
Abstract
Germ cells occupy a unique place in development through their requirement to maintain underlying totipotency while producing highly differentiated gametes. This is reflected in the expression of key regulators of pluripotency in the fetal germline and the ability of these cells to form pluripotent stem cells and germ cell tumors. Culture of whole fetal testes, including germ cells, provides a key model for studying gonad and germline development, but it is critical that such models mimic physiological development as closely as possible. We aimed to determine the effects of differing culture conditions, including serum-free and serum-containing conditions, on fetal germ cell and testis development. We tested a commonly used model that employs knockout serum replacement (KSR) to provide more defined culture conditions than media containing fetal bovine serum (FBS). In FBS conditions, cell cycle parameters in germ and Sertoli cells closely resembled normal development. In contrast, KSR significantly inhibited male germ cell entry into mitotic arrest, a key milestone in male germline development. Moreover, KSR disrupted molecular control of cell cycle and inhibited the transcription of a range of male germ cell differentiation markers. In the somatic compartment, KSR stimulated proliferation and inhibited differentiation in Sertoli cells. These data demonstrate that KSR substantially alters germ and somatic cell differentiation in fetal testis culture and should not be used to replicate normal gonadal development. In contrast, basal media with or without serum support germ and supporting cell differentiation and cell cycle dynamics that are in line with in vivo characteristics.
Introduction
L
Gonadal sex differentiation leads to the establishment of a sex-specific supporting environment that promotes male or female germline development and leads to the production of male and female hormones that direct sexual differentiation of the individual. In mice, male development is initiated in chromosomally XY individuals when the undifferentiated gonad commits to testicular development in response to sex-determining region Y (Sry) expression in pre-Sertoli cells at around embryonic day (E)11.5 [1,2]. This key event results in a cascade of gene expression changes that are required for testes development and germ cell commitment to a male sexual fate [3].
Sex-determining region Y-box 9 (Sox9) is upregulated shortly after Sry [4,5] and leads to the expression of masculinizing factors between E11.5 and E12.5, including fibroblast growth factor 9 (Fgf9) [6 –9], inhibin beta A (Inhba; encoding the activin A subunit) [10], anti-Müllerian hormone (Amh) [11], prostaglandin D2 synthase (Ptgds) [12], doublesex and mab-3-related transcription factor 1 (Dmrt1) [13], the steroid hormone pathway genes, Cyp11a1 (encoding P450scc) and Cyp17a1 [14,15], and the retinoic acid-metabolizing gene, Cyp26b1 [16 –18].
Germline development is initiated with specification of primordial germ cells at approximately E7.0 [19]. These early germ cells express core regulators of pluripotency and remain highly proliferative during migration along the hindgut toward the genital ridge and during their entry into the bipotential gonad at E10.5 [20]. At this stage, the germ cells express high levels of the pluripotency genes, Oct4, Sox2, and Nanog [21 –23], and begin to express mouse VASA homolog (MVH), which is specifically expressed in germ cells of both sexes from E10.5 until after birth [24 –28]. After entering the gonad, germ cells undergo sex-specific differentiation, which depends on male and female-promoting signals produced by the neighboring somatic cells in the differentiating gonad.
Male primordial germ cells (PGCs) begin to express determinants of male germline differentiation, including developmental pluripotency-associated 4 (Dppa4), which is maintained in male PGCs and repressed in female PGCs after E12.5 [22,29], and members of the piRNA pathway, including piwi-like RNA-mediated gene silencing 1 (Piwil1), Piwil4, and tudor domain containing 9 (Tdrd9) [30,31]. In addition, germ cells undergo extensive epigenetic programming after colonization of the gonad, which involves regulation of genes such as the histone-modifying genes, euchromatic histone-lysine N-methyltransferase 2 (Ehmt2) and enhancer of zeste 2 (Ezh2), and the DNA methyltransferase cofactor Dnmt3L [32].
Once the germ cells have entered the gonad, they continue to proliferate until such time that they receive cues to enter mitotic arrest (males) or meiosis I (females) [33]. Mitotic arrest occurs relatively synchronously between E12.5 and E14.5 in XY mouse germ cells, with some variation in timing according to the genetic strain [27,29]. Cell cycle genes are dynamically regulated during the time of male germ cell entry into mitotic arrest, which is associated with repression of Cyclin E1 (Ccne1) and Cyclin E2 (Ccne2), and upregulation of members of the cyclin-dependent kinase inhibitors, including p15INK4b (Cdkn2b) and p27KIP1 (Cdkn1b) [27]. During entry into mitotic arrest, Sox2 and Nanog are transcriptionally repressed, while OCT4 protein levels are substantially reduced [28,34]. The biological processes that regulate cell cycle, pluripotency, and differentiation in male germ cells are not only essential for normal spermatogonial development but are also considered important for preventing the formation of germ cell tumors [33].
