
Abstract
Heart disease is the leading cause of human death in the 21st century. Heart transplantation is a promising way to treat this. Because donor resources are limited, cell-based therapy has been developed as an alternative. Therefore, genes that trigger cardiogenesis could have potential in the treatment of heart disease. Fibroblast growth factor 1 (FGF1) is reported to stimulate cardiomyocyte proliferation under conditions of myocardial infarction, but little is known about its function during cardiac differentiation. In this study, we established an in vitro cardiogenesis model through a reliable chemical induction protocol to determine whether FGF1 and its gene expression are involved in cardiogenesis. Oxytocin, not only a well-known hormone but also a cardiac differentiation inducer, was used in a mouse embryonic stem cell line, E14Tg2a, to achieve cardiac differentiation. After differentiation, beating cell clusters appeared and the expression of FGF1B mRNA was upregulated in the late differentiation stage (differentiation days 8–14). Interestingly, FGF1B expression patterns during cardiac differentiation were similar to those of a mature cardiomyocyte marker, troponin T2, cardiac. The blockage of FGF1-FGF receptor (FGFR) signaling reduced not only the appearance of beating cluster formation but also the expression levels of cardiomyocyte-associated genes. Moreover, by investigating FGF1 downstream signaling cascades, we observed that the efficiency of beating cluster formation was mainly regulated through the FGF1-FGFR-PKC signaling axis. Taken together, we provide evidence to support that FGF1 could regulate cardiogenesis primarily through the protein kinase C signaling, but not through the mitogen-activated protein kinase signaling, pathway.
Introduction
C
The mammalian heart was formerly thought to permanently leave cell cycle turnover. After much labor in this field, however, it was discovered that the mammalian heart does have the ability to regenerate, although very slowly, especially when compared with the robust regenerative ability of amphibians and zebrafish [3]. In humans, the annual regeneration rate of a 25-year-old adult heart's cardiomyocyte is merely 1%–2% by radiocarbon detection [4]. A similar result in mouse heart was also observed in 2013 by Senyo et al. [5]. Although the turnover rate is very low, the truth that the mammalian heart can regenerate has been established. Furthermore, in 2011, Sadek and his team observed that the 1-day-old mouse, like adult zebrafish, could regenerate its heart in 21 days after amputation in ventricle site. However, this regenerating ability could be manifested only in neonatal mice, not in 7-day-old mice [6].
In part, because this regenerative ability is extremely low, many problems remain in using mice directly as a platform to study mammalian heart regeneration. Embryonic stem (ES) cells have been used as a powerful material for differentiation into multipotent lineage cells [7]. With the aid of reliable chemical inductions, cardiac differentiation can be achieved in mouse embryonic carcinoma/stem cell lines [8 –10]. Through the use of ES cells, scientists could use genetic modification to gradually reveal the mechanism of cardiogenesis.
The fibroblast growth factor (FGF) family, with diverse biological functions, comprises 22 members in mammals [11]. The acid FGF, also called fibroblast growth factor 1 (FGF1), could contribute to cell proliferation or differentiation, neural development, and angiogenesis [12,13]. Unlike other FGF members—such as FGF2, FGF8, and FGF10—FGF1 has rarely been discussed in terms of its role in cardiac systems [14 –16]. In humans, the FGF1 gene has at least four different promoters to control its tissue-specific expression [12]. Similar to the human FGF1 gene, the mouse FGF1 gene contains at least three untranslated exons—FGF1A, FGF1B, and FGF1G—each transcribed from a different promoter. The mouse FGF1A transcript is mainly found in the heart and kidney; the FGF1B transcript is predominantly located in the brain; and the FGF1G transcript is expressed mostly in the liver and kidney [17]. Our previous study found that FGF1B expression cells have a capacity to differentiate into all three neural lineages, including neurons, astrocytes, and oligodendrocytes [18]. It was shown that FGF1 mRNA is abundantly expressed in human fetal heart [19]. Jonker and colleagues have further demonstrated that FGF1B is expressed in the heart region [20]. However, the function of FGF1B in cardiogenesis is still not understood.
FGF1 has been reported to increase expression of cardiomyocyte-associated genes under retinoic acid induction in the mouse embryonal carcinoma P19 cell line [10]. In addition, FGF1 induces cardiomyocyte regeneration and improves function recovery in a myocardial infarction situation [21]. Thus, FGF1 is thought to play a role in enhancing cardiac regeneration [10,21]. However, the detailed mechanism of how FGF1 signaling transduction affects cardiogenesis is still unknown. The FGF1 signaling downstream member, the FGF receptor (FGFR), is reported to be essential for the appearance of contractile cardiomyocyte foci. In addition, the fgfr1-null mice-derived embryonic bodies displayed downregulation of cardiomyocyte marker myosin, light polypeptide 2, regulatory, cardiac, slow (Mlc2v) [22].
In the present study, we used oxytocin (OT) to induce cardiac differentiation in a mouse ES cell line, E14TG2a. After 14 days of induction, beating colonies were observed. By comparing the induction group with the control group, we found that the expression of FGF1B transcript and FGF1 was upregulated. The cardiac differentiation-associated genes were also elevated after OT induction. We further showed that the efficiency of beating cluster formation was correlated with activation of the FGF1-FGFR signaling pathway. In the FGF1 downstream signaling cascades, beating cluster formation was mediated primarily through the protein kinase C (PKC) signaling, but not through the mitogen-activated protein kinase (MAPK) signaling, pathway. Furthermore, PKC ɛ could be the molecular target responsible for these events.
Materials and Methods
Culture of mouse ES cells
The mouse ES cell line, E14Tg2a, was purchased from ATCC. The culture protocol was described in previous literatures [23,24]. Briefly, ES cells were cultured in Glasgow's minimum essential medium (GMEM; Gibco) supplemented with 10% knockout serum replacement (Gibco), 1% fetal bovine serum (Hyclone), 0.1 mM MEM nonessential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), 2 mM
In vitro cardiac differentiation
The cardiac induction procedure followed the protocol in a previous study [9]. Briefly, 2.5 × 105 ES cells were cultured in a Petri dish (Falcon) with Iscove's modified Dulbecco's medium (IMDM; Gibco) containing 20% fetal bovine serum (Hyclone), 10−7 M OT (Sigma), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco) for 4 days. On differentiation day 4, the cells were transferred to a gelatin-coated tissue culture dish or gelatin-coated tissue culture 24-well plate (Corning) for further differentiation in differentiation medium, which was IMDM containing 20% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The medium was replaced every 2–3 days until day 14. The efficiency of beating cluster formation was evaluated by calculating the percentage of wells containing beating cell clusters in a 24-well plate at day 14.
