
Abstract
Autophagy is required for hematopoietic stem cell multilineage differentiation, but the underlying mechanism is unknown. Using a conditional mouse model and human leukemia cells, we uncovered a mechanistic link between autophagy and hematopoietic stem cell differentiation. Loss of autophagy in mouse hematopoietic stem cells diminished the bone marrow generation of functional blood cells, in particular lymphocytes, and resulted in a leukemic phenotype and elevated Notch signaling. Physiological autophagy activity in mice was inversely correlated with Notch signaling during adult hematopoietic stem cell differentiation, while pathologically low autophagy was associated with upregulated Notch signaling in dysfunctional hematopoietic stem cells of acute leukemia patients. Furthermore, we show that autophagy directly degraded intracellular Notch, the active form of Notch receptor cleaved from the full-length Notch molecule by γ-secretase. Finally, we show that hematopoietic multilineage differentiation potential was restored in autophagy defective hematopoietic stem and progenitor cells when their Notch signaling was abrogated either pharmacologically with γ-secretase inhibitor DAPT or genetically with RNA interference of Notch effector RBPJ. Hence, we propose that autophagy sustains hematopoiesis by direct targeting Notch.
Introduction
H
Although an increasing number of studies have indicated a role for autophagy in conferring terminal differentiation in various cell types [7, 8], the role of autophagy in the differentiation of various stem cells, and in particular its underlying mechanism, is yet to be explored. Recent studies by others and us indicate the implications of autophagy in adult hematopoietic stem cell maintenance [9], fetal hematopoiesis [10, 11], and adult hematopoietic stem cell cycle regulation [12], as well as megakaryopoiesis, megakaryocyte differentiation, and thrombopoiesis [13]; however, a direct mechanistic link between autophagy and stem cell differentiation remains obscure.
Notch signaling has a complex, but unique mode of action and is implicated in many developmental and differentiation systems [14]. Accumulated studies demonstrate a highly context- and tissue-specific role of Notch signaling pathway in regulation of stem cell function. This context-dependent Notch activity can therefore drive numerous aspects of the development of multicellular eukaryotic organisms and has recently been linked to stem cell fate and maintenance in embryonic and adult tissues [15, 16]. In mammalian embryonic development, Notch signaling is dispensable in the self-renewal of hematopoietic stem cells in primitive hematopoiesis [17], but regulates definite hematopoiesis [18]; in mammalian adults, however, Notch signaling is found not essential for hematopoiesis, but could expand hematopoietic stem cells and undifferentiated hematopoietic progenitors [19,20]. Why Notch is not required in certain lineage differentiation or how Notch signaling is attenuated in the physiological differentiation of hematopoietic stem cells in adult mammals remains largely unresolved.
In this study, we uncover a mechanism by which autophagy sustains hematopoietic stem cell differentiation progression by direct targeting of intracellular Notch (IC-Notch). This finding provides a critical insight into our current mechanistic understanding of the physiological and pathological connections between autophagy and hematopoietic stem cell differentiation.
Materials and Methods
Reagents and antibodies
3-Methyladenine (3-MA), bafiliomycin A1 (Baf-A1), N-acetylcysteine (NAC), benzidine dihydrochloride, and retinoid acid were obtained from Sigma-Aldrich, N-[N-(3,5-Difluorophenacetyl-l-alanyl)]-S-phenylglycinet-butylester (DAPT) was from Merk, and rapamycin was from Calbiochem. Antibodies were from various sources, including Cell Signaling (anti-cleaved Notch1, anti-4EBP1, anti-p-4EBP1, anti-Numb, anti-Histone H3, anti-p-AMPKa, anti-AMPKa, anti-p-mTOR, anti-mTOR, and anti-GAPDH), Abcam (anti-activated Notch1), Novous (anti-LC3), and eBioscience (anti-CD11b, anti-CD45, and anti-Flk2), BD (Rat anti-mouse Ly6A/E-PE, CD117-APC, CD34-FITC, anti-CD3, and anti-CD19). Mouse lineage cell depletion kit was obtained from Miltenyi Biotech.
Animals
Atg7f/f mice kindly provided by Dr. Komatsu were crossed with Vav-Cre mice (from Jackson Laboratories) [21] to obtain Atg7f/f:Vav-Cre mice [12]. Histological and biological analyses were performed for both male and female mice at 8–11 weeks of age. The mouse experiments were performed according to the protocols approved by the Institutional Animal Use and Care Committee of Soochow University. In each experiment with mice, 6–10 mice for each treatment group were used.
Human samples
Bone marrow and peripheral blood samples were collected from patients and unaffected volunteer donors. The leukemia stem cells and progenitor cells were obtained by fluorescence-activated cell sorting (FACS) following magnetic-activated cell sorting (MACS) isolation of lineage negative cells. All samples were collected after obtaining informed consent, following the guidelines approved by the Ethics Committee on the Use of Human Subjects at the Soochow University.
Cell lines
Human leukemia cell lines NB4 and K562, obtained from ATCC, and GFP-LC3/K562, generated in this study, were maintained in the RPMI medium 1640 supplemented with 10% fetal bovine serum (Hyclone).
Treatment with chemical compounds
Rapamycin treatment
Rapamycin was reconstituted in dimethyl sulfoxide (DMSO) or ethanol as a carrier at 10 mg/mL and diluted in 5% Tween-80 and 5% PEG-400. For in vivo treatment, mice received 4 mg/kg rapamycin by intraperitoneal injection every other day; for in vitro treatment in premature hematopoietic cells, 1 μM of rapamycin was used.
3-MA treatment
K562 and/or NB4 cells were treated with early-phase autophagy inhibitor 3-MA at 5 mM to inhibit autophagy by blocking autophagosome formation through the inhibition of type III phosphatidylinositol 3-kinases.
Baf-A1 treatment
K562 and NB4 cells were treated with late-phase autophagy inhibitor Baf-A1 at 1 nM to prevent maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes.
DAPT treatment
The γ-secretase inhibitor DAPT was dissolved in DMSO: 40 μM for in vitro treatment of human cell lines K562 and NB4 and 10 μM for ex vivo treatment of lineage-negative cells isolated from mouse bone marrow of wild-type and autophagy-essential gene knockout mice.
Blood routine examination
The assay was done as we previously described [13]. Briefly, 20 μL mouse peripheral blood was added into 500 μL CPK-303A solution (37°C), and then, blood routine examination was performed using Sysmex KX-21N.
Benzidine staining of erythroid cells
The protocol was described previously [22]. Briefly, erythroid differentiation was scored by the benzidine staining method for determination of the percentage of hemoglobin-positive K562 cells induced by rapamycin. Briefly, cells were washed twice and then resuspended in 20 μL phosphate-buffered saline (PBS). Ten microliters of benzidine solution (0.2% benzidine, 0.6% H2O2, 0.5 M acetic acid) was added and incubated for 2 min at room temperature in the dark. Benzidine-positive cells (blue-black staining) were quantitated by light microscopy. At least 100 cells were counted in triplicate for each condition.
Wright-Giemsa staining
Morphology was evaluated by Wright-Giemsa staining as previously described [13]. Briefly, cytospin preparations of 2 × 105cells were incubated sequentially in solution A for 1 min and solution B for 7 min, washed with water, air-dried, and then examined under an Olympus microscope.
Flow cytometry and cell sorting
The flow cytometric protocol was modified from a previously described method [23]. Specifically, to determine the effects of DAPT or rapamycin treatment on the cell differentiation, human leukemia cells were pelleted by centrifugation for 5 min at 400 g. The cells were then washed twice in PBS before incubation with FITC-conjugated anti-CD11b antibody for 30 min and analyzed by the use of a FACS Calibur flow cytometer (BD Biosciences). Mouse bone marrow cells were isolated from the iliac crests, tibiae, and femurs of wild-type and Atg7 knockout mice. Lin-negative cells were purified by MACS, followed by cell sorting for isolation of pluripotent, multipotent, and oligopotent hematopoietic stem cells [24] with BD FACS AriaIII cell sorter.