We and others have previously used ex vivo whole gonad culture to study fetal gonad development, including the role of signaling pathways in regulating gonadal sex differentiation, germ cell mitotic arrest, and entry into meiosis [6,16,17,24,35 –39]. Importantly, whole gonad culture preserves gonadal tissue integrity, maintaining cell contact and signaling capability between somatic and germ cells. Nevertheless, the surrounding molecular environment is likely to be significantly altered ex vivo. Therefore, it is important to establish the best possible cellular environment in culture to obtain data that are most relevant to the in vivo cell situation.
Clearly, it is also advantageous to develop a culture system using defined conditions where possible, allowing for more certain interpretation and increased reproducibility of experimental outcomes. The use of fetal bovine serum (FBS) to supplement culture medium with essential growth factors, macromolecular proteins, and nutrients is common. However, this practice introduces the possibility that batches vary (and the requirement to continually monitor this) and the caveat that sera contain unknown components that cannot be accounted for in experimental outcomes [40].
Past studies of germ cell development, the culture of whole gonads, or whole embryos have employed a range of conditions, including the use of FBS, serum replacement, or simple Dulbecco's modified Eagle's medium (DMEM) in which FBS or serum replacement is omitted [6,16,17,24,35 –39,41 –46]. Media containing serum replacement typically include knockout serum replacement (KSR), a synthetically produced alternative to FBS that is widely used to provide a more defined, serum-free culture environment. However, KSR is not fully chemically defined and contains a considerable quantity of animal protein (albumin) and natural and synthetic micronutrients [47].
KSR has been widely used to enhance the in vitro derivation of undifferentiated mouse embryonic germ (EG) cell colonies from E8.5 to E12.5 migrating and gonadal germ cells [48 –51]. Typically, KSR is used at concentrations ranging from 2% to 20% in manufacturer-recommended complementary medium, knockout DMEM (KO DMEM). The cellular mechanisms whereby KSR exerts improved EG colony derivation are not known, yet its positive effects on both derivation and maintenance of EG colonies indicate a role, or roles, in the control of cell cycle, maintenance of pluripotency, and/or inhibition of differentiation.
In this study, we examine the effect of using media supplemented with FBS or KSR in ex vivo fetal gonad cultures over a 72 h developmental period from E12.5 that is characterized by well-defined developmental milestones in germ and Sertoli cells [24,27,29]. Using flow cytometric and molecular approaches, we demonstrate that the transcriptional and developmental profiles of germ and somatic cells are substantially altered under different culture conditions, especially those containing KSR.
Materials and Methods
Ethics
All animal experiments were conducted under approval from Monash University Animal Ethics Committees.
Gonad collection and culture
Fetuses were collected at E12.5 from Swiss females mated to 129T2svJ Oct4-GFP transgenic males. Mice were housed under 12 h light/12 h dark cycles. The presence of a vaginal plug indicated mating and was used to determine E0.5 of pregnancy. Fetuses were collected on E12.5 and sexed based on gonadal morphology, with males identified by the presence of clearly visible testis cords. Embryos were morphologically examined to confirm developmental stage consistent with E12.5 [29]. All cell culture reagents were purchased from Life Technologies unless otherwise stated. Mesonephros was removed and whole testes were cultured on 30-mm organotypic cell culture inserts (PICM03050; Merck Millipore) in 1,200 μL of the following seven culture treatment media: DMEM:F12K with GlutaMAX containing 0%, 2%, or 10% FBS, or KO DMEM supplemented with 1× GlutaMAX and containing 0%, 2%, 10%, or 15% KSR. In follow-up experiments, male E12.5 gonads were cultured in KO DMEM supplemented with 15% KSR or
Flow cytometry
Single gonads were collected from each condition and prepared for flow cytometry for each biological replicate. A detailed protocol for gonad collection, dissociation, fixation, and antibody staining for flow cytometry has been previously published [35]. For cell cycle analyses, 0.02 mM ethynyl deoxyuridine (EdU; Click-iT® EdU Alexa Fluor® 647 Flow Cytometry Assay Kit; Life Technologies) was added to the culture medium 2 h before culture cessation. After culture, gonads were washed in phosphate-buffered saline (PBS), dissociated to single-cell suspension in 0.25% trypsin containing ethylenediaminetetraacetic acid (EDTA), and passed through a 35-μm nylon mesh filter (BD Biosciences) to remove tissue debris.
