenesis was more effective in the 8-Bromo cAMP-treated mESC group. Because angiogenesis is critical to successful wound repair, the therapeutic efficacy of 8-Bromo cAMP-treated mESCs for faster wound healing was evaluated in a mouse model of full-thickness dermal wound. Based on the hypothesis that cAMP is able to accelerate tissue regeneration through stem cells, we investigated the secreted cytokines/wound repair-related molecules by screening the 8-Bromo cAMP-treated mESCs and skin wound. VEGF and IL-6, the angiogenesis-related cytokines having important roles in wound healing, were increased by transplantation of 8-Bromo cAMP-treated mESCs. Furthermore, components of ECM were changed by mESC transplantation with/without 8-Bromo cAMP. The ECM increased at the early phase (first phase) of the wound healing process, such as fibronectin, type I and V collagen (Col I and V) were decreased by 8-Bromo cAMP-treated mESCs, but Col III (which increased at the proliferative phase of the wound healing process) was increased. Interestingly, 8-Bromo cAMP treatment elicited exactly opposite effects of the 8-Bromo cAMP-treated mESC group (Fig. 1E).
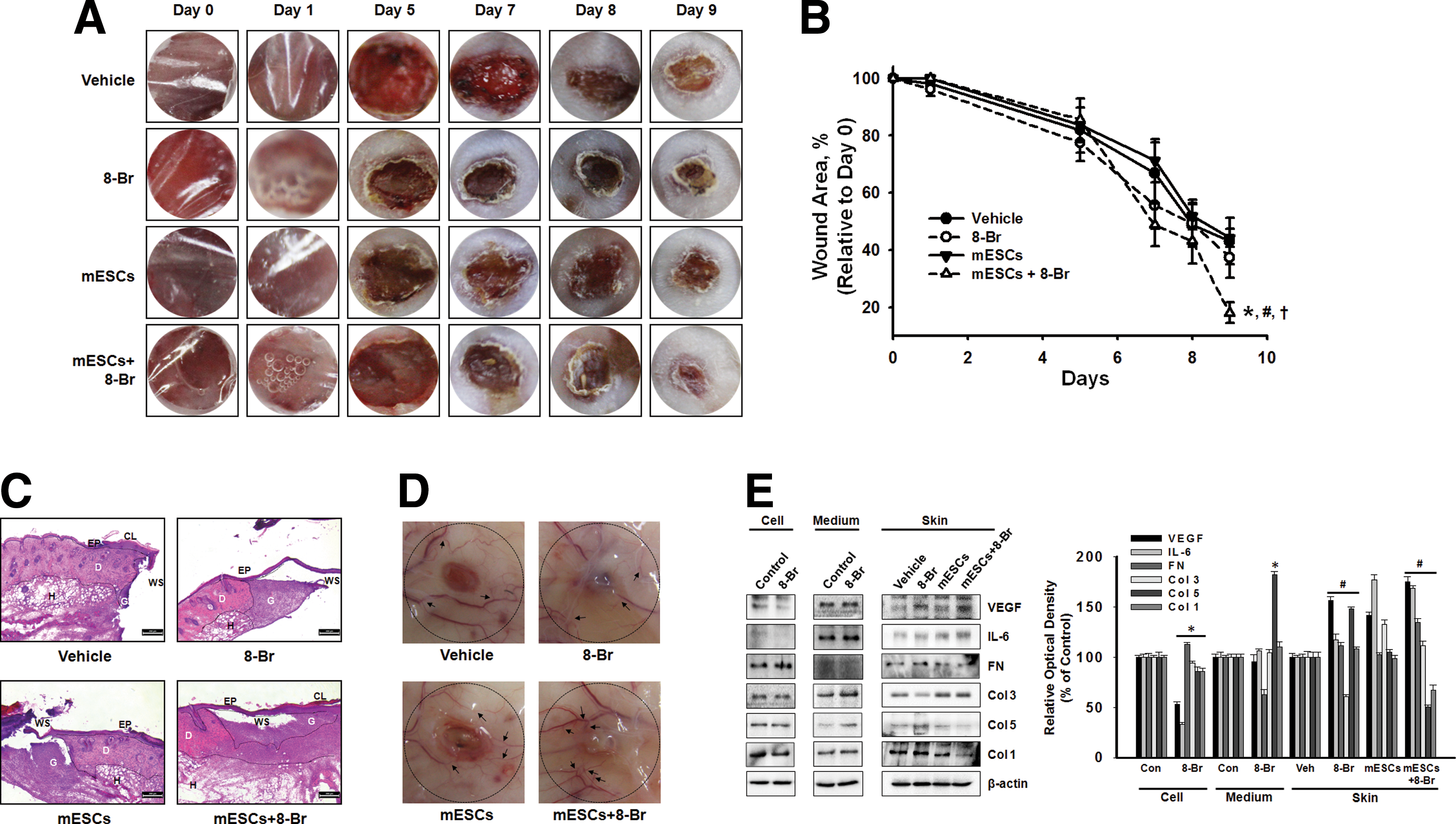
Effect of 8-Bromo cyclic adenosine 3′,5′-monophosphate (cAMP) and mouse embryonic stem cells (mESCs) on skin wound healing. Experimental animals were divided into four groups: vehicle group (n = 5), which received 60 μL of 1:1 mixture of DMEM and growth factor-reduced Matrigel (BD Biosciences), 8-Bromo cAMP group (n = 5), mESCs (n = 5), and 8-Bromo cAMP (10−5 M)-treated mESCs (n = 5). Vehicle, 8-Bromo cAMP, mESCs, or mESCs with 8-Bromo cAMP (mESCs + 8-Br) were applied onto the skin wound.