For the activated-Notch 1 analysis, K562 cells were fixed with a fix/permeabilization buffer (ADG) and then incubated with anti-activated-Notch1 antibody and PE-conjugated secondary antibody (eBiosciences), respectively, for 30 min at room temperature. The samples were analyzed by flow cytometry after the removal of unbound antibodies. In addition, mouse LSK and LK cells were sorted by costaining with anti-Sca-1 and anti-c-Kit. Lineage-negative cells were isolated with a lineage depletion kit (Miltenyi Biotech).
Immunoblotting
Cells were cultured and were lysed in a lysis buffer (Cell Signaling) containing protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail (Roche) on ice for 30 min. Crude lysates were obtained by centrifugation, centrifuged at 13,000 g for 20 min, and heated at 95°C for 5 min at 4°C. The protein was detected by immunoblotting. Equal amounts of protein were loaded on a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidenedifluoride membrane (Millipore). Anti-cleaved Notch1, anti-4EBP1, anti-p-4EBP1, anti-Numb, or anti-LC3 polyclonal antibodies were revealed using an appropriate horseradish peroxidase-conjugated secondary antibody (Cell Signaling) and detected by an enhanced chemiluminescence kit (Pierce). In conjunction, blots were probed with anti-GAPDH or anti-Histone H3 Monoclonal antibody (Cell Signaling) to confirm equal loading of protein.
Semiquantitative reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA from K562 cells was extracted with Trizol (Life Technologies, Inc.), and 1 μg used to synthesize cDNA using the RevertAid First Strand Kit (Fermentas). The RT-PCR primers used in this study were human β-actin, the forward primer was 5′-AAGAGCTACGAGCTGCCTGACG-3′, and the reverse primer was 5′-CGCCTAGAAGCATTTGCGGTGG-3′; human a-globin, the forward primer was 5′-CTGGAGAGGATGTTCCTGTCCTTG-3′, and the reverse primer was 5′-CAGCTTAACGGTATTTGGAGGTCAT-3′; human r-globin, the forward primer was 5′-ACTCGCTTCTGGAACGTCTGA-3′, and the reverse primer was 5′-GTATCTGGAGGACAGGCACT-3′; human Notch1, the forward primer was 5′-GCGGCCGCCTTTGTGCTTCTGTTC-3′, and the reverse primer was 5′-GCCGGCGCGTCCTCCTCTTCC-3′; human Notch2, the forward primer was 5′-TCGTGCAAGAGCCAGTTACCC-3′, and the reverse primer was 5′-AATGTCATGGCCGCTTCAGAG-3′; human Notch3, the forward primer was 5′-AAGTTACCCCCAAGAGGCAAGTGTT-3′, and the reverse primer was 5′-AAGGAAATGAGAGGCCAGAAGGAGA-3′; human Notch4, the forward primer was 5′-AGCAGACAAACTGCAGTGGA-3′, and the reverse primer was 5′-CTGTTGTCCTGGGCATCTTT-3′; human Jagged1, the forward primer was 5′-AGCGACCTGTGTGGATGAG-3′, and the reverse primer was 5′-GGCTGGAGACTGGAAGACC-3′; human Jagged2, the forward primer was 5′-TCTCTGTGAGGTGGATGTCG-3′, and the reverse primer was 5′-CAGTCGTCAATGTTCTCATGG-3′; human DLL1, the forward primer was 5′-CCACGCAGATCAAGAACACC-3′, and the reverse primer was 5′- TTGCTATGACGCACTC>ATCC-3′; human DLL2/4, the forward primer was 5′-CCAACTATGCTTGTGAATGTCC-3′, and the reverse primer was 5′-TGTGGAGAGGTCGGTGTAGC-3′; mouse Atg7, the forward primer was 5′-ATGCCAGGACACCCTGTGAACTTC-3′, and the reverse primer was 5′-ACATCATTGCAGAAGTAGCAGCCA-3′; and mouse GAPDH, the forward primer was 5′-GAGCTGAACGGGAAGCTCAC-3′, and the reverse primer was 5′-ACCACCCTGTTGCTGTAGC-3′.
Real-time PCR analysis
As described previously, total RNA was extracted using the Trizol reagent (Invitrogen) and real-time PCR analysis was performed. The real-time PCR primers used in this study were human Notch-1, the forward primer was 5′-CAC TGT GGG CGG GTC C-3′, and the reverse primer was 5′-GTT GTA TTG GTT CGG CAC CAT-3′; human Cyclin D1, the forward primer was 5′-CTGGCCATGAACTACCTGGA-3′, and the reverse primer was 5′-CTCCGCCTCTGGCATTTTGG-3′; human c-myc, the forward primer was 5′-GGTGCTCCATGAGGAGACAC-3′, and the reverse primer was 5′-GCAGAAGGTGATCCAGACTC-3′; human Hey2, the forward primer was 5′-AGATGCTTCAGGCAACAGGG-3′, and the reverse primer was 5′-CAAGAGCGTGTGCGTCAAAG-3′; human Atg5, the forward primer was 5′-CAATCTGTTGGCTGTGGG-3′, and the reverse primer was 5′-GCTGTTTCGTCCTGTGGC-3′; human Atg7, the forward primer was 5′-GGCCTTTGAGGAATTTTTTGG-3′, and the reverse primer was 5′-ACGTCTCTAGCTCCCTGCATG-3′; human Beclin1, the forward primer was 5′-ACCGCAAGATAGTGGCAGAA-3′, and the reverse primer was 5′-GCGACCCAGCCTGAAGTTAT-3′; human p62, the forward primer was 5′-ATCGGAGGATCCGAGTGT-3′, and the reverse primer was 5′-TGGCTGTGAGCTGCTCTT-3′; human LC3, the forward primer was 5′-ACCATGCCGTCGGAGAAG-3′, and the reverse primer was 5′-ATCGTTCTATTATCACCGGGATTTT-3′; human β-actin, the forward primer was 5′-CATCGAGCACGGCATCGTCA-3′, and the reverse primer was 5′-TAGCACAGCCTGGATAGCAAC-3′; mouse Hes1, the forward primer was 5′-GAGCACAGAAAGTCATCAAAGCC-3′, and the reverse primer was 5′-TCTCTAGCTTGGAATGCCGG-3′; mouse Hey1, the forward primer was 5′-AATGGAAACTTGAGTTCGGCG-3′, and the reverse primer was 5′-TGTTATTGATTCGGTCTCGTCG-3′; mouse Hey2, the forward primer was 5′-TGAAGATGCTCCAGGCTACAGG-3′, and the reverse primer was 5′-CCACTTCTGTCAAGCACTCTCG-3′; mouse Atg5, the forward primer was 5′-GCCAAGAGTCAGCTATTTGACGTTG-3′, and the reverse primer was 5′-CTTGGATGGACAGTGTAGAAGGTCC-3′; mouse Atg7, the forward primer was 5′-CAGTTTCCAGTCCGTTGAAGTCCT-3′, and the reverse primer was 5′-GGGTCCATACATCCACTGAGGTTC-3′; mouse Beclin1, the forward primer was 5′-GGCCAATAAGATGGGTCTGA-3′, and the reverse primer was 5′-CACTGCCTCCAGTGTCTTCA-3′; mouse p62, the forward primer was 5′-TGAAACATGGACACTTTGGCT-3′, and the reverse primer was 5′-ACATTGGGATCTTCTGGTGGA-3′; mouse LC3, the forward primer was 5′-ACTGCTCTGTCTTGTGTAGGTT-3′, and the reverse primer was 5′-TCGTTGTGTGCCTTTATTAGTGCATC-3′; mouse cyclin D1, the forward primer was 5′-TTGTGCATCTACACTGACAAC-3′, and the reverse primer was 5′-GAAGTGTTCGATGAAATCGT-3′; mouse c-myc, the forward primer was 5′-GCTGTAGTAATTCCAGCGAGAGACA-3′, and the reverse primer was 5′-CTCTGCACACACGGCTCTTC-3′; mouse p21, the forward primer was 5′-ACCAGCCTGACAGATTTCTA-3′, and the reverse primer was 5′-TGACCCACAGCAGAAGAG-3′; mouse p27, the forward primer was 5′-AGTGTCCAGGGATGAGGA-3′, and the reverse primer was 5′-GGGAACCGTCTGAAACAT-3′; mouse DLL1, the forward primer was 5′-GGTTGCTCTGTGTTCTGCCG-3′, and the reverse primer was 5′-GTTGGTCATCACACCCTGGC-3′; mouse DLL4, the forward primer was 5′-AAGAATAGCGGCAGTGGTCG-3′, and the reverse primer was 5′-GATGAGAGAGTTTCCTGGCG-3′; mouse Jagged1, the forward primer was 5′-TGGATTCAAGTGTGTGTGCC-3′, and the reverse primer was 5′-GGAAGGCAATCACAGTAGTAGC-3′; mouse Jagged2, the forward primer was 5′-GCTTTGAATGCCACTGTCCG-3′, and the reverse primer was 5′-AGATGCACTCGAAGCCGTCC-3′; and mouse GAPDH, the forward primer was 5′-ACAGTCAAGGCCGATAATGG-3′, and the reverse primer was 5′-TCTCCATGGTGGTGAAGACA-3′. Real-time PCR was performed with SYBR Green Master Mix Reagent (ABI and Roche) using ABI 7500 and LightCycler480® Real-Time PCR detector.