RNA preparation, reverse transcription–polymerase chain reaction, and real-time polymerase chain reaction
We used the FavorPrep Tissue Total RNA Mini Kit (Favorgen) to extract cell RNA. The reverse transcription (RT) step was performed using the Thermo Scientific RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific). A total of 1 μg RNA was used in the RT in a final 20 μL volume. Next, real-time polymerase chain reaction (Q-PCR) was carried out in the ABI 7500 Fast Real-Time PCR System machine using SYBR FAST Universal 2× qPCR Master Mix reagent (KAPA). In Q-PCR analysis, 1 μL of cDNA was used. Thermal protocol was set according to the recommendations of KAPA SYBR FAST Universal 2× qPCR Master Mix reagent manufacturer's datasheet. Gene expression detection and data analysis were performed using ABI 7500 1.41 version software (ABI). The specific primer sequences for the mouse gene were as follows:
POU domain, class 5, transcription factor 1 (Oct4)—FW: 5′-GGATGCTGTGAGCCAAGG-3′, RV: 5′-GAACAAAATGATGAGTGACAGACAG-3′ [25]; mesoderm posterior 1 (Mesp1)—FW: 5′- TTTCCTTTGGTCTTGGCACCTTCG-3′, RV: 5′- TCCAAGG AGGGTTGGAATGGTACA-3′ [26]; GATA-binding protein 4 (Gata4)—FW: 5′-CTGTCATCTCACTATGGGCAC-3′, RV: 5′-CCAAGTCCGAGCAGGAATTTG-3′ [27]; troponin T2, cardiac (cTnT)—FW: 5′-ATCCCCGATGGAGAGAGAGT-3′, RV: 5′-TTCCCACGAGTTTTGGAGAC-3′ [28]; myosin, heavy polypeptide 6, cardiac muscle, α (α-MHC)—FW: 5′-AACCAGAGTTTGAGTGACAGAATG-3′, RV: 5′-ACTCCGTGCGGATGTCAA-3′ [29]; Mlc2v—FW: 5′-ACTTCACCGTGTTCCTCACGATGT-3′, RV: 5′-TCCGTGGGTAATGATGTGGACCAA-3′ [30]; and FGF1A and FGF1B primer sequences were as described previously [17,31]. The β-actin expression levels were used as internal controls (We generated a standard curve in which coefficient of determination R 2 = 0.9999 and determined the numbers of molecules of β-actin under various experimental conditions. We demonstrated that the β-actin expression levels were almost the same in different experimental conditions).
Immunostaining, western blotting, and fluorescence-activated cell sorting
For immunofluorescence staining, differentiated cells (day 14) were dissociated and plated onto an eight-well Lab-Tek II chamber slide (Nunc) at a density of 1 × 104 cells per well overnight. The adherent differentiated cells were washed with phosphate-buffered saline (PBS) solution twice and fixed with 4% paraformaldehyde (Sigma) for 20 min. Then, cells were washed with PBS solution thrice and put into 0.1% bovine serum albumin (BSA) in PBS for 1 h as blocking. After blocking, the primary antibody recognized for mouse α-actinin was diluted in 0.01% BSA in PBS and used in 1:1000 dilution (Sigma) and incubated at 4°C overnight. Subsequently, the cells were washed thrice in PBS for 10 min each time, and then incubated in FITC-conjugated secondary antibody (Millipore) (diluted to 1:1000) for 1 h; 4′,6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei; and sample slides were observed using a fluorescent microscope (Olympus) or a confocal microscope (Leica).
For western blotting, protein samples were harvested using radioimmunoprecipitation assay (RIPA) buffer (Millipore) containing protease inhibitor (Merck). The concentrations of protein samples were assessed by protein assay dye reagent (Bio-Rad). Forty-microgram protein samples were loaded onto sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE) and then separated by gel electrophoresis. Thereafter, the gel was transferred to polyvinylidene fluoride (PVDF) membranes (Amersham) and blocked in blocking solution (5% skim milk in phosphate buffered saline Tween-20 [PBST]) for 1 h at room temperature, followed by incubation at 4°C overnight with specific primary antibody. The primary antibodies and application titers we used in western blotting were as follows: Oct4 1:1000 (NB100-2379; Novus), Nanog 1:1000 (ab5731; Millipore), FGF1 1:1000 (produced by Genetex), cTnT 1:2000 (10214; Abcam), α-tubulin 1:8000 (GTX628802; Genetex), β-actin 1:5000 (MAB1501; Millipore), PKC ɛ 1:500 (sc-214; Santa Cruz), and PKC δ 1:500 (sc-213; Santa Cruz). After primary antibody incubation, the membrane was washed thrice for 10 min each in PBST. Next, secondary antibodies conjugated with horseradish peroxidase (Millipore) were used for 1 h of incubation at room temperature. Next, the membrane was washed thrice for 10 min each in PBST; the immunoreactive signals were enhanced through chemiluminescence (ECL) reagent (Millipore). Finally, the signals were detected and developed on photographic films (Fujifilm).
For fluorescence-activated cell sorting (FACS), 1 × 106 cells from day 0 control and day 14 differentiated cells were collected separately. The staining protocol followed the procedure according to a previous literature [32]. After permeabilization, the control or differentiated cells were incubated with cTnT primary antibody in 1:200 dilution (10214; Abcam) and then with a secondary antibody that was conjugated with R-phycoerythrin (Invitrogen). The mouse IgG was used as an isotype control. The contents of cTnT-positive cells were measured by using FACS Calibur (BD Bioscience).
Blockage of FGF1 and its downstream signaling cascades
For blocking the signaling of endogenous FGF1, either the FGF1 siRNA or FGF1-neutralizing antibody was used. In the FGF1 siRNA experiment, FGF1 siRNA or negative control siRNA (Invitrogen) was incubated with Lipofectamine RNAiMAX (Invitrogen) in Opti-MEM I reduced serum medium (Gibco), and then added to differentiation medium to transfect cells on day 8. The transfection was performed as we replaced the medium every 2–3 days until day 14. On day 14, the efficiency of beating cluster formation was evaluated. Next, in the FGF1-neutralizing antibody administration test, the FGF1-neutralizing antibody (49996; Abcam) or PBS control was added into differentiation medium from day 8 to 14 as we replaced the medium every 2–3 days. The efficiency of beating cluster formation and Q-PCR analysis was analyzed on day 14.
To block and rescue FGF1 downstream signaling pathways, we supplemented the following chemicals with differentiation medium during days 8–14: FGFR inhibitor, SU5402 (20 μM; Sigma); MEK inhibitors, U0126 and PD98059 (10 and 50 μM, respectively; Millipore); Akt inhibitor, Akt 1/2 kinase inhibitor (1 or 10 μM, as indicated; Sigma); PKC inhibitor, staurosporine (STA; 1 or 10 nM, as indicated; Cell Signaling); and PKC activator, phorbol-12-myristate-13-acetate (PMA; 30 or 300 nM; Merck). Dimethyl sulfoxide (DMSO) was used as vehicle control. On day 14, the efficiency of beating cluster formation was evaluated, then the cells were collected either for gene expression analysis or for western blotting.