Role of 8-Bromo cAMP and cell junctions in ESC migration
In vivo bromodeoxyuridine (BrdU) labeling of day 9 full-thickness wounds showed migrating cells in the wounds (Fig. 2A). To identify the mechanisms responsible for the influence of 8-Bromo cAMP on mESC recruitment, we assessed the migratory activity of mESCs in vitro using wound healing migration assay and the Oris cell migration assay. In contrast to the control, 8-Bromo cAMP evoked a substantial migration of cells into the denuded area (Fig. 2B). These results were confirmed by observations of increased migration of 8-Bromo cAMP-stimulated cells directly in the Oris cell migration assay (Fig. 2C). In addition, 8-Bromo cAMP induced the translocation of junctional proteins from the plasma membrane to the cytosol (Fig. 2D). We confirmed this result with immunofluorescence staining. We did immunofluorescence staining with Cx43, E-cadherin, occludin, and ZO-1 (Fig. 2E). To confirm the effect of 8-Bromo cAMP-induced disruption of cellular junctions on its function, we performed SL/DT and cell adhesion assay. 8-Bromo cAMP reduced the transfer distance of Lucifer Yellow dye, which moved through gap junction intracellular communication (GJIC) (Fig. 2F) and minimized the role of cell adhesion in the cell adhesion assays, which tested the strength of the adherent junction (Fig. 2G).
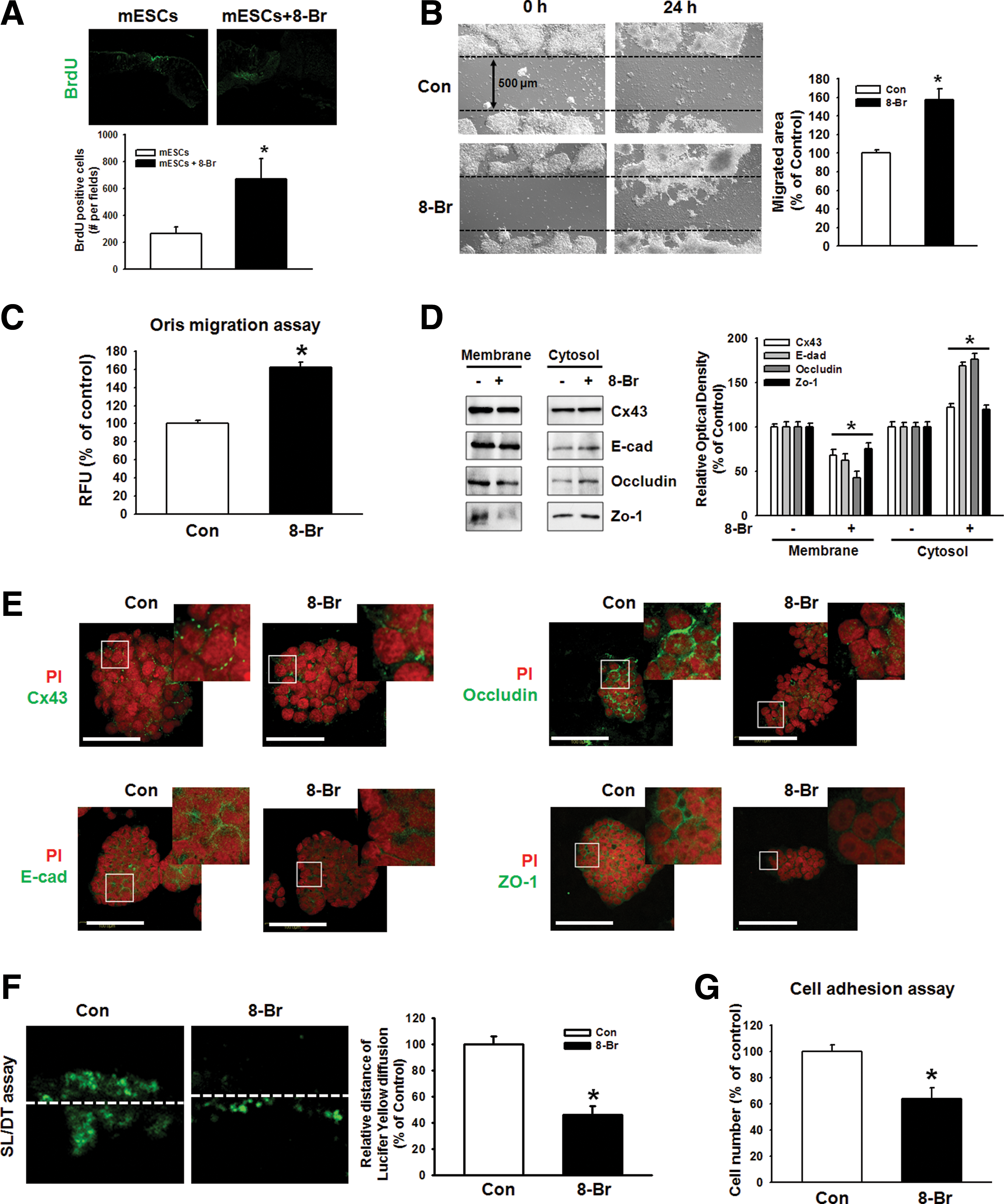
Effect of 8-Bromo cAMP in ESC migration and involvement of cell junctions.
8-Bromo cAMP induces junctional disruption through Rho GTPase