Hierarchical clustering analysis
Hierarchical clustering and heat map of a subset of genes were performed using MEV software, according to a previously described method [25]. Red refers to upregulation of gene expression and green refers to downregulation of gene expression. The Pearson correlation coefficients were computed after principal component analysis using SPSS software.
Cell transfection
To knock down the Notch effector RBPJ-1 expression by RNA interference, lineage negative cells were cultured in StemSpan SFEM (Stem Cell Technologies) supplemented with 20 ng/mL mouse stem cell factor and 20 ng/mL human thrombopoietin. siRNAs for RBPJ-1 and its corresponding control were transfected into lineage negative cells using Lipofectamine RNAiMAX (Invitrogen). The effects of siRNA on the expression of RBPJ-1 were evaluated by semiquantitative RT-PCR 48h after transfection. The target sequences for siRNA were designed and synthesis by GenePharma.
Colony-forming cell assay
The colony-forming cell (CFC) assay was performed as previously described [26]. Briefly, lineage negative cells were first isolated from wild-type, Atg7f/+:Vav-Cre, and Atg7f/f:Vav-Cre mice. Cells were then seeded in methylcellulose (Stem Cell, Canada) with DMSO (carrier), 10 μM DAPT at a density of 3,000 cells per dish, and cultured for 7 days. At the end, cells were counted under light microscopy. The graph shows the mean number of colonies counted on day 7. *P < 0.05;**P < 0.01.
Confocal microscopy analysis
Confocal microscopy was conducted primarily according to a previously described method [27]. Briefly, K562 GFP-LC3 cells were subjected to cytospin centrifugation and before incubation with antibodies, cells were fixed for 15 min with 4% paraformaldehyde at room temperature, permeabilized for 30 min with 0.25% Triton X-100 (Sigma-Aldrich), and blocked for 30 min in 1% bovine serum albumin. Incubation overnight at 4°C with the primary antibodies was followed by the secondary antibody for 1 h at room temperature. After incubation with 10 μg/mL Hoechst, confocal laser scanning of fixed cells was performed on an Olympus Multiphoton Laser Scanning Microscope. The experiments were repeated three times.
Statistical analyses
Statistical analyses were performed with a two-tailed unpaired t-test. P < 0.05 was considered statistically significant. *P < 0.05; **P < 0.01.
Results
Autophagy defect-caused bone marrow hematopoietic stem cell differentiation failure is associated with upregulation of Notch signaling
To explore the mechanistic connection between autophagy and hematopoietic stem cell differentiation, we generated a conditional knockout mouse model using a Vav promoter-directed Cre gene expression to conditionally delete atg7, an autophagy-essential gene [21], in the hematopoietic system (Atg7f/f:Vav-Cre). This resulted in an autophagy defect in hematopoietic stem cells and spleen cells, manifested by the failure in LC3 lipidation in the targeted cells (Fig. 1A).
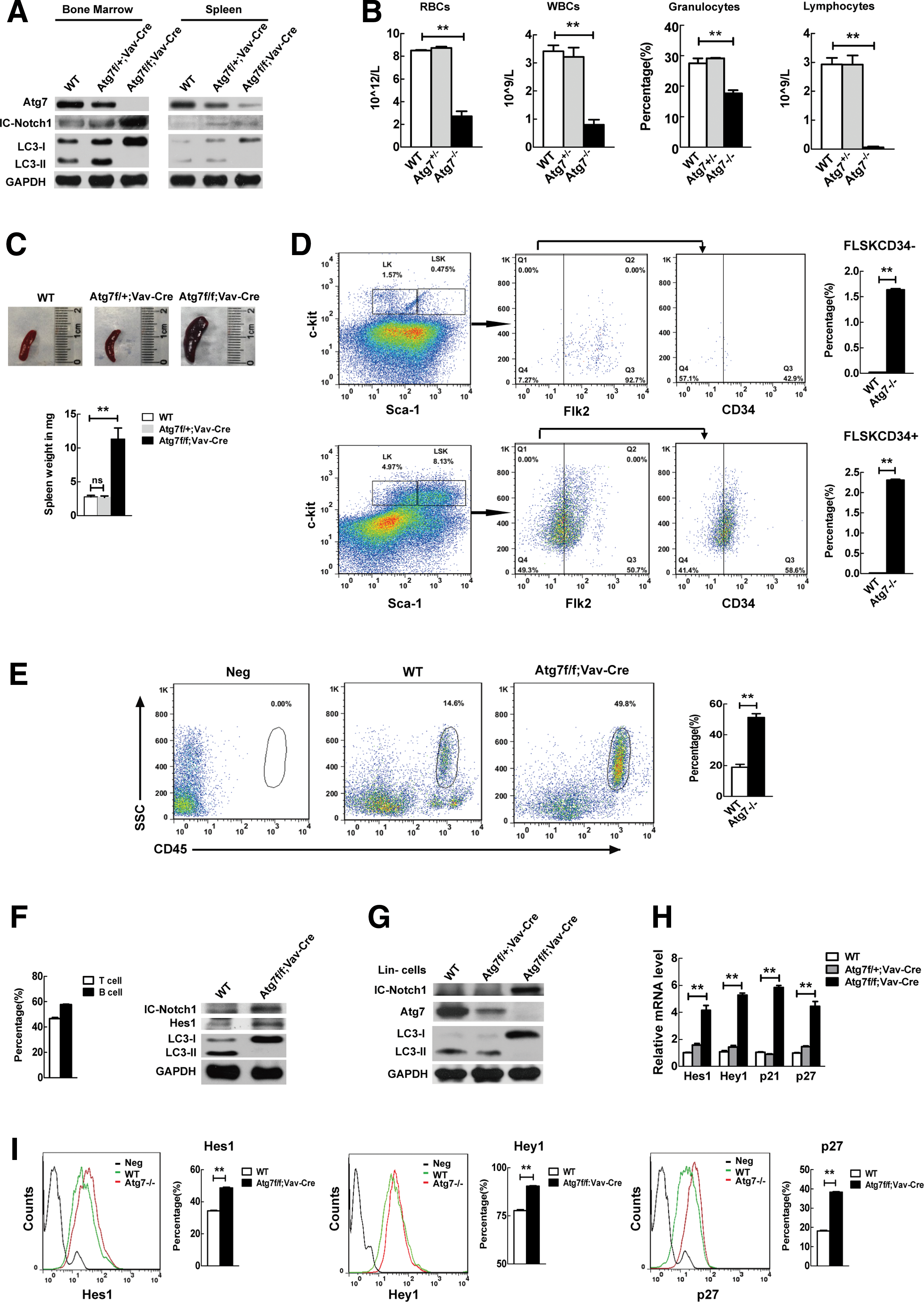
Autophagy defect-caused hematopoietic stem cell differentiation failure is associated with upregulation of Notch signaling.