Statistical analyses
Data are expressed as mean ± SEM. The results were analyzed at least in triplicates. Analysis of variance with Tukey's multiple comparisons test was used to analyze data for statistical evaluation as previously described [33,34]. Statistical significance was scored when P < 0.05.
Results
OT treatment of mouse ES cells resulted in cardiac differentiation
OT is not only a reproductive hormone but also an inducer of cardiac differentiation [9]. In addition, the E14Tg2a mouse ES cell line has been reported as a suitable model for cardiac differentiation study [35]. To test the ability of OT in inducing cardiac differentiation of E14Tg2a cells, we analyzed cardiac-associated gene expression after OT treatment (Fig. 1A, B). Cardiogenesis is a complicated procedure under multiple gene regulations. Therefore, estimating cardiac-associated gene expression levels at various differentiation time points could be used to monitor cardiac differentiation. Using Q-PCR, the gene expression levels of Oct4, a well-known pluripotency marker, showed their highest expression in undifferentiation condition and gradually decreased as differentiation progressed. Mesp1, a mesoderm cell fate marker, presented its highest expression at day 8. Gata4, a transcription factor involved in cardiac development, is a cardiovascular progenitor cell marker. At day 10, Gata4 expression significantly increased and showed the highest expression level at day 14. Matured cardiomyocyte markers, cTnT, Mlc2v, and α-MHC, displayed stronger expression from day 8 to 14 and peaked at day 14 (Fig. 1B). These data illustrated that cardiac-associated gene expression levels were upregulated after cardiac induction, demonstrating that our in vitro ES cell induction system was valid. Furthermore, we calculated the ratio of beating cluster formation, which is a characteristic of in vitro functional cardiomyocytes (Fig. 1C). We observed that the beating cell clusters began to form on day 12 and reached 98.0% ±2.0% on day 14. Using a separate ES cell line, 46C, we showed that these cells could display beating clusters with 47.5% ±4.8% efficiency on day 18. These results validated that OT could be used as a cardiogenesis inducer in ES cells and that our in vitro cardiac induction protocol was feasible for producing functional cardiomyocytes.
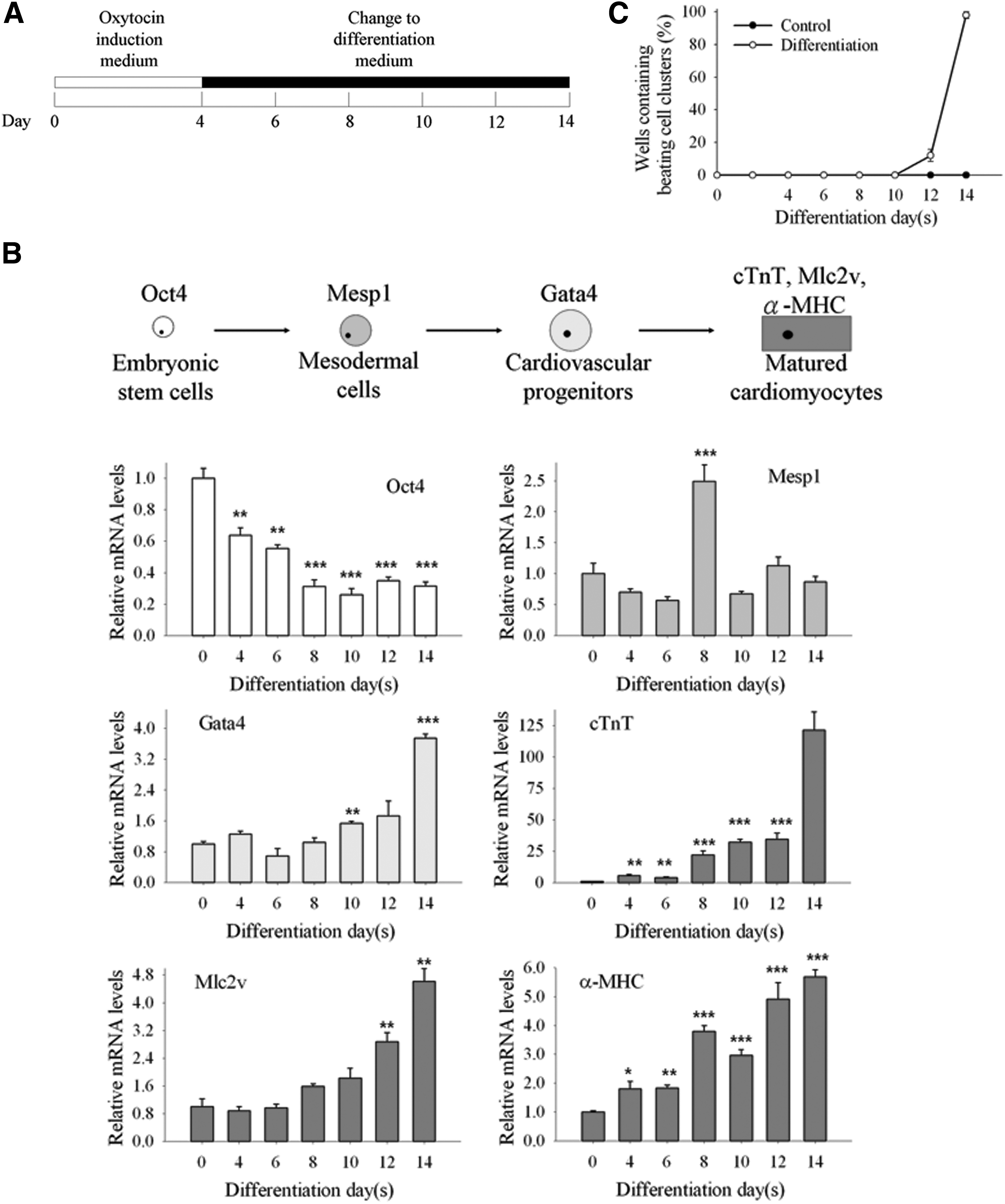
Oxytocin (OT) treatment of mouse embryonic stem (ES) cells resulted in cardiac differentiation.