Next, we proceeded to clarify the molecular mechanisms responsible for the regulation of the junctional disruption by 8-Bromo cAMP in mESCs. Elevation of cAMP activates both PKA-dependent and Epac-dependent signaling pathways, which in turn stimulate the small GTPases of the Rho family. 8-Bromo cAMP increased the activation of Rac1/Cdc42, which was blocked by a PKA inhibitor (PKI) and Epac siRNA (Fig. 3A). Rho family of GTPases modulates cell–cell junctions mainly by controlling the assembly and contractility of the actin cytoskeleton, an essential structural scaffold of the junctions. To examine the role of myosin in the junctional disruption, we exposed cells to 8-Bromo cAMP. Pretreatment with Rac1 and Cdc42 siRNAs blocked the 8-Bromo cAMP-induced myosin light chain (MLC) phosphorylation and increase in MLCK expression (Fig. 3B). Next, we noticed that 8-Bromo cAMP induced the interaction of ZO-1 with junctional proteins in the membrane fractions in the immunoprecipitation assays. The 8-Bromo cAMP and calyculin A (Cal A, 10−6 M, phosphatase inhibitor) decreased Cx43, claudin, occludin, and E-cadherin binding with ZO-1, which were inhibited by MLCK inhibitor (ML-7, 10−5 M), suggesting that the MLCK-mediated MLC phosphorylation is closely associated with 8-Bromo cAMP-induced cellular junction disruption (Fig. 3C). Because the phosphorylated MLC could be regulated by degradation, we investigate whether the 8-Bromo cAMP-induced disruption of cell junction depends on the degradation of MLC. As shown in Fig. 3D, a lysosomal degradation inhibitor (chloroquine, 10−6 M) and proteasome inhibitor (lactacystin, 10−6 M) inhibited 8-Bromo cAMP-mediated translocation of Cx43, claudin, occludin, E-cadherin, and ZO-1. In addition, the inhibition of protein degradation by chloroquine and lactacystin was blocked by 8-Bromo cAMP-induced mESC migration in the Oris cell migration assay and in an in vitro wound healing migration assay (Fig. 3E, F).