Blood routine examination and flow cytometric analysis of cell surface markers on the bone marrow hematopoietic cells have demonstrated that the terminally differentiated mature blood cell types, including red blood cells, white blood cells, granulocytes, and lymphocytes, were strikingly reduced in number in the Atg7−/− mice, apparently due to the conditional autophagy defect in hematopoietic stem cells (Fig. 1B). The autophagy defect in hematopoietic stem cells resulted in bone marrow hematopoiesis failure, which in turn leads to extramedullary hematopoiesis, shown by abnormal spleen (Fig. 1C) and strikingly elevated percentage of long-term hematopoietic stem cells (FLSKCD34−) and short-term hematopoietic stem cells (FLSKCD34+) in the spleen of Atg7f/f:Vav-Cre mice (Fig. 1D). Furthermore, an abnormal blast population (leukocyte common antigen CD45+) was detected in the Atg7−/− mouse and constituted more than 50% of the total leukocytes (Fig. 1E), indicating a leukemic phenotype due to the autophagy defect.
Notably, the autophagy defect almost completely abolished the generation of lymphocytes (Fig. 1B), suggesting that the hematopoietic stem cell lineage differentiation toward lymphocytes is more dependent on autophagy. It has been reported that abnormal activation of Notch signaling causes impaired lineage differentiation toward T lymphocytes and occurrence of T-cell acute lymphoblastic leukemia (ALL) [28], and acute promyelocytic leukemia (APL) [29]. Surprisingly, the autophagy defect in hematopoietic stem cells also disclosed an upregulated IC-Notch1 protein, an active form of Notch (Fig. 1A, second lane of the left panel) in total bone marrow cells. To further examine whether the autophagy defect caused multilineage differentiation failure links to upregulated Notch signaling, we next measured the Notch level in spleen that is rich in lymphocytes, since T and B cells account for the majority of all spleen cells (Fig. 1F, left panel), as opposed to the lymphocytes virtually undetectable in peripheral blood in the conditional autophagy-defective mice (Fig. 1B). The results showed that the lymphocytes isolated from the splenocytes of Atg7−/− mice also had an elevated IC-Notch1 (Fig. 1F, right panel). Hes 1, a direct target of IC-Notch, was also upregulated due to the autophagy defect (Fig. IF, right panel). Similarly, IC-Notch was again upregulated in the bone marrow lineage negative cells of autophagy-defective mice (Fig. 1G). To further examine whether upregulated IC-Notch enhances Notch signaling, we measured the transcription levels of Notch target genes of hematopoietic stem and progenitor cells by RT-PCR, and the results show that Hes1, Hey1, p21, and p27, previously all reported as Notch targets [30 –32], were upregulated when autophagy was defective in hematopoietic stem cells (Fig. 1H). The elevated protein levels of the Notch targets Hes1, Hey1, and p27 in the atg7 −/− hematopoietic stem and progenitor cells detected by flow cytometry (Fig. 1I) are consistent with the above transcriptional results.
Therefore, the above data indicate that autophagy defect causes upregulated Notch signaling, which is associated with bone marrow hematopoietic stem cell differentiation failure.
Physiological upregulation of autophagy-essential genes correlates with reduced Notch signaling during mouse hematopoietic stem cell differentiation progression
Our data from the conditional knockout mouse model prompted us to investigate a possible physiological correlation between autophagy and Notch signaling in the regulation of hematopoietic stem cell differentiation. To test this possibility, we measured autophagy potential activity by detecting the expression levels of a group of autophagy-essential genes and also measured Notch signaling activity by detecting the expression level of a group of IC-Notch target genes, during the sequential stages of stem cell differentiation in the hematopoietic hierarchy in mice. This included hematopoietic stem cells (Lin−Sca-1+c-Kit
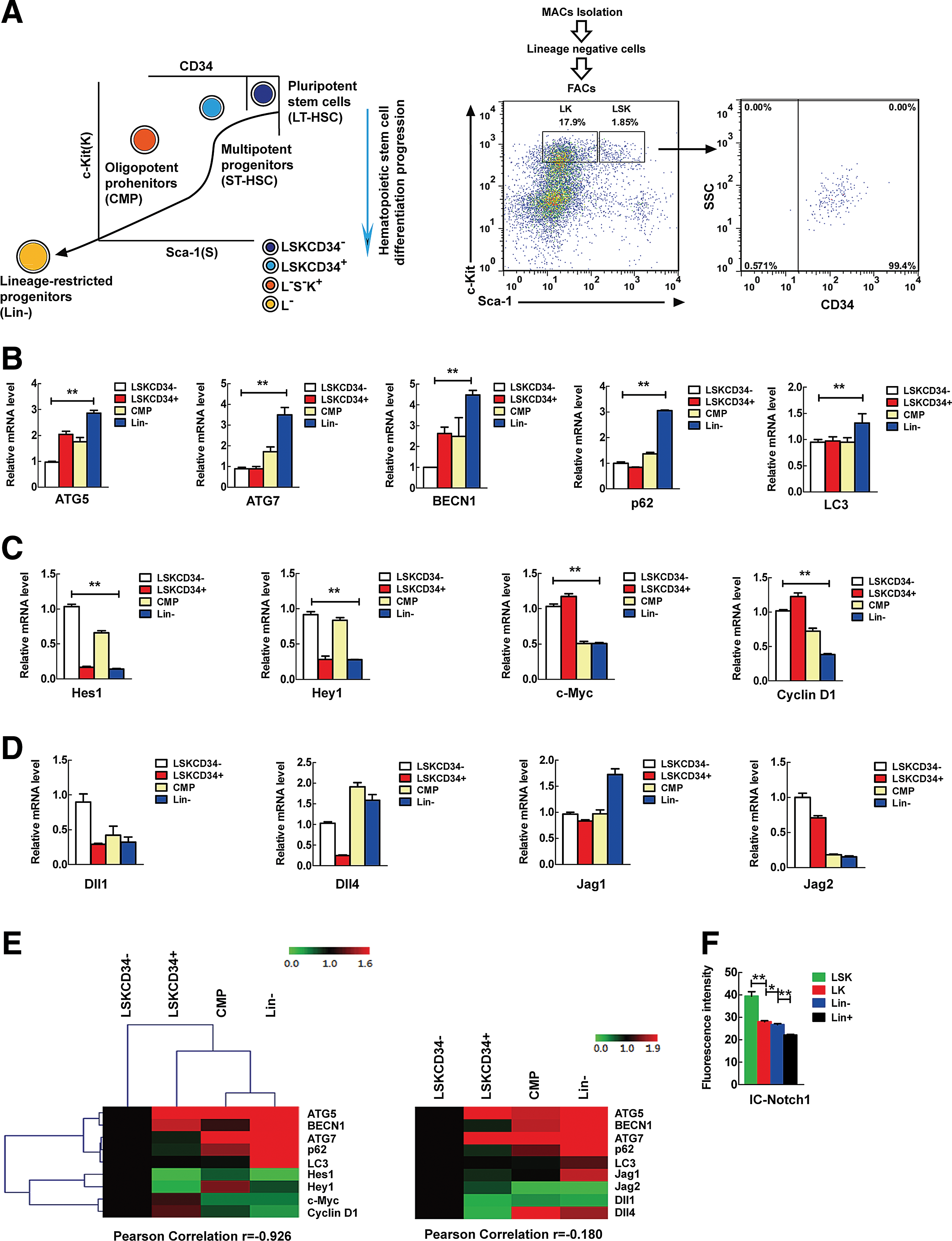
Upregulation of autophagy-essential gene expression correlates with downregulation of Notch signals during hematopoietic differentiation progression.
Meanwhile, we observed that Notch signaling, represented by the expression of Notch target genes, was progressively downregulated in general along with hematopoietic stem cell differentiation progression, although each Notch target gene displayed a different expression pattern (Fig. 2C). However, the Notch ligand genes did not show consistent expression changes during hematopoietic stem cell differentiation (Fig. 2D). This data suggest that the downregulation of Notch signaling is more likely required for stem cell differentiation progression.
Based on the above data, we speculated whether autophagy upregulation is linked to the physiological downregulation of Notch signaling and performed correlation analysis by means of hierarchical clustering to investigate this possibility. The results show that autophagy-essential gene expression is negatively correlated with the expression of Notch target genes (Fig. 2E, left panel). In contrast, the expression of autophagy-essential genes does not obviously correlate with that of Notch ligands in a consistent manner (Fig. 2E, right panel).