Immunostaining analysis, western blotting, and FACS revealed terminal cardiac differentiation
In addition to conducting functional tests and cardiac-associated gene profile analyses, we also verified cardiac differentiation using immunostaining and western blotting. Through the detection of alkaline phosphatase activity, a standard method to detect cell pluripotency, we observed stronger alkaline phosphatase activity in undifferentiation status (day 0) and decrease in the differentiation group (day 14) (Fig. 2A). On the other hand, α-actinin (a sarcomeric structure marker of mature cardiomyocytes) [36] -positive cells were found only in the differentiated cells, not in the day 0 ES cells (Fig. 2B). Similar results were found using western blotting (Fig. 2C). In day 0 ES cells, the intensities of Oct4 and Nanog immunoreactive signals were much higher than in the differentiated cells. On the contrary, the intensities of cTnT and FGF1 were upregulated in day 14 differentiated cells. To find out the percentage of cells that are differentiated in the beating clusters, we used the FACS to show that cTnT-positive cells increased from 0.4% of the day 0 control group to 18.0% of the day 14 differentiated group (Fig. 2D). These data demonstrated that the cardiac differentiation was achieved using our protocol.

Immunostaining analysis, western blotting, and fluorescence-activated cell sorting (FACS) revealed terminal cardiac differentiation.
FGF1B mRNA levels increased during cardiac differentiation
After establishing an in vitro cell model for cardiogenesis, we observed that the expression level of FGF1 was upregulated after cardiac differentiation. Thus, we next wanted to know whether FGF1 transcriptional machinery participated in the cardiac differentiation event. In a study of FGF1 regulation, FGF1A was reported to express in the heart [17]. FGF1B was first described to express mainly in the brain [37] and could be used to select neural stem/progenitor cells in the brain [18,38]. A recent study further reveals that FGF1B is also expressed in the heart, although its function there is still unknown [20]. We used Q-PCR to estimate the mRNA expression levels of FGF1 transcripts, FGF1A and FGF1B, at different differentiation time points (Fig. 3). We defined the differentiation time procedure into three stages: days 0–4 represented the early differentiation stage; days 4–8 meant the middle differentiation stage; and days 8–14 indicated the late differentiation stage. In the early and middle differentiation stages, FGF1A and FGF1B expression remained at constant levels. However, when the late differentiation stage began, FGF1B started to increase its expression and showed stronger expression levels during days 10–14. In contrast, FGF1A displayed similar expression levels from the beginning of differentiation to the end stage. These data revealed that FGF1B increased its expression levels during cardiac differentiation and suggested that the upregulation of FGF1 in cardiogenesis was mediated mainly through FGF1B.
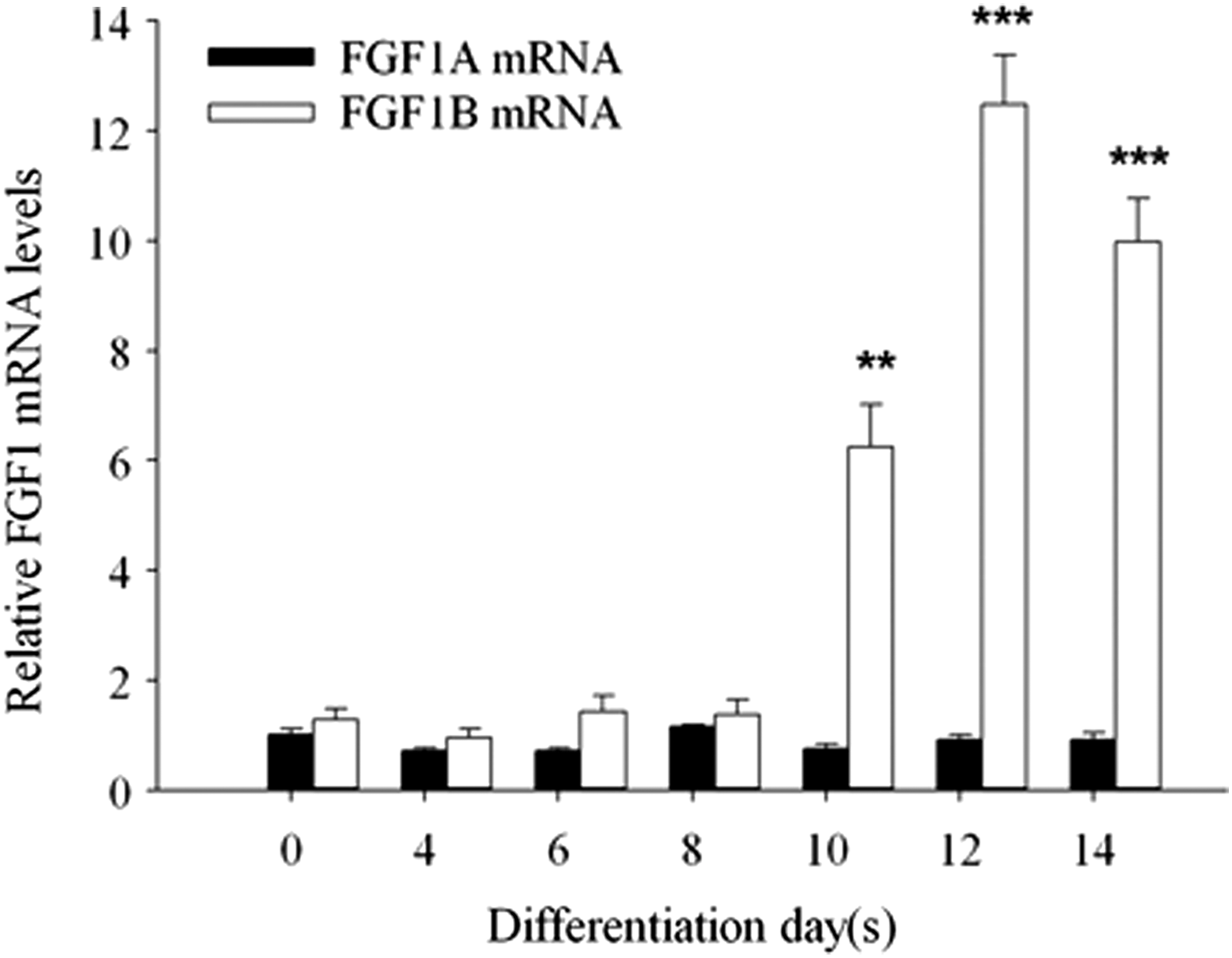
FGF1B mRNA levels increased during cardiac differentiation. Temporal FGF1 gene regulation machinery expression profiles during cardiogenesis. The expression levels of FGF1A mRNA remained at constant levels through all differentiation time points. On the other hand, FGF1B expression was upregulated after day 8 and reached its apex on day 12. In addition, FGF1B expression levels were higher than those of FGF1A after day 8, which suggested that FGF1B mediated FGF1 expression during cardiac differentiation. The figure showed relative expression levels normalized to day 0 FGF1A expression level. Data are presented as mean ± SEM, N ≥ 3. **P < 0.01; and ***P <0.001 versus the day 0 FGF1A control.