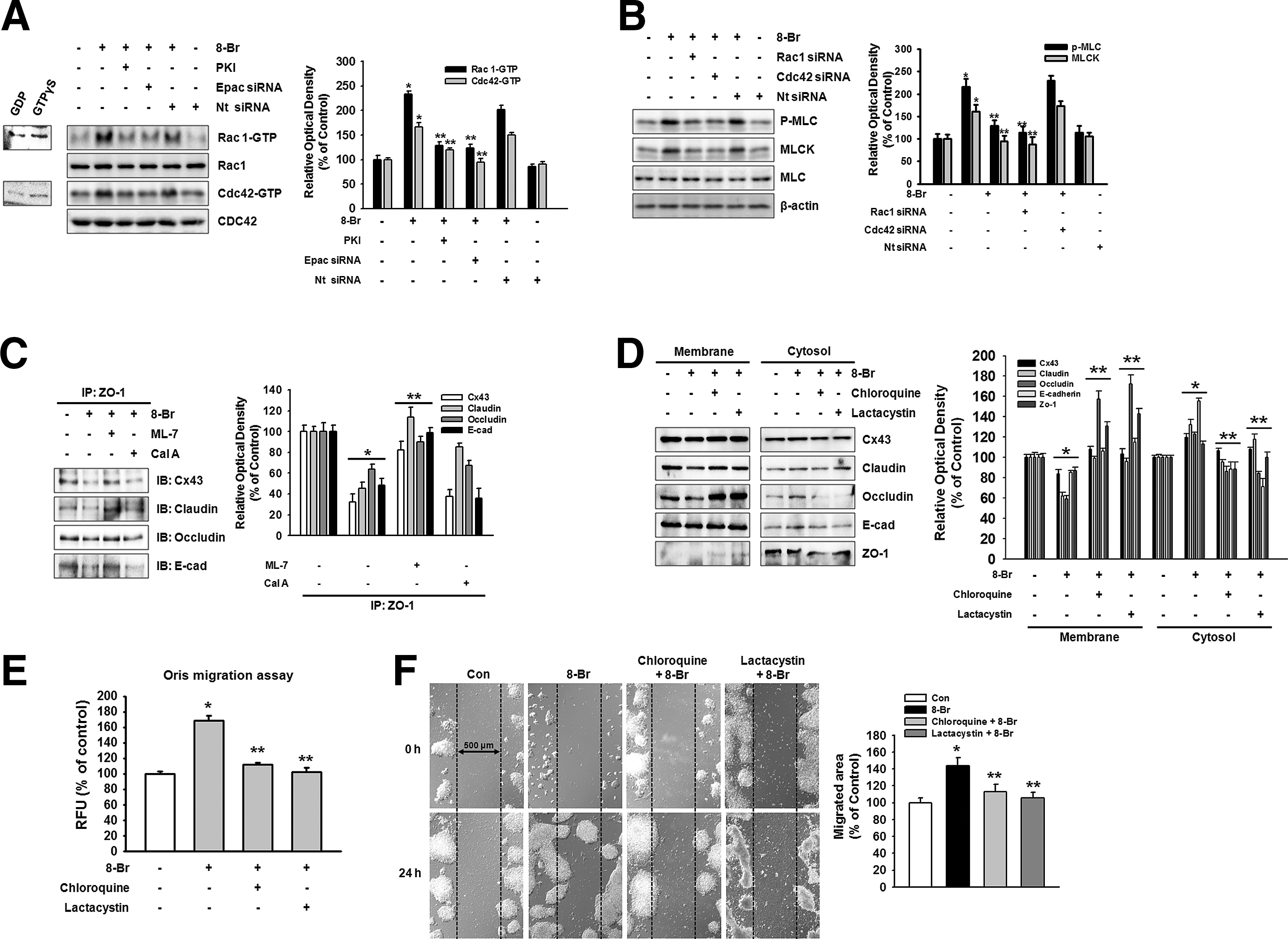
Effect of Rho GTPase on junctional disruption.