Furthermore, flow cytometric analysis disclosed that IC-Notch protein level is progressively reduced in hematopoietic stem cell differentiation progression from LSK to Lin + population (Fig. 2F), which is contrary to the dynamic pattern of autophagy activity (Fig. 2B) and agrees with the Notch target expression detected by RT-PCR (Fig. 2C).
These physiological data suggest that autophagy may downregulate Notch signaling, but not through a negative regulation of Notch ligands, during the physiological differentiation of hematopoietic stem cells.
Pathological downregulation of autophagy-essential genes correlates with elevated Notch signaling in impaired hematopoietic system
The blockage of stem cell differentiation in hematopoietic system may lead to leukemia [33]. Recent studies have shown that Notch is abnormally activated in T-cell ALL [28], and abnormal activation of Notch1 signaling maintains APL [29]. We thus speculated that abnormal activation of Notch signaling may be involved in the inhibition of hematopoietic stem and progenitor cell differentiation. Given the observations that autophagy defects caused differentiation failure and upregulated Notch signaling (Fig. 1), we evaluated whether autophagy in the stem cells of leukemia patients is downregulated and if the upregulation of Notch signaling is associated with this downregulation. Specifically, we measured the expression of a group of autophagy-essential genes collectively reflecting the autophagy activity and also a group of Notch target genes in hematopoietic stem cells from patient samples, including acute myeloid leukemia (AML) and ALL. Both these leukemia types are the result of a stem cell differentiation blockade. Quantitative PCR analysis revealed that, compared with human normal bone barrow mononuclear cells, autophagy-essential genes are significantly downregulated in hematopoietic stem cells (CD34+CD38−) as well as mature B cells (CD34−CD19+) from AML and ALL patients (Fig. 3A, B), demonstrating a markedly reduced pathological autophagy activity in dysfunctional hematopoietic differentiation in AML and ALL patients, which is in contrast to the autophagy activity in chronic leukemia cells such as primary CD34+ CML cells [34], suggesting that different types of leukemias and different stem cell populations may differ in the autophagy activity. Importantly, these cells that were defective in both autophagy and differentiation also displayed upregulated c-myc or Hey2 (Fig. 3C), both of which are targets of Notch 1 [35,36].
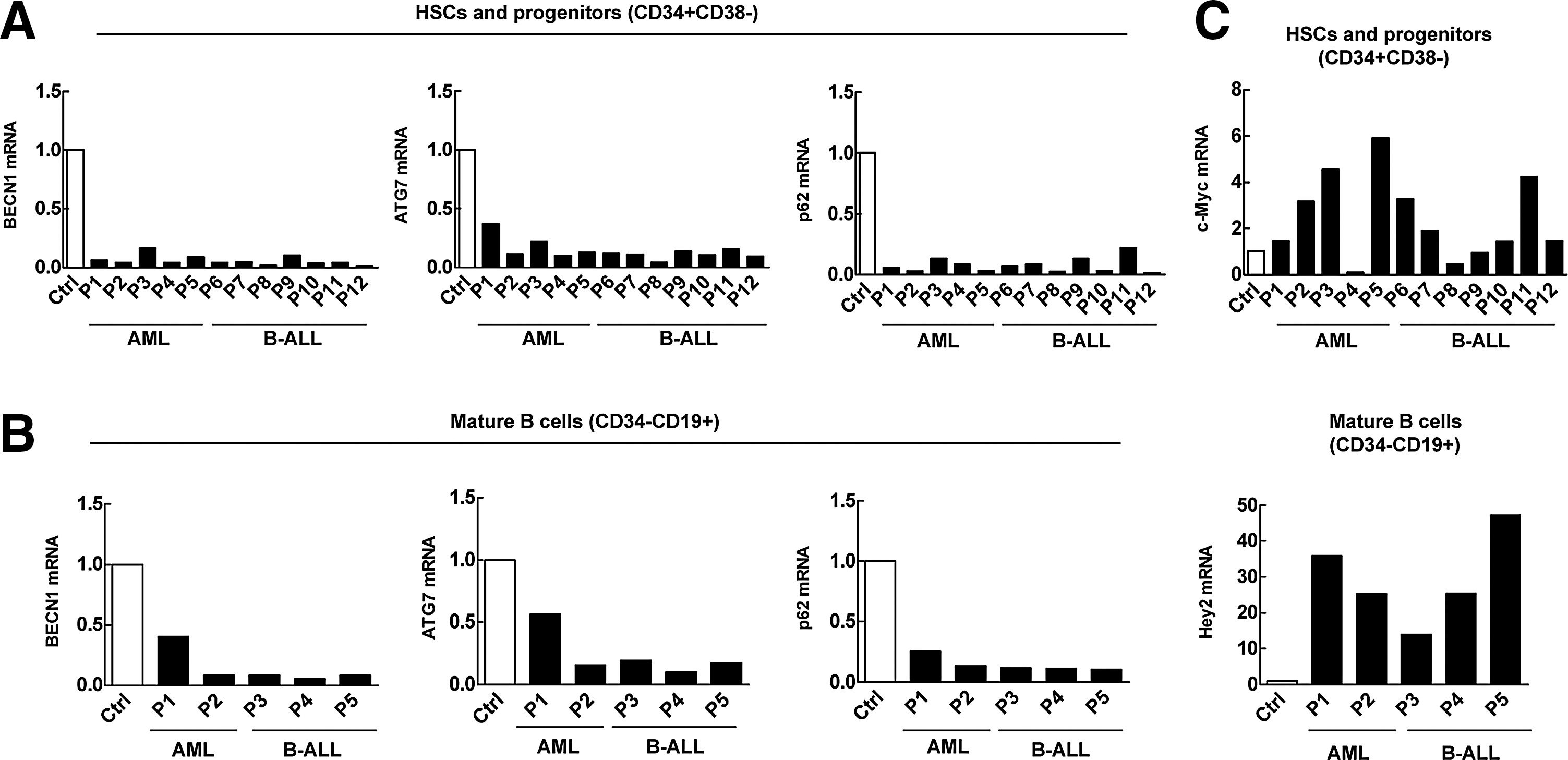
Low autophagy-essential gene expression is associated with upregulation of Notch targets in differentiation-blocked malignant hematopoietic cells.
The above data, thus suggested the pathological relevance of autophagy in the regulation of Notch signaling and hematopoietic stem cell differentiation. Impaired autophagy in hematopoietic stem and progenitor cells of dysfunction is pathologically associated with upregulation of Notch signaling and may underlie the differentiation blockage that is a major cause of acute hematological malignancies.
Autophagy directly targets IC-Notch
While Notch signaling is necessary for embryonic hematopoietic stem cell development, it is considered dispensable for its role in adult hematopoietic stem cells or to be inhibitory to adult hematopoietic stem cell multilineage commitment when it remains abnormally activated [19, 20, 37]. How Notch is reduced or Notch signaling is weakened, but not diminished, in hematopoietic stem cells during differentiation progression remains unknown. Our finding that autophagy was inversely correlated with Notch signaling during hematopoietic stem cell differentiation progression (Figs. 1 and 2) and pathologically low autophagy is associated with upregulated Notch signaling (Fig. 3) prompted us to examine if Notch signaling is directly attenuated by autophagy in the multilineage differentiation cascade. Notch signaling is known to be initiated when the Notch receptor is activated by ligand-induced proteolysis [38]. The hierarchical nature of normal hematopoiesis is well conserved in hematopoietic malignancies such as human AML or chronic myelogenous leukemia (CML), two types of hematopoietic stem cell malignancies. K562, derived from a CML patient, is a multipotent hematopoietic malignant cell line capable of differentiating into lineage-restricted progenitors of erythrocytes, granulocytes, and monocytes; whereas NB4 is a malignant oligopotent progenitor cell line and has a capacity to mature into functional polynuclear cells. They can thus be used as in vitro models to study the biology of hematopoietic differentiation. We thus used DAPT, a γ-secretase inhibitor, which prevents Notch cleavage and activation, to treat these immature hematopoietic cell line models. Treatment with DAPT increased the γ-globin expression (Fig. 4A) and significantly increased the CD11b expression (Fig. 4B), both of which are myeloid differentiation markers. DAPT treatment of these immature hematopoietic cells caused a decreased nuclear–cytoplasmic ratio, which is indicative of mature cells (Fig. 4C). Thus, a blockade of Notch signaling by the γ-secretase inhibitor DAPT induced the myeloid differentiation.