Cells were collected by centrifugation, fixed in 4% paraformaldehyde for 15 min, and washed in wash reagent (supplied in the EdU Click-iT Kit). Nonspecific binding sites were blocked in 10% normal donkey serum (Sigma) for 20 min at room temperature (RT). As the buffers used in EdU staining abolish enhanced green fluorescent protein (eGFP) fluorescence, we used an MVH-specific antibody to separate germ cells for cell cycle analyses performed with EdU [24,29,35]. Cells were incubated in rabbit anti-MVH (1:200, D10C5; Cell Signaling Technology) and goat anti-AMH (1:50, SC-6886; Santa Cruz) primary antibodies for 1 h at RT. Cells were washed 3× in wash reagent solution and incubated in donkey anti-rabbit Alexa Fluor 488 (1:300, A21206; Life Technologies) and donkey anti-goat biotin (1:500, A16009; Life Technologies) secondary antibodies for 45 min at RT. Cells were washed 2× in wash reagent and incubated in EdU Click-iT reaction cocktail containing Alexa Fluor 647 azide fluorescent dye for 30 min at RT. Cells were washed 2× in wash reagent and incubated in streptavidin-Pacific Blue (1:1,000, S-11222; Life Technologies) for 30 min at RT. Cells were resuspended in wash reagent containing 0.05 mg/μL propidium iodide (PI; Sigma) and subjected to flow cytometry using the BD FACSCanto II analyzer (BD Biosciences). Controls included single-channel gonad-positive samples for the MVH and AMH antibodies, a single-channel cultured mesonephros (+EdU) sample for EdU staining, and a negative uncultured mesonephros sample for the MVH and AMH antibodies and EdU staining.
Flow cytometry was additionally used to determine phospho Histone 2AX (γH2AX) expression intensity in male germ cells exposed to KSR. Germ cells were isolated using goat anti-MVH (1:100, AF2030; R&D Systems), and γH2AX expression in germ cells was measured using rabbit anti-γH2AX (1:50, #2577; Cell Signaling Technology). Respective secondary antibodies used were donkey anti-goat Alexa Fluor 488 (1:300, A11055; Life Technologies) and donkey anti-rabbit biotin (1:500, A16027; Life Technologies)/streptavidin-Pacific Blue (1:1,000, S-11222; Life Technologies). To provide a positive control for staining, cultured XX control gonads (E12.5 + 72 h) were also included in the analysis of the meiotic marker, γH2AX. Flow cytometry was also used to measure the germ cell levels of OCT4 and E-cadherin (CDH1) protein across culture conditions. Germ cells were gated based on eGFP signal detected from the Oct4-GFP transgene. OCT4 and E-cadherin mean intensity levels were measured in the gated germ cell population using goat anti-OCT4 (1:100, SC-8628; Santa Cruz) and rabbit anti-E-cadherin (24E10, 1:100, 3195; Cell Signaling Technology). Respective secondary antibodies used were donkey anti-goat biotin (1:500, A16009; Life Technologies)/streptavidin-Pacific Blue (1:1,000, S-11222; Life Technologies) and donkey anti-rabbit Alexa Fluor 647 (1:300, A31573; Life Technologies). Flow cytometric analysis was performed using FlowJo 5.4+ Software (FlowJo, LLC.).
Fluorescence-activated cell sorting
For each media condition, two male gonads were pooled per biological replicate (n = 5/media condition) for FACS purification. After the culture period, gonads were washed in PBS, dissociated to a suspension of single cells in 0.25% trypsin containing EDTA, and passed through a 35-μm nylon mesh filter (BD Biosciences). The cell suspension was FACS purified into viable cells that were GFP+ (>95% purity; germ cell) and GFP− (somatic cell) using the BD Influx™ Cell Sorter (BD Biosciences). Cell viability determined by PI staining was >99% in germ cell populations and >95% in somatic cell populations across all treatment groups (Supplementary Fig. S1; Supplementary Data are available online at
Gene expression analyses
FACS-sorted germ and somatic cells were processed using the Cells-to-cDNA™ II Kit (Invitrogen). Briefly, sorted cells were resuspended in Cell Lysis II Buffer to a density of 2,500 GFP+ germ cells per microliter and 5,000 GFP− somatic cells per microliter. Cell suspensions were subjected to DNAse I treatment to remove genomic DNA before reverse transcription. For cDNA synthesis, 5 μL of cell suspension was added to each reverse transcription reaction, including 1× reverse transcription buffer, 5 μM random decamers, 0.5 mM dNTPs, 0.5 U/μL RNase inhibitor, and 0.5 μL M-MLV reverse transcriptase in a 10 μL final volume. Reactions were incubated at 42°C for 1 h, then 95°C for 10 min, and cDNA samples were stored at −20°C.