8-Bromo cAMP induces cell cytoskeleton dynamics through Rho GTPase
Several recent studies have shown that the regulation of cell migration is induced by cell cytoskeleton dynamics. Rac1/Cdc42 stimulating actin polymerization through its interaction with WASP and N-WASP, leading to the activation of the Arp2/3 complex, has been well characterized. PAK inhibits cofilin-mediated actin depolymerization, thereby promoting F-actin accumulation. The effect of 8-Bromo cAMP on Arp3, TOCA, PAK, and N-WASP protein expression, which is believed to be essential in the dynamic regulation of actin cytoskeleton, was investigated to confirm the effect of 8-Bromo cAMP on mESCs. Arp3, TOCA, PAK, and N-WASP protein expression was significantly increased, and phospho-cofilin was decreased in response to treatment with 8-Bromo cAMP for 4 h, which was inhibited by Rac1 and Cdc42 siRNAs (Fig. 4A). In addition, we confirmed the localization of Arp3, TOCA, PAK, and N-WASP in the mESCs through immunofluorescence staining (Fig. 4B). We further explored the ability of 8-Bromo cAMP to induce actin cytoskeleton remodeling in mESCs with immunofluorescence staining (Fig. 4C). The 8-Bromo cAMP-induced increases in the Oris and the wound healing migration assays were blocked by the siRNAs of the signaling pathway-related molecules (Fig. 5A, B). To further examine this, we quantified the migration distance by live-cell imaging and observed that the mESCs showed a significant increase in the migration distance for the 8-Bromo cAMP-treated cells (Fig. 5C).

Effect of Rho GTPase on cell cytoskeleton dynamics. Cells were transfected with Rac1 and Cdc42 siRNA (20 nM) or Nt siRNA for 1 day before treatment of 8-Bromo cAMP for 24 h and expression of PAK, Arp3, N-WASP, TOCA, and p-cofilin was detected by western blot
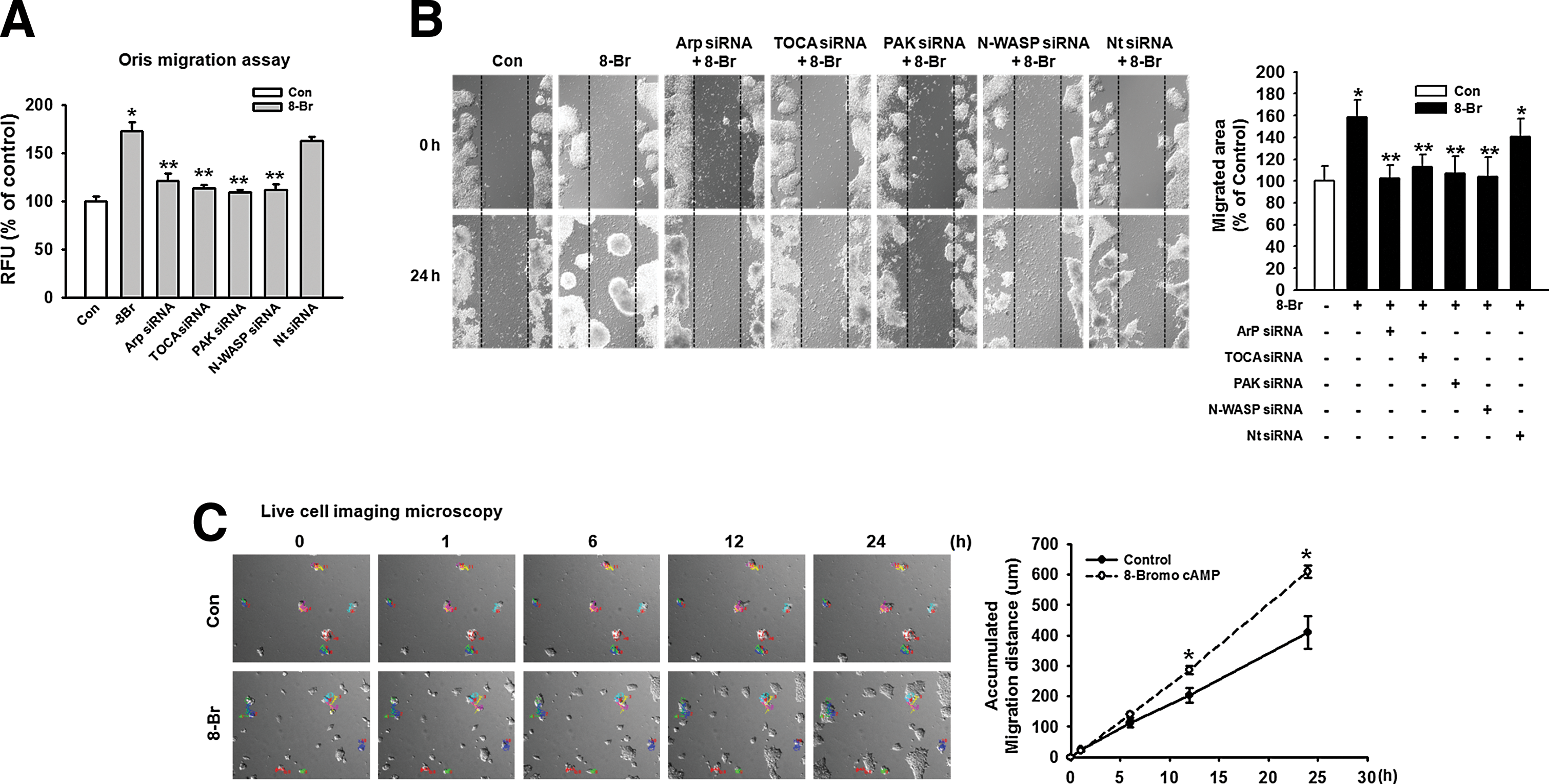
Involvement of actin cytoskeleton dynamics on mESC migration. mESCs were transfected with Arp3, TOCA, PAK, N-WASP siRNA (20 nM), or Nt siRNA (20 nM) for 24 h before 8-Bromo cAMP treatment for 24 h.