Blockage of FGF1 signaling decreased the efficiency of beating cluster formation and reduced the expression levels of cardiomyocyte marker genes
Since the time of beating cell cluster appearance was coincident with upregulation of FGF1 and FGF1B, we treated the differentiating ES cells with FGF1 siRNA from day 8 to 14. We found the efficiency of beating cluster formation dropped to almost half compared with that of control cells (95.0% vs. 45.0%, respectively). To further corroborate this result, we showed that FGF1-neutralizing antibody reduced FGF1 protein expression levels (Supplementary Fig. S1A; Supplementary Data are available online at
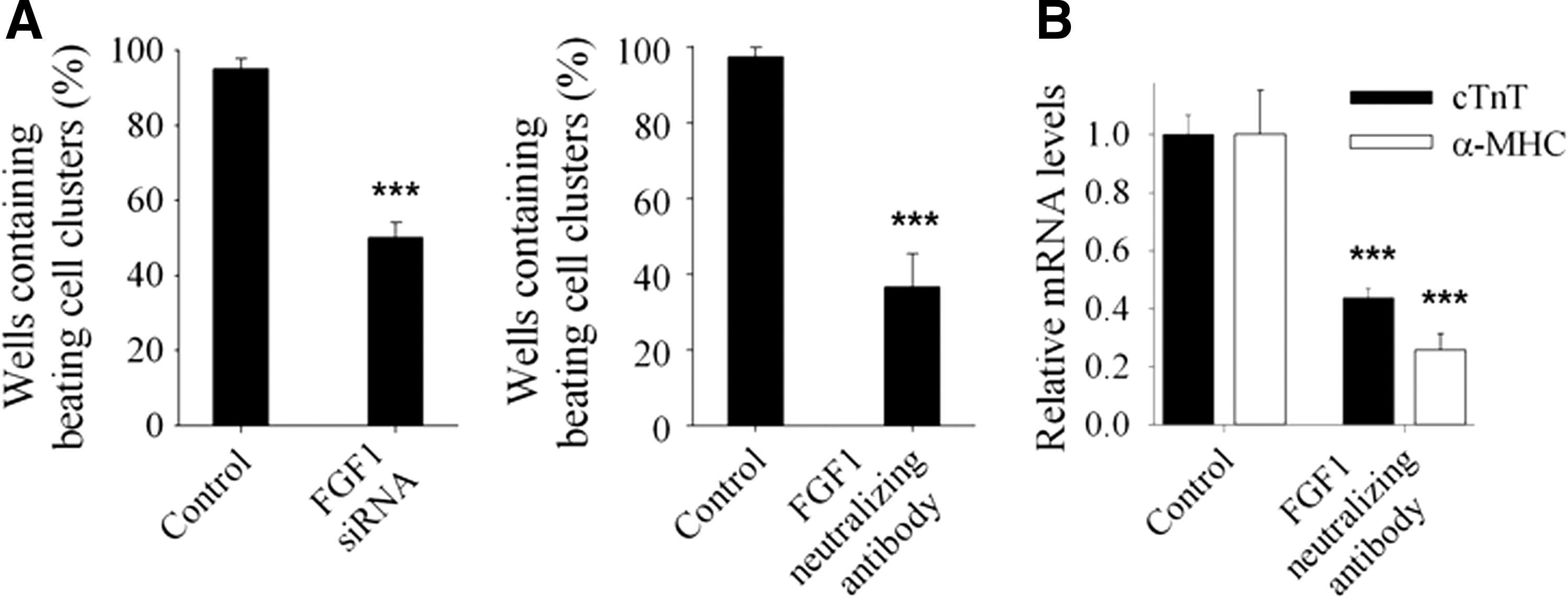
Blockage of FGF1 signaling decreased the efficiency of beating cluster formation and reduced the expression levels of cardiomyocyte marker genes.
FGFR inhibitor reduced the efficiency of beating cluster formation and the mRNA levels of cardiomyocyte marker genes, and the reduction was rescued by PMA
The above results demonstrated that FGF1 could regulate beating cluster formation and cardiomyocyte differentiation. To ascertain that FGF1-mediated cardiomyocyte differentiation and beating cluster formation was through FGFR downstream signaling, we used FGFR inhibitor, SU5402, to test the efficiency of beating cluster formation. When treated with the FGFR inhibitor in the late differentiation stage, beating cell colonies were reduced to 8.3%. A previous study revealed that FGFR downstream activator, PMA, could rescue SU5402-elicited beating loss effect during cardiac differentiation [22]. We used PMA and SU5402 cotreatment and found that 300 nM PMA rescued the efficiency of beating cluster formation completely (Fig. 5A and Supplementary Movie 1). Moreover, a lower concentration of PMA (30 nM) could not rescue the beating phenotype. These findings indicated that PMA could restore the SU5402-caused adverse effect in beating cluster formation in a dose-dependent manner (Fig. 5A). Similar outcomes could be observed in bright-field micrograph images. Under morphological observation, the differentiation structure of SU5402-treated cells became looser than control cells. However, coadministration of SU5402 and PMA could restore the differentiation structure (Fig. 5B). These results demonstrated that FGF1-FGFR signaling regulated beating performance. We then analyzed the cardiomyocyte marker gene expression levels in the DMSO, SU5402, and SU5402/PMA administration groups. The cTnT and Mlc2v expression levels were significantly decreased after SU5402 treatment, whereas SU5402/PMA could recover their expression levels. Moreover, α-MHC expression was largely reduced through SU5402 treatment and the coadministration group could also efficiently restore its expression (Fig. 5C). These results demonstrated that FGF1 could regulate cardiomyocyte differentiation and beating cluster formation through FGFR signaling transduction.