Gene expression was compared using real-time quantitative polymerase chain reaction (qPCR) with BioMark HD technology (Fluidigm) between DMEM, KO DMEM, KO DMEM 2% KSR, and KO DMEM 15% KSR. GFP+ and GFP− cDNA samples were run separately on two 48.48 Dynamic Array IFCs (Fluidigm). A full list of TaqMan assays used in the Biomark HD runs is presented in Supplementary Tables S1 and S2. A total of 33 and 24 assays were included in each Biomark HD run for germ cell (GFP+) and somatic cell (GFP−) cDNAs, respectively. These assays included cell type-specific markers, cell cycle genes, and a selection of epigenetic modifiers we have shown to be expressed in fetal germ cells at these stages of development [27,28]. To confirm germ cell purity, a Sertoli-specific negative control gene, Sox9, was included in the germ cell assays. Similarly, to confirm somatic cell purity, the germ cell-specific marker, Ddx4 (encodes MVH), was included in the somatic cell assays. In both runs, these negative control assays were measured below the limit of detection. Reference genes included Canx, Sdha, and Mapk1, which we have shown previously to be expressed at stable levels in E12.5–E15.5 fetal germ cells and somatic cells [26]. CT values obtained from each reference gene for each sample were highly correlated (R > 0.95 in all cases); therefore, the geometric mean of all three reference genes was used to normalize gene expression data using the ΔΔCT method.
Statistical analysis
A one-way ANOVA plus Tukey's post hoc test was used to statistically analyze the effect of medium preparation on biological outcome (PRISM software v6.0e; GraphPad). Where data are presented graphically, statistically significant (P < 0.05) post hoc outcomes between each group are represented by differing letters (a, b, and c).
Results
Testis morphology is normal across culture conditions
Gonads were cultured for a period of 72 h from E12.5 and photographed under bright-field and fluorescence optics to detect GFP (Supplementary Fig. S2). Testis cords containing GFP+ germ cells were clearly visible across medium conditions and testis morphology and cord architecture did not appear to differ among groups (DMEM:F12K 0% and 10% FBS, and KO DMEM 0% and 15% KSR shown; Supplementary Fig. S2). We observed that gonads cultured without the presence of protein (FBS or KSR) tended to adhere and lie more flatly on the membrane, thereby occupying a larger surface area, whereas the addition of protein to the culture led to a more rounded three-dimensional constitution.
Male germ or Sertoli cell cycle dynamics are not affected by FBS
Cell cycle is strictly regulated in both the germ cell and pre-Sertoli cell populations in the developing testis, with germ cells entering mitotic arrest and pre-Sertoli cells undergoing rapid proliferation [27,29,52]. To determine the cell cycle state in germ, pre-Sertoli and other somatic cells in the developing testis cell proliferation and cell cycle stage were analyzed by measuring a combination of EdU incorporation during DNA synthesis and PI staining to identify DNA content (ie, 2N or 4N), allowing G0/G1, S-phase, and G2/M stages of the cell cycle to be distinguished (Fig. 1A). In vivo, >98% of male germ cells (from mice of a similar genetic background) enter mitotic arrest by E14.5 [27,29]. In this study, cell proliferation in E12.5 gonads cultured ex vivo for 72 h in the presence of 0%, 2%, or 10% FBS in DMEM:F12K medium was similar to proliferation levels at E14.5 in vivo. Germ cells (MVH-labeled) entered mitotic arrest, with just 0%–2% germ cells incorporating EdU in DMEM:F12K media and no significant change with the addition of FBS (Fig. 1A-i, ii, and B-ii). Consistent with this, there were no significant alterations in cell proportions in G0/G1 (Fig. 1B-i) or G2/M stages in these treatments (Supplementary Fig. S3A). These data confirm that under these culture conditions, all germ cells enter mitotic arrest as expected from in vivo analyses. In addition, Sertoli cell (AMH-labeled) proliferation (15%–20%) was similar across all DMEM:F12K culture conditions (15%–20%; Fig. 1C-ii). Significantly, this proliferation index is similar to that observed for Sertoli cells in vivo [52] (P.S. Western, unpublished data). Moreover, the proportion of Sertoli cells in G0/G1 (Fig. 1C-i) and G2/M (data not shown) was not changed in the DMEM:F12K background ± FBS. Interestingly, the presence of FBS in the culture medium had a minor stimulatory effect on proliferation of the mixed interstitial cell population (MVH/AMH double-negative; Supplementary Fig. S4). These data indicate that the addition of FBS as a medium supplement for the ex vivo culture of the mouse fetal testes generally provides cell proliferation and cell cycle state outcomes that are similar to those that occur in vivo.