Discussion and Conclusions
In the present study, our data show that transplantation of mESCs activated with 8-Bromo cAMP, which resulted in enhancement of cellular motility through cellular adherent junction disruption and cytoskeleton protein rearrangement, accelerated the wound healing process (Fig. 6). In the mouse wound healing model, transplantation of mESCs activated with 8-Bromo cAMP most potently stimulated wound healing at 9 days after wounding, which provides a mechanistic explanation of the important relationship between 8-Bromo cAMP and stem cells in skin wound healing. In addition, transplantation of mESCs with 8-Bromo cAMP exhibited a higher capillary density in the wound site. Furthermore, our results suggest that the 8-Bromo cAMP-treated mESCs enhanced skin wound healing through regulation of paracrine effects. Indeed, the paracrine effect of stem cells is now widely accepted and involved in the repair of damaged tissue [20]. These results suggest the possible application of mESCs activating with cytokine or chemical in cell-based therapy. Furthermore, 8-Bromo cAMP stimulated mESC migration to the wound site, suggesting the possibility that the 8-Bromo cAMP-induced mESC migration enhances the wound healing progress. To our knowledge, this is the first observation that 8-Bromo cAMP-treated ESCs stimulate the wound healing through migration into the wound site and augment the paracrine function of mESCs. Thus, the molecular mechanisms for cAMP-induced wound repair should be clarified to understand the regulation of stem cell fate in regenerative medicine. Cell migration is a complex process induced by diverse stimuli that take place during certain stages of embryonic development, organogenesis, and wound healing [30]. In addition, animated migration to the wound site is important for the stem cell in cell therapy. Actually, many studies have recently announced the improvement of the stem cell migration to the wound site in directly treated stem cell therapy for the tissue regeneration [31]. In this respect, dissecting the regulatory pathways and deciphering underlying molecular mechanisms of stem cell migration by cAMP would be necessary to advance stem cell therapy in the management of wound healing.
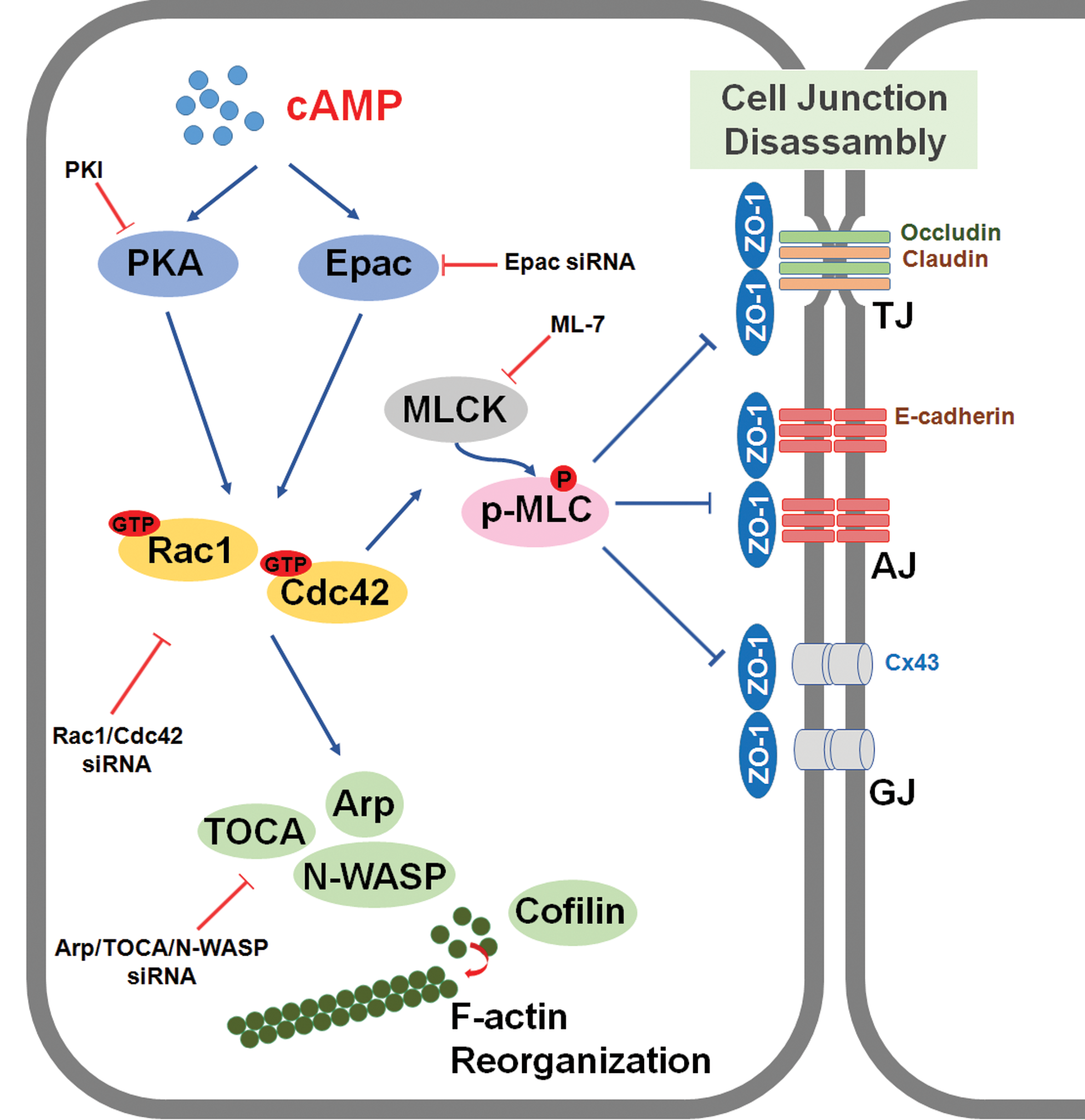