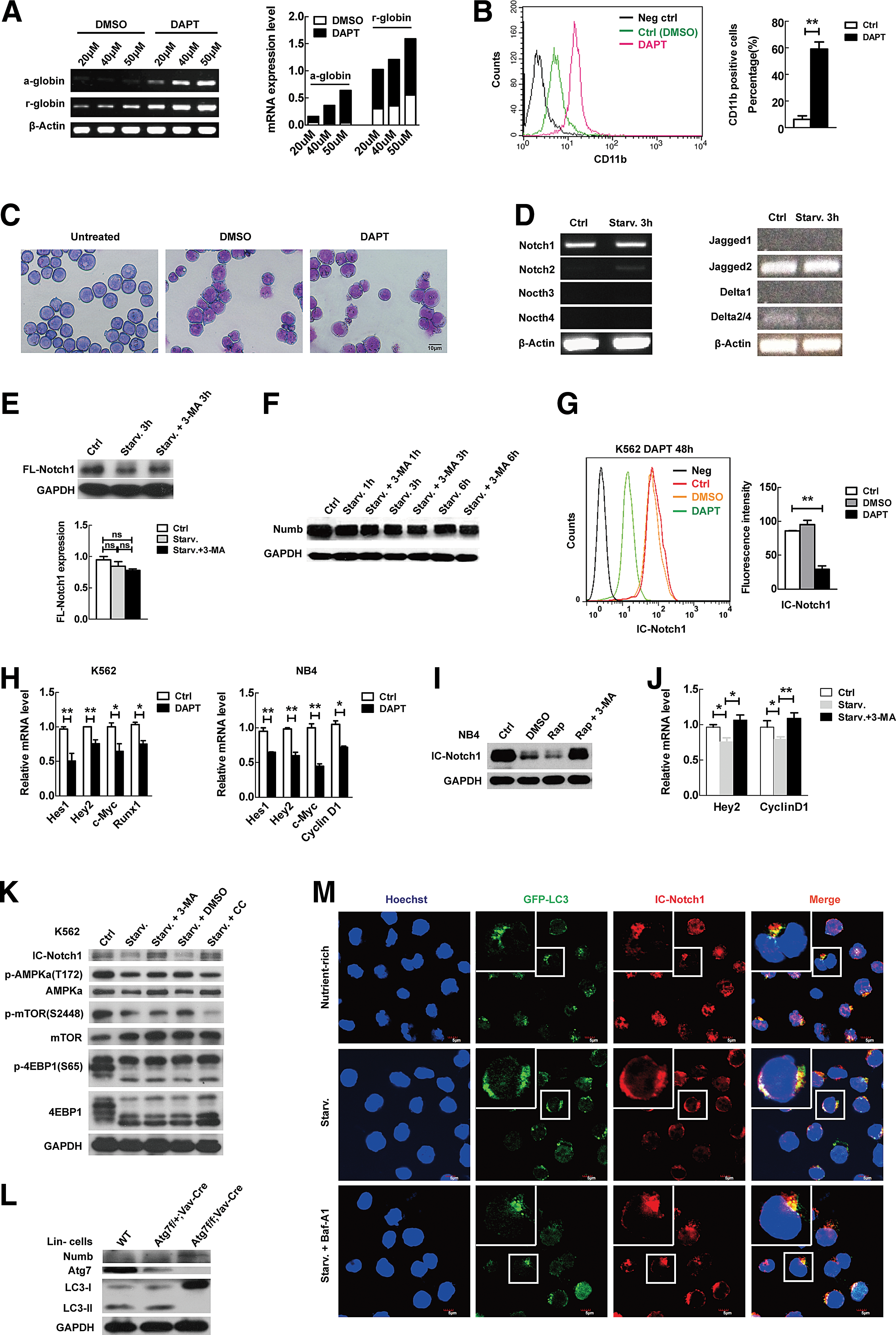
Autophagy targets Notch signaling by degradation of IC-Notch.
Given that the autophagy defect caused upregulated Notch signaling (Fig. 1A, F–I) and the inhibition of Notch signaling caused an enhancement of differentiation (Fig. 4A–C), the latter being mimicking the progressive decrease in Notch signaling that occurs in tandem with physiological hematopoietic stem cell differentiation (Fig. 2), we investigated whether autophagy regulates differentiation through the control of the Notch signaling pathway. Although four Notch receptors have been reported in the mammalian system, only Notch 1 is expressed in K562 cells (Fig. 4D, left panel), and the activation of autophagy by starvation did not significantly affect the transcription level of Notch 1 or its ligands (Fig. 4D), suggesting that the regulation of Notch signaling by autophagy is not at the transcriptional level of Notch and its ligands. Examination of full-length Notch 1 protein when autophagy was activated by starvation or inhibited by 3-MA disclosed that autophagy does not significantly regulate the full-length Notch protein level (Fig. 4E). This suggests that autophagy may not directly target the full-length Notch either by degradation or by inducing the cleavage of full-length Notch to active IC-Notch. Likewise, the activation of autophagy by starvation or its inhibition by 3-MA did not significantly alter the protein level of Numb (Fig. 4F), the upstream regulator of Notch signaling, which prevents the nuclear localization and targets the IC-Notch for degradation through E3 ligase [39]. Taken together, these data from premature hematopoietic cell models suggest that autophagy regulates Notch signaling not through full-length Notch or its upstream regulator, or through the ligands for Notch receptors, but possibly through IC-Notch, the active form of the Notch receptor.
If autophagy regulates multilineage differentiation by regulating IC-Notch, we surmised that the Notch downstream target genes should be responsive to autophagy stimuli. A group of Notch targets has been identified, including the Hes and Hey family of transcriptional repressors, cyclin D1 and c-Myc [30,40,41]. To examine the connection between IC-Notch and its targets in the premature cell line models, we first determined a proper DAPT dose with which the activation of Notch could be efficiently inhibited by blocking Notch processing (Fig. 4G). Indeed, inhibiting Notch inhibited Hes1, Hey2, c-Myc, and cyclin D1 in both NB4 and K562 cells, confirming these genes as bona fide targets of Notch signaling in the immature hematopoietic cell models (Fig. 4H).
To seek for the direct link between autophagy and Notch signaling in the context of hematopoietic differentiation, we next examined the IC-Notch protein level in response to autophagy stimuli. Activation of autophagy of NB4 premature hematopoietic cells with rapamycin clearly reduced IC-Notch, which was reversed by 3-MA (Fig. 4I). We also examined the expression of the Notch targets in response to autophagy. Our data with these premature hematopoietic cells show that activation of autophagy by nutrient starvation causes reduced transcription of Hey2 and cyclin D1 genes, which is again reversed by the inhibition of autophagy with 3-MA (Fig. 4J).
Similarly, IC-Notch protein was significantly reduced in response to starvation, which was reversed by the autophagy inhibitor 3-MA in K562 premature hematopoietic cells (Fig. 4K). Of interest, inhibition of AMPK signaling, an upstream signaling cascade for controlling autophagy, with compound C attenuated the phosphorylation of mTOR and reversed the reduction of IC-Notch caused by autophagy stimulus starvation (Fig. 4K). These signaling data indicate that IC-Notch may be a direct target of autophagic regulation.
Furthermore, in vivo data from bone marrow progenitor cells (Lin− cells) of the mouse model show that, unlike significant upregulation of IC-Notch1 in the autophagy-defective cells (Fig. 1A, G), Numb maintains a relatively lower level in both autophagy intact cells and autophagy-defective cells, the latter being evidenced by the absence of atg7 and failure of LC3 lipidation (Fig. 4L), which is consistent with immature hematopoietic cells revealing an unchanged Numb level (Fig. 4F). The data from the conditional knockout mouse model further suggest that autophagy regulates stem cell differentiation through the regulation of IC-Notch, but not its upstream Numb or its ligands.