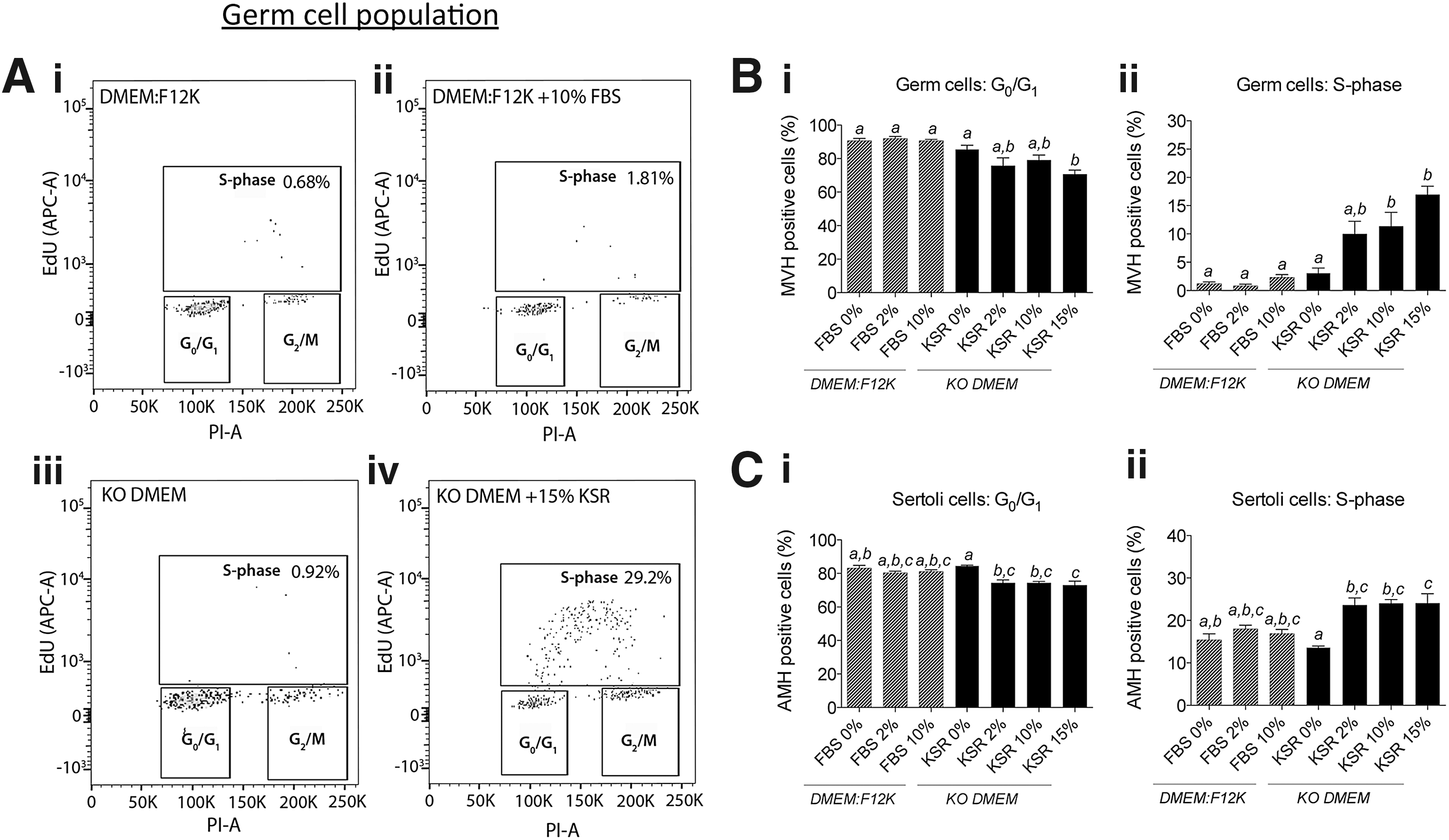
Knockout serum replacement (KSR) stimulates both male germ cell and Sertoli cell proliferation in ex vivo fetal testis culture. Germ cell cycle was assessed using flow cytometry where S-phase was determined using ethynyl deoxyuridine (EdU) incorporation during culture, and G0/G1 and G2/M stages were identified by propidium iodide (PI) staining to give DNA content. Representative images of flow cytometry analyses in germ cells [mouse VASA homolog (MVH)-labeled] isolated from E12.5 male gonads cultured for 72 h in
Entry of male germ cells into mitotic arrest is disrupted by KSR
To determine the suitability of KSR for ex vivo gonad cultures, E12.5 male gonads were cultured in the presence of 0%, 2%, 10%, and 15% KSR, concentrations that are widely used in culture in other studies [48,50,51,53]. Flow cytometric analyses of the cell cycle stage revealed a significant role for KSR in promoting gonadal cell proliferation in cultured fetal testes. KSR supplementation of KO DMEM culture medium resulted in a dose-dependent and highly significant increase in the proportion of proliferating male germ cells after 72 h of culture (Fig. 1B-ii). There was a robust shift in germ cells incorporating EdU (S-phase) with the addition of 2% KSR (10.0% germ cells incorporating EdU; non-significant), 10% KSR (11.4% germ cells incorporating EdU; P < 0.05), and 15% KSR (17.0% germ cells incorporating EdU; P < 0.0001) compared with DMEM:F12K (1.2% germ cells incorporating EdU) and KO DMEM (3.1%; not significantly different to DMEM:F12K) media (Fig. 1B-ii). The observed increase in germ cell proliferation in 15% KSR conditions was mirrored by reciprocal losses in the proportion of germ cells in G0/G1 (Fig. 1B-i).