The proposed model for the signaling pathways involved in 8-Bromo cAMP-induced mESC migration through adherent junction disruption and actin cytoskeleton rearrangement. Elevated intracellular cAMP by 8-Bromo cAMP stimulated both PKA and Epac, which subsequently activated Rac1 and Cdc42. 8-Bromo cAMP-induced activation of Rac1/Cdc42 increased MLCK expression and MLC phosphorylation, which resulted in disruption of cellular junctions (adherens junction, tight junction, and gap junction). In addition, 8-Bromo cAMP-induced activation of Rac1/Cdc42 also elicited activation of PKA, Arp, TOCA, and N-WASP and stimulated dephosphorylation of cofilin, which evoked the F-actin rearrangement. This 8-Bromo cAMP-induced disruption of cell junctions and cytoskeleton remodeling stimulated mESC migration. Color images available online at
The effects of cAMP are mediated by two ubiquitously expressed intracellular cAMP receptors: the classic PKA and the recently discovered exchange protein directly activated by Epac [15,32]. Although the target for PKA/Epac, which controls small GTPase activation, is currently unknown, PKA/Epac appears to be required for the activation of small GTPases. The Rho family of small GTPases, consisting of Rho, Rac, and Cdc42, is a critical regulator of actin dynamics [33] and controls the adhesion and migration of stem and progenitor cells, including HPCs and MSCs [34]. The evidence, at present, indicates that the small GTPases, Rac1 and Cdc42, are the most significant effectors of PKA and Epac. In addition, it has been reported that the PKA directly phosphorylates Ser-188 of RhoA, which inhibited RhoA activity through decrease in binding with its effector, ROKα [35,36]. In contrast to its inhibitory effects on Rho, cAMP/PKA signaling has been shown to be required for Rac activation and function in carcinoma [37]. Additionally, unlike Rho, Rac and Cdc42 are not directly phosphorylated by PKA and the target for PKA that controls Rac and Cdc42 activation is currently unknown [7]. These facts reveal that while Rho is a major important target for PKA, it is very clear that it is not the only target. Furthermore, RhoA positively and negatively affects cell migration depending on gradient sensing, polarization, and orientation, as well as specific cell types [38]. In addition to these previous findings, this study showed that the activation of Rac1 and Cdc42 leads to cell structural changes in mESC migration. Therefore, to understand the stimulating effect of small GTPases on cell migration, we separately studied the effects of 8-Bromo cAMP on cell junction distribution and actin cytoskeleton remodeling in mESCs.