To visually examine whether the downregulation of IC-Notch by autophagy is caused by direct autophagic degradation, we constructed a stable transfectant K562 cell line expressing the GFP-LC3 marker. In the absence of autophagy stimuli (nutrient-rich), IC-Notch is localized to the nucleus after cleavage in the cytoplasm. Autophagy stimulation with starvation resulted in an increased distribution of IC-Notch in the cytoplasm and a Notch reduction in the nucleus. Furthermore, autophagy stimulation with starvation caused colocalization of active Notch with GFP-LC3 punctuate dots. This colocalization between GFP-LC3 punctuate dots and IC-Notch could be accumulated by the lysosomal vacuolar H+-ATPase inhibitor Baf-A1 (Fig. 4M), confirming that autophagy machinery directly degrades IC-Notch in its autolysosomes upon an autophagy trigger. The above data support our notion that autophagy promotes hematopoietic stem cell differentiation progression by directly targeting IC-Notch.
The differentiation potential of stem cells is restored when Notch signaling is blocked in a conditional autophagy defective system
To verify whether autophagy promotes stem cell differentiation by targeting Notch signaling, we first performed CFC assay that reflects the reconstitution potential of hematopoietic stem cells, a property which is essential for the differentiation of hematopoietic stem and progenitor cells. We isolated Lin− cells from the bone marrows of the mouse models (Atg7f/f:Vav-Cre, Atg7f/+:Vav-Cre) for a CFC assay (Fig. 5A, left panel). The results showed that partially autophagy-impaired Lin− cells from the atg7+/− mice had a reduced colony-forming ability (Fig. 5A, right panel, dark grey column), and fully autophagy-impaired Lin− cells from the atg7−/− mice show a much more significant colony forming failure (Fig. 5A, right panel, black column). We then abrogated Notch signaling using DAPT, an inhibitor of γ-secretase that processes the full-length Notch precursor to IC-Notch, its active form. The results showed that the inhibition of Notch signaling with DAPT fully restored the colony-forming potential in partially autophagy-impaired mouse Lin− cells (Fig. 5A, right panel, orange column), and the inhibition of Notch signaling also significantly restored the colony-forming potential in fully autophagy-impaired Lin− cells of mice (Fig. 5A, right panel, red column), comparable to the partially impaired Lin− cell level (Fig. 5A, right panel, dark grey column). These CFC data indicate that the restoration of the differentiation potential is more difficult if there has been a homologous ablation of autophagy in extensively dysfunctional stem and progenitor cells. Nevertheless, the CFC data suggest that autophagy regulates hematopoietic stem and progenitor cell differentiation specifically through the targeting of Notch signaling.
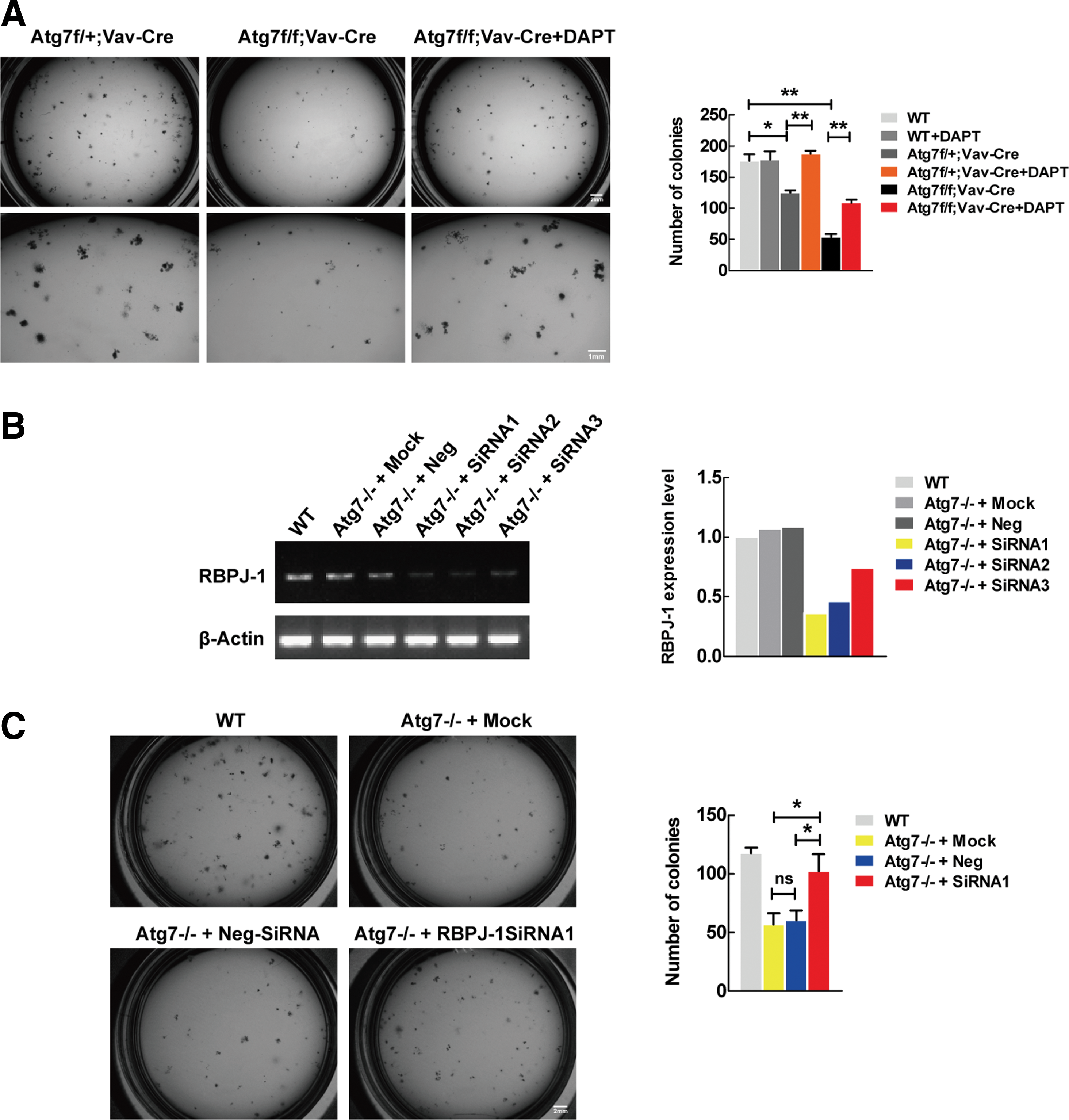
Differentiation capability was restored when Notch signaling was blocked in conditional autophagy defective systems.
To activate its downstream targets, upon cleavage, IC-Notch in the cytoplasm has to translocate to the nucleus, where it forms an active transcriptional complex with transcription factor RBPJκ and coactivators [38,42]. Therefore, the removal or inhibition of RBPJ can more specifically block the Notch signaling. To further confirm the rescued differentiation by pharmacological inhibition on Notch signaling, we performed an RNA silencing experiment against RBPJ gene, and the results show a successful inhibition of RBPJ expression (Fig. 5B). The autophagy defective hematopoietic stem and progenitor cells (sorted against LSK markers) regained the differentiation potential when the RBPJ expression was blocked with the siRNA1 against RBPJ gene (Fig. 5C). Clearly, the genetic data confirmed that it is autophagy that regulates hematopoietic stem cell differentiation progression specifically through the targeting of Notch signaling.
Discussion
Leukemia is mostly derived from hematopoietic stem cell differentiation blockage. Understanding the regulatory mechanisms by which hematopoietic stem cells undergo multilineage differentiation is thus pivotal for the prevention and treatment of leukemia. Autophagy is required for hematopoietic stem cell differentiation, but how autophagy regulates the stem cell differentiation has remained elusive. Our present study addresses a critical aspect of this question. Conditional inactivation of autophagy in mouse hematopoietic stem cells disclosed an upregulated Notch signaling, associated with multilineage differentiation failure (Fig. 1). The physiological autophagy activity in mice is correlated with the downregulation of Notch signaling during the progression of hematopoietic stem cell differentiation (Fig. 2). Screening human leukemia patient samples, which are characterized as having a differentiation blockage at the stem cell level, revealed that a pathologically low autophagy activity is associated with upregulated Notch signaling in dysfunctional hematopoietic stem cells (Fig. 3). These observations together prompted us to investigate whether autophagy truly regulates stem cell differentiation through the downregulation of Notch signaling.