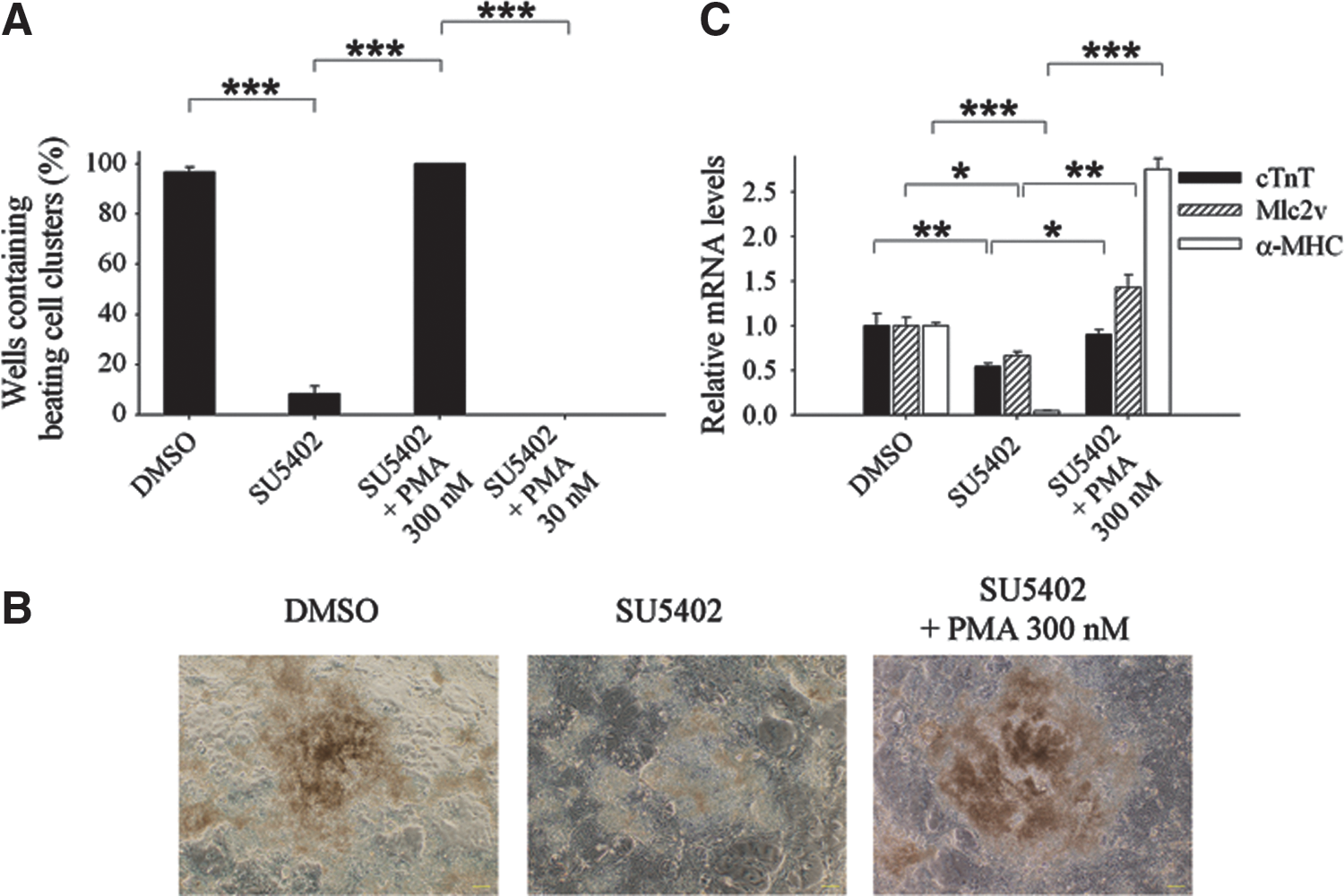
FGF receptor (FGFR) inhibitor reduced the efficiency of beating cluster formation and the mRNA levels of cardiomyocyte marker genes, and the reduction was rescued by phorbol-12-myristate-13-acetate (PMA).
PKC was the main regulator downstream of FGF1-FGFR to mediate beating cluster formation
Previous results demonstrated that the FGF1-FGFR signaling pathway was involved in the regulation of beating phenomenon and cardiomyocyte differentiation. The FGF1-FGFR downstream signaling cascades comprise MAPK, Akt, and PKC signaling pathways, which also affect beating cluster formation and cardiomyocyte differentiation [22,39 –42]. Hence, we used FGFR inhibitor (SU5402), MEK inhibitor (PD98059 or U0126), Akt inhibitor (Akt ½ kinase inhibitor), or PKC inhibitor (STA) to treat differentiated cells in the late differentiation stage to identify which signaling pathway was important to regulate beating cluster formation and cardiomyocyte differentiation. The results showed that FGFR inhibitor, SU5402, reduced the beating cluster formation efficiency by 91.7%, while MEK inhibitors (PD98059 and U0126) reduced the efficiency by only 15% and 16.7%, respectively. Akt inhibitor reduced the efficiency by 50%. Administration of SU5402, PD98059, or U0126 reduced Erk phosphorylation (Supplementary Fig. S1B) and administration of Akt inhibitor reduced Akt phosphorylation (Supplementary Fig. S1C). Furthermore, under these treatments, no cell proliferation changes could be observed (Supplementary Fig. S2B). Notably, PKC inhibitor STA reduced the efficiency by 83.3% and 96.7% with different concentrations (Fig. 6A). It was fascinating to find that the PKC inhibitor caused a beating loss effect similar to the FGFR inhibitor results. These findings suggested that FGF1 affected beating cluster formation in the late differentiation stage predominantly through the FGF1-FGFR-PKC signaling axis.