Male germ cell entry into G2/M was not altered by culture media containing FBS or KSR, and unlike meiotic female germ cells, the proportion of XY PGCs in G2/M remained very low (Supplementary Fig. S3A). In addition, quantitative flow cytometric analysis demonstrated that meiotic marker, γH2AX, remained at baseline levels in XY PGCs of gonads cultured in any of the FBS or KSR conditions, while robust levels of γH2AX were detected in PGCs of control XX gonads (Supplementary Fig. S3B). To further examine whether basal medium background might influence male germ cell development, we tested the effect of KSR in DMEM:F12K on cell cycle parameters. To our surprise, KSR was much less effective at maintaining male germ cell proliferation in DMEM:F12K (Supplementary Fig. S5A) than in KO DMEM (Fig. 1B-ii).
KSR promotes proliferation of Sertoli cells
The proliferation-promoting effects of KSR on germ cells were also observed in Sertoli cells of fetal testes cultured with KSR in KO DMEM. In E12.5 gonads cultured for 72 h in KO DMEM, the proportion of Sertoli cells in S-phase was 13.6%; however, in the presence of 2%, 10%, and 15% KSR, this increased to ∼24% in each case (Fig. 1C-ii). Consistent with the increased proportion of cells in S-phase, significant reciprocal decreases in Sertoli cells residing in G0/G1 were also observed after gonad culture in KSR compared with KO DMEM alone (Fig. 1C-i). Very few Sertoli cells resided in the G2/M stage of the cell cycle and these proportions were not significantly altered by the culture condition studied (data not shown).
Consistent with these findings, while there were no detectable effects of basal medium type on Sertoli cell cycle dynamics (Fig. 1C), we assessed the effect of KSR in DMEM:F12K base media (Supplementary Fig. S5B) and observed similar effects to using KO DMEM base media. In DMEM:F12K, KSR increased Sertoli cell proliferation from 12.8% (DMEM:F12K alone) to 21.7% (15% KSR; P < 0.05; Supplementary Fig. S5B).
KSR alters male germ and somatic cell cycle transcriptional profiles
We next determined whether gene expression profiles were affected in germ (GFP+) and somatic (GFP−) cells FACS purified from fetal testes cultured in basal media or KSR-containing medium. Consistent with the cell cycle data, culture of gonads with KSR resulted in significant dysregulation of cell cycle genes in both germ and somatic cells (Fig. 2 and Supplementary Tables S1 and S2). Proliferation of germ cells is marked by the G1-S cyclins, Ccne1 and Ccne2, and these genes are repressed as germ cells enter mitotic arrest [27]. In germ cells cultured with 15% KSR, Ccne1 transcription was 5-fold higher than in germ cells cultured with KO DMEM alone (P < 0.001; Fig. 2A-i). Similarly, Ccne2 was significantly higher in germ cells exposed to 15% KSR (P < 0.05; Fig. 2A-ii) compared with basal media. In addition, transcription of the S-phase marker, proliferating cell nuclear antigen (Pcna), was higher in germ cells cultured in 2% KSR (P < 0.05) and 15% KSR (P < 0.001; Fig. 2A-iii) compared with basal media. Furthermore, transcription of Cdkn2b encoding p15INK4b, an inhibitor of G1-S progression, was lower in germ cells isolated from gonads cultured in 2% and 15% KSR (P < 0.01; Fig. 2A-iv). A number of other cell cycle genes were not altered by KSR in germ cells, including Cyclin D2 (Ccnd2) and Cyclin D3 (Ccnd3), Cdkn1b, encoding the G1-S inhibitor p27KIP1, and cyclin-dependent kinase inhibitor 1c (Cdkn1c; encoding the G1-S inhibitor p57KIP2; Supplementary Table S1).