Although studies in other systems have identified Notch as a key player in stem cell maintenance [43], several reports have presumed that the Notch activity is dispensable for this function in the hematopoietic system, while other studies have described opposite observations. Nevertheless, in mammalian species, Notch signaling is not required in the primitive hematopoiesis, but required in the definitive hematopoiesis in embryonic development [17] and has been shown to expand adult hematopoietic stem cells [20,44]. These findings revealed a significant impact of Notch signaling in maintaining a pool of hematopoietic stem cells. Some early studies reported that Notch inhibits myeloid differentiation [37,45], but others reported the opposite [46,47]. Previous studies have also reported that T- versus B-cell fate specification in the thymus depends on Notch signaling [48 –51], but genetic approaches have not identified an obligatory role for Notch signaling in adult hematopoietic stem cells [48,52,53]. A recent study reported that the inhibition of Notch signaling in in vitro hematopoietic stem cells promotes T- or B-cell lineage differentiation [16], together reflecting a complex yet unresolved role of Notch in hematopoietic multilineage differentiation.
Despite the controversy surrounding the role of Notch in adult hematopoietic stem cells in mammalian systems over the years, the prevailing opinion is that Notch signaling is dispensable for certain types of lineage differentiation at certain stages, or abnormally activated Notch signaling is inhibitory to adult mammalian hematopoietic stem cell differentiation. However, this notion may have resulted in less attention being paid to why Notch signaling is not required or weakened in this context and how Notch signaling in hematopoietic stem cells is somehow downregulated in the course of cell fate decisions. Also unanswered has been the biological significance of silenced or reduced Notch signaling in adult hematopoiesis. Notch inhibits myelopoiesis and ex vivo activation of Notch in hematopoietic stem cells blocks myeloid differentiation [54], and the ectopic expression of IC-Notch1 in mouse bone marrow cells caused T-ALL [55]. Recent studies have shown that Notch is highly expressed in T-ALL [28,56,57], and Notch1 signaling is required for the maintenance of APL [29]. Collectively, these results indicate that excessive Notch signaling blocks hematopoietic stem cell differentiation and leads to hematological malignancies. Hence, the overall dynamic picture of the Notch cascade from literatures appears to be such that Notch signaling is active in the definitive hematopoietic stem cells during embryonic development and becomes less active in adult definitive hematopoiesis, particularly in the differentiation progression of hematopoietic stem cells to functional blood cells, and when overactivated, it leads to the blockage in hematopoietic stem cell differentiation, ultimately acute leukemogenesis.
To test whether silencing or downregulation of Notch signaling is necessary for hematopoietic stem cells to undergo differentiation, we first established an assay with which all canonical Notch pathways can effectively be manipulated by specifically blocking the processing of intact Notch to its intracellular active form and subsequently validated bona fide Notch targets in the hematopoietic system (Fig. 4H). Our results indicated that autophagy downregulates Notch signaling (Fig. 4I–L). Furthermore, the conditional autophagy defect in our mouse model led to an elevated IC-Notch (Fig. 1A, F, G), and inhibition of AMPK-mTOR, a major signaling pathway inhibiting autophagy, reversed the reduction of IC-Notch by autophagy stimulus starvation (Fig. 4K). Confocal microscopic data indicate the colocalization of IC-Notch in autolysosomes in response to starvation, which was further pronounced by inhibition on autophagic degradation (Fig. 4M), suggesting that autophagy machinery directly degrades IC-Notch. Furthermore, we applied an ex vivo reconstitution assay with a conditional autophagy defect mouse model to verify the autophagy-Notch-differentiation axis. Indeed, our dada revealed that the differentiation potential of stem cells was restored when Notch signaling was blocked pharmacologically (with DAPT) or genetically (with RBPJ knockdown) in the conditional autophagy defective system (Fig. 5).
It should be noted that autophagic downregulation of IC-Notch attenuates, but does not diminish Notch signaling, and Notch signaling remains and continues along with multilineage differentiation progression (Fig. 2C, F), which is believed to confer certain lineage differentiation that requires relatively low level of Notch signaling. Nevertheless, we can conclude that it is autophagy that regulates hematopoietic stem cell differentiation, specifically through its targeting of IC-Notch to limit Notch signaling. To our knowledge, this is the first evidence that autophagy targets a critical signaling protein to regulate stem cell function.
In summary, we propose a mechanism by which autophagy sustains hematopoietic stem cell differentiation through the direct targeting of IC-Notch (Fig. 6). Our results indicate that in the cause of hematopoietic stem cell differentiation after birth, autophagy targets Notch signaling to sustain adult hematopoiesis. Autophagy dysfunction is attributed to the differentiation blockades, which are often the cause of hematological malignancies. Therefore, our present findings provide a critical insight into the current mechanistic understanding of physiological and pathological connections between autophagy and hematopoietic stem cell differentiation, thereby proposing a novel mechanism by which autophagy sustains hematopoiesis and thus protects against leukemogenesis.
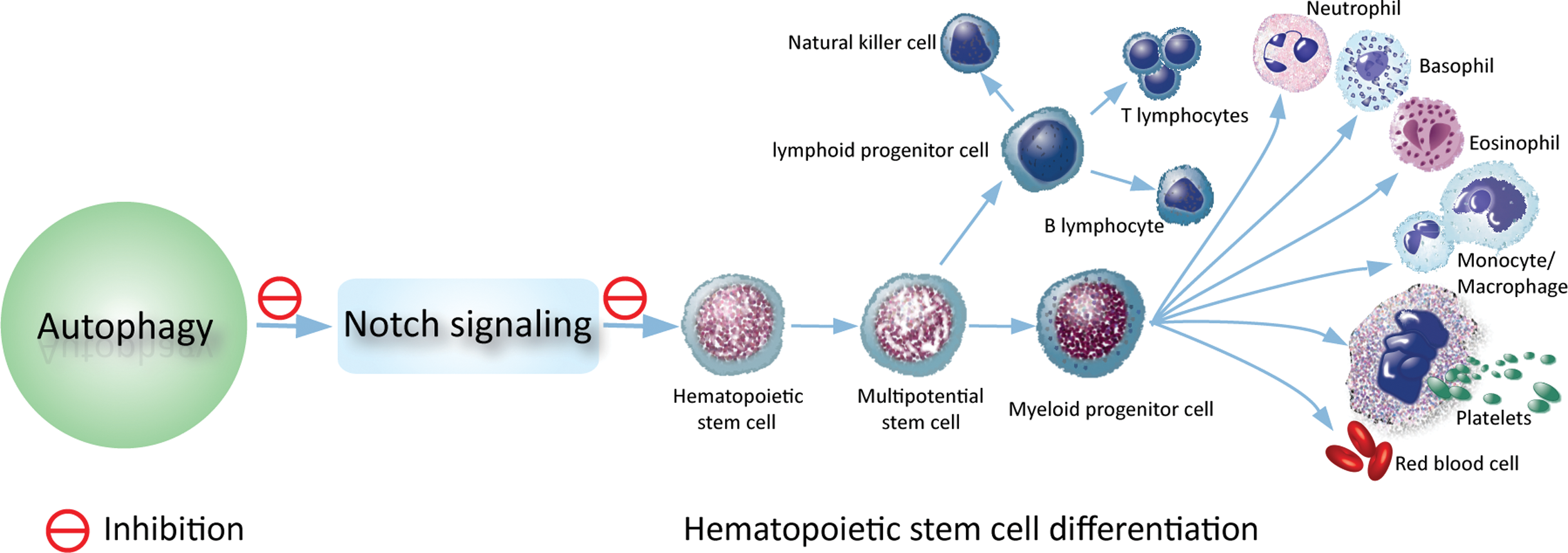
Autophagy-Notch axis regulating hematopoietic stem cell differentiation. A carton illustrating the mechanism by which autophagy regulates hematopoietic stem cell differentiation. In the cause of hematopoietic stem cell differentiation, autophagy is progressively upregulated to limit the IC-Notch level, thereby sustaining bone marrow hematopoiesis. Color images available online at
Footnotes
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (no. 31071258, no. 81272336, no. 31271526 and no. 31201073), The Ministry of Science and Technology of China (no. 2011CB512101), The Department of Science and Technology of Jiangsu Province of China (no. BK20130333), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author Disclosure Statement
The authors declare no competing financial interests.