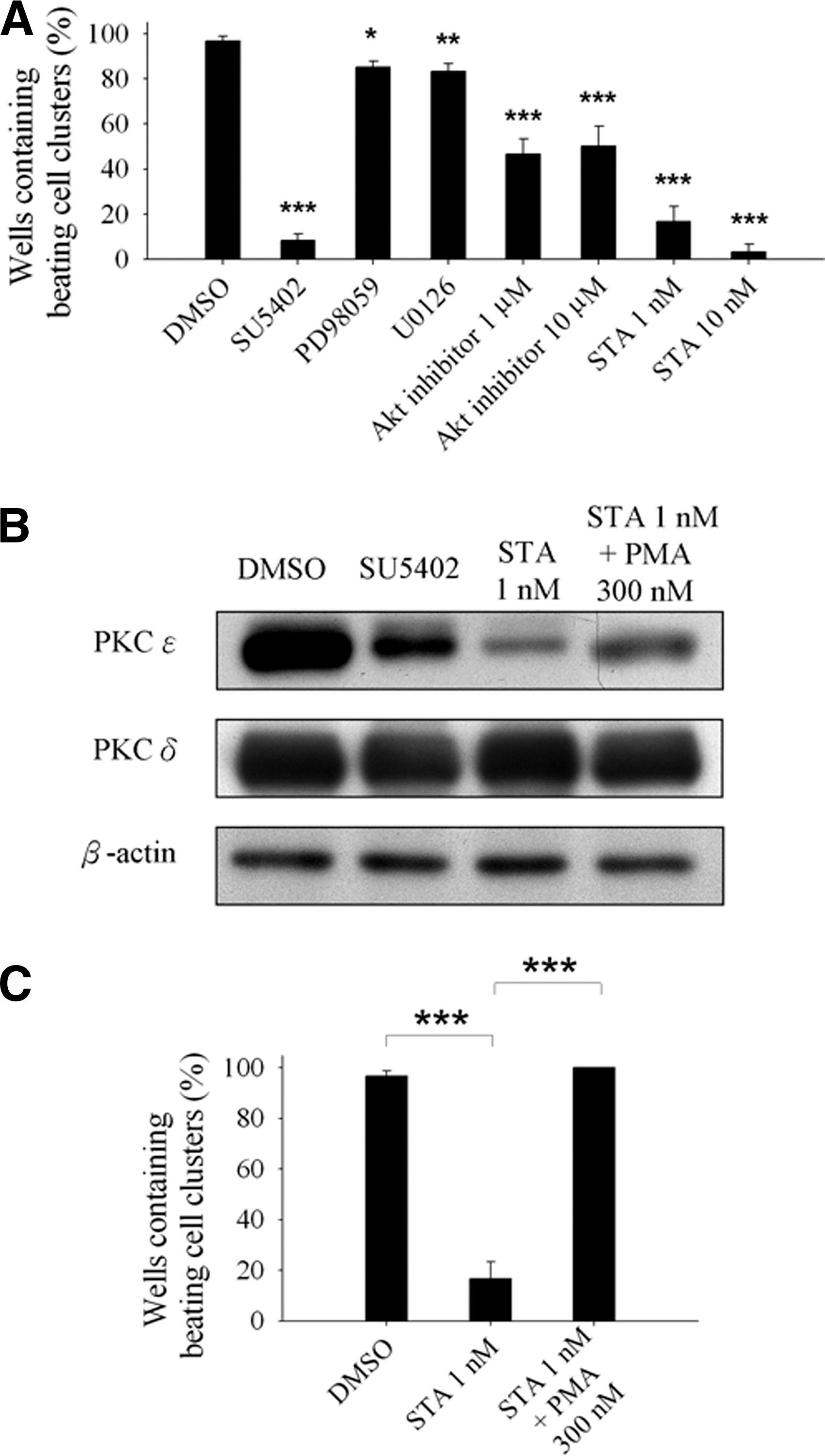
Protein kinase C (PKC) was the main regulator downstream of FGF1-FGFR to mediate beating cluster formation.
A previous investigation reported that PKC ɛ is an important factor to mediate beating cluster formation [40]. This prompted us to determine the expression levels of PKC ɛ in the DMSO, SU5402, STA, and STA/PMA treatment groups. Using western blotting assay, we observed that PKC ɛ expression was slightly decreased in SU5402 treatment cells and largely reduced in STA treatment cells compared with control cells (Fig. 6B). However, when we used STA/PMA combination to treat differentiated cells, the PKC ɛ expression levels were increased 3.7-fold over the STA group (Fig. 6B). In contrast, PKC δ expression levels remained the same (Fig. 6B). We also tested the efficiency of beating cluster formation and found that the beating cluster loss in the STA treatment group was completely recovered through PMA coadministration (Fig. 6C and Supplementary Movie 2). From these data, we concluded that the FGF1-FGFR-PKC ɛ signaling axis was a crucial regulator in beating cluster formation throughout the late differentiation stage of cardiogenesis.
Discussion
OT is a cardiac differentiation inducer [9] and its signaling in cardiac cells has been discussed. When OT binds to OT receptor, its downstream signaling will upregulate PKC and Erk signaling [43]. On the other hand, we also found that FGF1 expression was activated during cardiac differentiation (Fig. 2C). Hence, it is possible that OT could enhance PKC and Erk signaling through FGF1 upregulation during cardiac differentiation.
FGF1 is an essential cytokine in organisms and has many essential biological functions. So, the expression of FGF1 is under a delicate regulation control. Previous studies have verified that FGF1 expression is tissue specific; for example, FGF1A is expressed mainly in the heart, whereas FGF1B is expressed mostly in the brain [12]. Furthermore, FGF1B has also been detected to express in the heart, although the function there remains unknown [20]. Our analysis of time course gene expression levels showed that FGF1B could be the important transcript to regulate FGF1 expression during cardiogenesis. In addition, FGF1A transcript maintained its expression at constant levels throughout all the cardiac differentiation time points. Consistent with previous research, Madiai and Hackshaw described that FGF1A mRNA displays unwavering expression levels from mouse embryonic day 9.5 to 15.5 in the heart by in situ hybridization [44]. In the present study, we initially identified that FGF1 and FGF1B were involved in cardiac differentiation and cardiomyocyte functional maturation. The efficiency of beating cluster formation was reduced when either FGF1 siRNA or FGF1-neutralizing antibody was used to treat the ES cells in the late differentiation stage. This was also the stage when FGF1 levels increased through the FGF1B transcript. Using Q-PCR, we showed that FGF1B mRNA expression patterns were similar to mature cardiomyocyte marker cTnT expression levels, indicating that FGF1B could be a novel factor contributing to cardiomyocyte differentiation. These data, taken together with the results of previous investigations, indicated that FGF1B transcript may be the major modulator to regulate FGF1 protein expression in cardiogenesis.
Since the connection between FGF1 and cardiac differentiation was verified, the next issue we wanted to address was which FGF1 downstream members were essential for beating formation. We first used an FGFR inhibitor, SU5402, to treat cells in the late differentiation stage. The reduction of efficiency in beating cluster formation outcome confirmed that FGF1 was through FGFR to regulate beating cluster formation. Moreover, the blockage of FGF1 or FGFR signaling also decreased cardiomyocyte marker gene expression levels. Thus, downregulating the expression of cardiomyocyte-associated proteins could contribute to the beating formation loss effect. This concept was further strengthened by a study, which has stated that exogenous FGF1 administration could activate the cardiac-related gene α-MHC and inhibit expression of skeletal muscle-related genes under skeletal muscle-inducing conditions [10]. After establishing that the FGF1-FGFR signaling transduction affects cardiogenesis in the late differentiation stage, we investigated FGF1-FGFR downstream signaling cascades to uncover which signaling pathway is crucial for beating cluster formation. Using FGFR downstream signaling inhibitors, we found that PKC inhibitor could cause severe beating formation loss similar to the levels rendered by the FGFR inhibitor, SU5402. Furthermore, one of the candidates in PKC family proteins that could contribute to beating cluster formation was PKC ɛ. Therefore, we provided a thorough investigation to validate that the role of FGF1-FGFR involving in beating cluster formation and cardiomyocyte differentiation was mainly through PKC, but not the MAPK pathway (Fig. 7).