KSR promotes expression of proliferation genes and repression of cell cycle inhibitor genes in germ and somatic cells in ex vivo fetal testis culture. Gene expression analyses were conducted using Biomark microfluidic quantitative polymerase chain reaction (qPCR) in fluorescence-activated cell sorting (FACS)-purified germ [green fluorescent protein (GFP+)] and somatic (GFP−) cell populations from E12.5 male gonads cultured for 72 h in DMEM:F12K medium or KO DMEM supplemented with 0%, 2%, or 15% KSR (n = 5/group). Cell cycle genes altered in the presence of KSR included
In the somatic cell population, genes promoting proliferation, including Ccnd2 (P < 0.01) and Ccne1 (P < 0.05), were upregulated in the presence of 15% KSR (Fig. 2B-i, ii, respectively). Moreover, transcription of both Ccnd2 and Ccne1 appeared to increase in the presence of 2% KSR, although this did not reach statistical significance. Ccne2 transcription was detected at relatively low levels (compared with Ccne1) in somatic cells and was unchanged in the presence of 2% and 15% KSR (Fig. 2B-iii). Unlike germ cells, Cdkn2b (encoding p15INK4b) was not expressed in somatic cells (below the level of detection). However, transcription of Cdkn1c (encoding the cell cycle inhibitor p57KIP2) was 2-fold reduced in KO DMEM and KO DMEM +2% KSR compared with DMEM:F12K medium (P < 0.01), but paradoxically was not affected in the presence of 15% KSR (Fig. 2B-iv). In somatic cells, other genes involved in the cell cycle, including Ccnd3, Cdkn1b, and Pcna, were not affected by KSR in culture medium (Supplementary Table S2).
KSR inhibits male germ and somatic cell differentiation, but does not impact pluripotency factors
Combined, the flow cytometric data and the molecular analyses of cell cycle state demonstrate that germ cells do not enter mitotic arrest properly in the presence of KSR. Since entry into mitotic arrest is a highly robust and key event in male fetal germline differentiation in mice, these data indicate that KSR inhibits male germ cell development. In addition, these data also suggest that KSR affects Sertoli cell differentiation in the male fetal gonad.
To determine whether this is the case, we performed transcriptional analyses of a range of key differentiation markers in germ and somatic cells isolated from gonads cultured in DMEM and KO DMEM +0%, 2%, or 15% KSR. Indeed, KSR repressed the normal transcriptional upregulation of a number of male germ cell differentiation markers. Transcription of male germ cell markers, Dppa4 (Fig. 3A), Piwil1 (Fig. 3B), Piwil4 (Fig. 3C), Tdrd9 (Fig. 3D), Map2k7 (Fig. 3E), and Dnmt3L (Fig. 3F), was significantly and dose-dependently lower in germ cells isolated from gonads cultured in 2% and 15% KSR compared with KO DMEM alone. However, paradoxically Dmrt1, which is required for male germline differentiation [54], was upregulated 3-fold in 2% KSR (P < 0.001) and 5-fold in 15% KSR (P < 0.001; Fig. 3F) compared with KO DMEM without KSR. Other germ cell-specific markers, including Ddx4 (Mvh), Nanos2, Nodal, Pou6f1, Piwil2, Tdrd5, and Map3k5, were unaffected by the selected culture conditions (Supplementary Table S1). Notably, there was no effect of the basal medium type (DMEM:F12K or KO DMEM) on the transcription of any of the germ cell markers used in this study (Fig. 3).

KSR represses markers of male germ cell differentiation and alters epigenetic modifiers in ex vivo fetal testis culture. Gene expression analyses were conducted using Biomark microfluidic qPCR in the FACS-purified germ (GFP+) cell population from E12.5 male gonads cultured for 72 h in DMEM:F12K medium or KO DMEM supplemented with 0%, 2%, or 15% KSR (n = 5/group). Germ cell markers that were significantly altered in the presence of KSR included
As KSR enhanced the proliferation of male germ cells and inhibited their differentiation in this study and has been associated with enhanced pluripotency in other studies [48,50,51], we hypothesized that KSR would maintain expression of pluripotency factors in the male germline. However, neither OCT4 protein levels nor transcription of Oct4, Nanog, or Sox2 was enhanced in gonads cultured in KSR-containing media compared with controls (Supplementary Fig. S6A–D). As retention of pluripotency might indicate that germ cells were being reprogrammed to an overtly pluripotent state, we tested whether expression of the reprogramming marker, E-cadherin [55], was enhanced in germ cells of gonads cultured with cultured KSR. However, E-cadherin levels remained unchanged (Supplementary Fig. S6E). Combined, these data indicate that KSR blocks male germline differentiation and enhances proliferation, but does not maintain the key markers of pluripotency or promote high expression of the reprogramming marker, E-cadherin.