Adherent junction molecules, such as E-cadherin, connexin, and occludin, have been known to influence the migration of stem cells as well as proliferation [39]. The loss of cell–cell junctions elicited increase in migration of stem cells, including ESCs and adult stem cells [40 –43], suggesting that the cell junction molecules are essential for regulation of stem cell migration. Especially, cAMP has long been shown to exert cell–cell and cell–matrix association. Thus, it is possible that the cAMP-dependent signal pathway could be associated with regulation of the mESC junction. In this study, 8-Bromo cAMP induced a deficiency in cell junctions, including the tight junctions, gap junctions, and adherens junctions, which suggests the possibility that the cAMP signaling could stimulate mESC migration through regulation of cell junctional complexes. We therefore asked whether specific alterations in cell junction distribution could be induced by small GTPases. Small GTPases capture key functions to preserve structural and functional properties of cell junctions. ZO-1, a common adaptor of tight junction proteins (occludin, claudin, and JAMs), directly or through their adaptor proteins, links to the cell junctions (eg, GJs; connexin 43, AJs; E-cadherin, and TJs; occludin), and the actin cytoskeleton manifests structural associations with the cell junctions through ZO-1 [44 –46]. The actin–myosin-based cytoskeleton is essential for contraction [47] and the process is triggered by the binding of the MLC, which is activated by phosphorylation through kinases, such as Rho-associated kinase (ROCK) or PAK [48]. Rac can induce focal complex/adhesion turnover both directly, through PAK, and indirectly, by antagonizing Rho activation [49]. We tested whether MLC could be regulated through 8-Bromo cAMP-induced activation of small GTPases. Our study revealed that MLC phosphorylation is mediated by PAK, the effector molecule of Rac1/Cdc42. Rac and Cdc42 can bind and activate PAK. PAK activates myosin II by phosphorylating its regulatory light chains [50,51]. In the present study, our observations demonstrate that the 8-Bromo cAMP-induced disruption of the junctional complexes reduced by the MLCK inhibitor, which suggests that MLC phosphorylation can reorganize cell junctions. Through the cytoplasmic tail of MLC, junctional proteins bind to cytoskeletal and signaling proteins, which enable the junctional proteins to anchor to the actin microfilaments and the transfer of intracellular signals inside the cell [52]. The association with actin is required not only for the reorganization of the junctions but also for the dynamic regulation of junction opening and closing, which were confirmed by our results through the immunoprecipitation and cell junction assays.
In addition, recent experiments have clarified some of the pathways by which Rho family members regulate cytoskeletal alterations. These cytoskeletal changes are brought about by small GTPases directly related to the general changes in cell morphology that are associated with migration. Following these studies, 8-Bromo cAMP-activated small GTPases are able to directly regulate actin cytoskeleton remodeling. Actin dynamics is regulated by activating upstream signaling molecules such as Arp3, TOCA, and N-WASP [53 –55]. In our results, Rac1/Cdc42 GTPases acted as regulators of Arp3, N-WASP, and TOCA-1, which nucleate actin branches and lead to a processive elongation of actin. Especially, TOCA-1 is an adaptor protein that may coordinate actin assembly through both the Cdc42 and Rac pathways based on observations. It has been shown that both N-WASP and Arp2/3 complexes are required for Cdc42 to trigger actin filament assembly. It is well established that N-WASP is a Cdc42-specific effector that regulates actin organization, which promotes a more motile phenotype [56]. Taken together, the findings of this study show that cAMP has an important role in two key processes in mESC migration: the turnover of cell junctions and the remodeling of the actin cytoskeleton. cAMP involvement in these processes makes it a potential target for stem cell therapy, which could help increase the rate of wound healing. 8-Bromo cAMP-based interventions to create a favorable microenvironment for mESC migration could be a promising and practical approach for controlling stem cell functions in clinical applications. In conclusion, 8-Bromo cAMP-induced increase in mESC migration accelerated the skin wound healing process through junctional disruption and actin cytoskeleton remodeling.
Footnotes
Acknowledgments
This research was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (A120216), and a grant from the Next-Generation BioGreen 21 Program (no.PJ011141), Rural Development Administration, Republic of Korea.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.