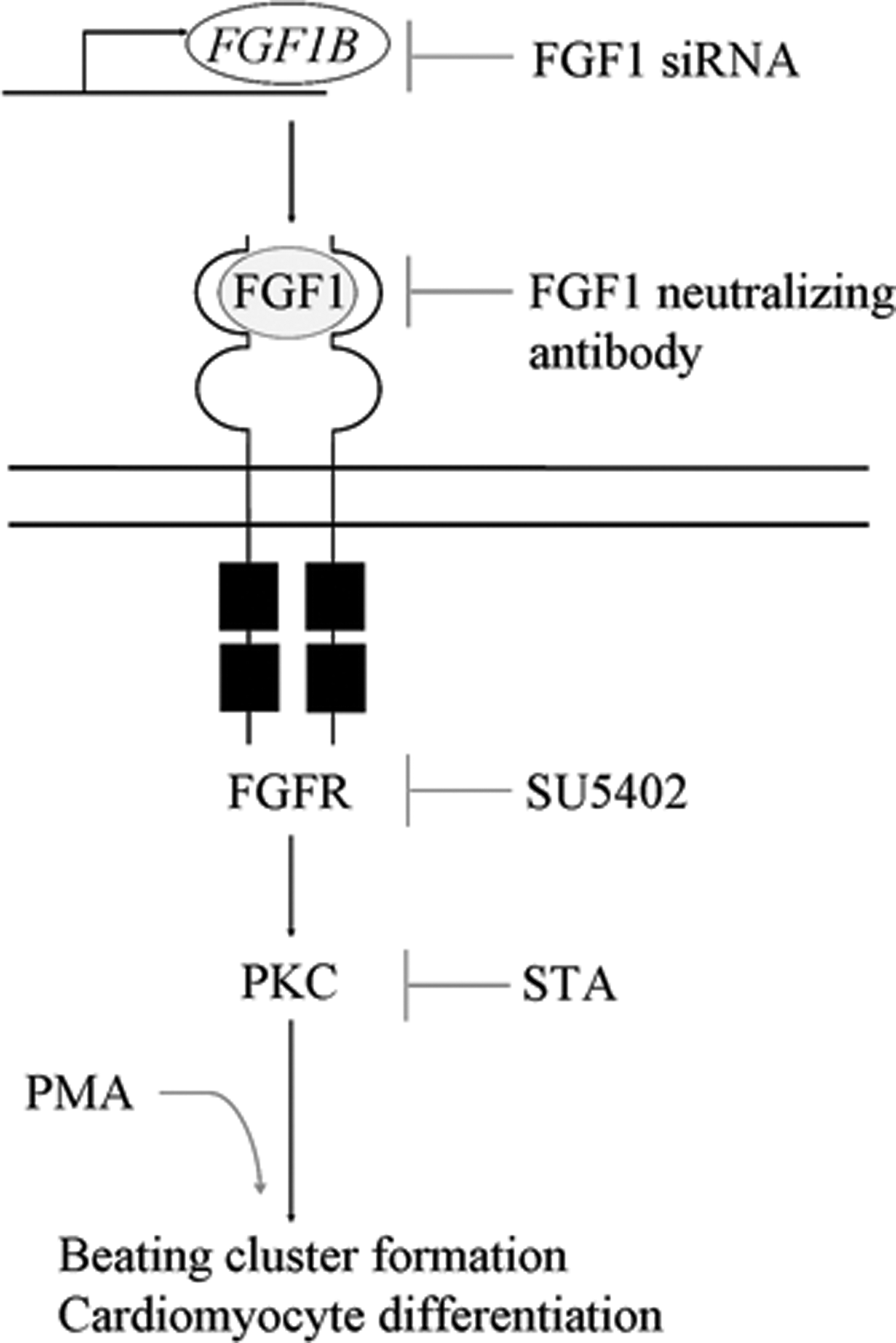
FGF1-FGFR-PKC signaling axis regulates beating cluster formation and cardiomyocyte differentiation in the late differentiation stage. FGF1B transcript and FGF1 protein are involved in cardiac differentiation regulation. The modulator of FGF1 protein expression is likely to be FGF1B transcript during cardiomyocyte differentiation. By investigating FGF1-FGFR downstream signaling members—MAPK, Akt, and PKC—in the late differentiation stage to identify which signaling pathway is critical for beating cluster formation and cardiomyocyte differentiation, we have demonstrated that the PKC signaling pathway is the main regulator downstream of FGF1-FGFR signaling.
A previous study reported that inhibition of FGFR signaling diminishes beating cluster formation and that this effect is reversible under SU5402/PMA coadministration. Furthermore, it has reported that the FGFR-PKC-MAPK signaling axis regulates these events [22]. However, in the current study, we found that FGF1 acted through the FGFR-PKC signaling axis to regulate beating cluster formation and cardiomyocyte differentiation during cardiogenesis without the involvement of MAPK signaling. There are two likely reasons for the differences. First, the earlier research emphasizes on FGFR signaling, which could be turned on by many FGF family protein ligands, including FGF1. However, we focus on the FGF1 role in cardiac differentiation. Second, the inhibition of FGFR signaling is from the beginning of the differentiation procedure to the end in the earlier report. The cardiogenesis time course contains all kinds of FGF members activating during cardiac differentiation. In our experimental protocol, we emphasized how FGF1 signaling regulates cardiogenesis, so we attempted to block FGF1 signaling during the period in which endogenous FGF1 was upregulated during cardiogenesis. Due to the FGF1 expression and FGF1B transcript temporal expression patterns, we concluded that FGF1-FGFR-PKC signaling axis cascades are more likely to address FGF1 signaling transduction during cardiac differentiation. We showed that the FGF1 upregulation occurs in the late differentiation stage. Furthermore, the postnatal cardiomyocytes are terminally differentiated cells resembling our ES cells during the late differentiation stage. Therefore, if FGF1 signaling can be turned on again in adult cardiomyocytes, a reasonable assumption might be that the adult cardiomyocyte turnover rate could be enhanced. Therefore, exploring how FGF1 signaling pathways function at different time points could help reveal how to maximize FGF1 effect in cardiogenesis.
In conclusion, we integrated FGF1 signaling in cardiac differentiation that is mainly through FGFR-PKC signaling transduction. Additionally, FGF1B could be a novel factor involving FGF1 regulation during cardiac differentiation. Within the PKC family proteins, PKC ɛ is an important candidate to modulate cardiogenesis. Based on these findings, further investigations to uncover how PKC ɛ works for beating cluster formation and cardiomyocyte differentiation regulation could benefit understanding of how to increase in vivo cardiomyocyte differentiation efficiency and use FGF1 and PKC ɛ as candidates for cardiomyocyte regeneration drug development and diseases.
Footnotes
Acknowledgments
This project is supported by grants from the Ministry of Science and Technology, Taiwan, and the Ministry of Health and Welfare, Taiwan. Hung-Yu Lin carried out his thesis research under the auspices of the Graduate Program of Biotechnology in Medicine, National Tsing Hua University and National Health Research Institutes.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.