Since male germ cells undergo extensive epigenetic reprogramming during the sex differentiation period [56], we also studied a number of genes involved in adding and removing epigenetic modifications to chromatin and that we have identified to be expressed in the germ cell at this time (P.S. Western, unpublished data). The male germ cell expressed the DNA methyltransferase gene, Dnmt3L, and was particularly impacted in germ cells in the presence of 15% KSR, as evidenced by 13-fold lower transcriptional levels relative to KO DMEM alone (P < 0.001; Fig. 3F). Interestingly, KSR also altered transcription of epigenetic genes encoding histone modifiers, including 2-fold lower levels of the histone 3 lysine 9 (H3K9)-specific methyltransferase, Ehmt2 (P < 0.05; Fig. 3H), and 2-fold higher levels of the histone 3 lysine 27 (H3K27me3)-specific methyltransferase, Ezh2 (P < 0.01; Fig. 3I). Other candidate epigenetic genes selected for analysis, but not altered by the culture conditions, included members of the polycomb repressive complex 2, embyronic ectoderm development (Eed), and suppressor of zeste 12 homolog (Suz12) (Supplementary Table S1). Basal medium type did not significantly influence expression of any epigenetic-related genes studied in germ cells (Fig. 3).
In somatic cells, KSR impacted transcription of Sertoli cell differentiation markers, with significantly lower levels of Amh, Fgf9, and Ptgds in 15% KSR-treated gonads compared with gonads treated with KO DMEM alone (Fig. 4A–C, respectively). Ptgds was not only dose-dependently reduced by KO DMEM +2% KSR (P < 0.01) and 15% KSR (P < 0.0001) compared with KO DMEM alone but also decreased in KO DMEM compared with DMEM:F12K media (P < 0.001; Fig. 4C). These effects appear to be mediated downstream of Sox9 transcription [57] as Sox9 was not significantly altered in KSR compared with control media (Fig. 4D). Despite decreased expression of male Sertoli cell markers, including Fgf9, Ptgds, and Amh, in KSR-exposed gonads, there was no increase in the female gene R-spondin 1 (Rspo1), which is required for ovarian development (Fig. 4E) [58]. Another Sertoli cell marker, Inhba, encoding the activin A subunit, which is upregulated between E12.5 and E15.5 and linked to Sertoli cell proliferation [10,24], was elevated in somatic cells exposed to 15% KSR compared with KO DMEM alone (P < 0.01; Fig. 4F). Interestingly, the Sertoli cell-specific gene, Cyp26b1, which is responsible for metabolizing retinoic acid in the fetal testis and thought to prevent premature male germ cell entry into meiosis [16], was downregulated 2-fold in 2% KSR (P < 0.05) and 15% KSR conditions (Fig. 4G).

KSR dysregulates markers of male somatic cells in ex vivo fetal testis culture. Gene expression analyses were conducted using Biomark microfluidic qPCR in the FACS-purified somatic (GFP−) cell population from E12.5 male gonads cultured for 72 h in DMEM:F12K medium or KO DMEM supplemented with 0%, 2%, or 15% KSR (n = 5/group). Somatic cell markers that were significantly altered in the presence of KSR included
Genes encoding steroidogenic enzymes and therefore representing pre-Leydig cells in the GFP− somatic population were also modified by KSR in ex vivo gonad culture. KO DMEM containing 2% and 15% KSR resulted in a 2-fold increase in Cyp11a1 (encoding P450scc; P < 0.05; Fig. 4H). In contrast, Cyp17a1 was decreased by 2-fold in all KO DMEM, 2%, and 15% KSR conditions compared with DMEM:F12K (Fig. 4I). Genes regulating the epigenetic state, which were studied in the germ cell population, were not altered in the somatic population by any of the culture conditions tested in this study (Supplementary Table S2).
Vitamin C is not the active ingredient promoting KSR-mediated germ cell proliferation
Vitamin C is a known ingredient of KSR [47] that is reported to exert the KSR stimulatory effects in maintaining the stem cell state in embryonic stem (ES) cells [59 –61]. Therefore, in an effort to address the mechanism by which KSR maintains male germ cell proliferation in ex vivo culture, we elected to treat gonads with vitamin C. E12.5 male gonads were cultured for 72 h in KO DMEM containing 15% KSR or vitamin C (Fig. 5) at concentrations consistent with those reported to be present in KSR [47]. Although KSR promoted germ cell proliferation in this experiment, vitamin C did not block entry of male germ cells into mitotic arrest and did not alter any other cell cycle parameter (Fig. 5A–C).
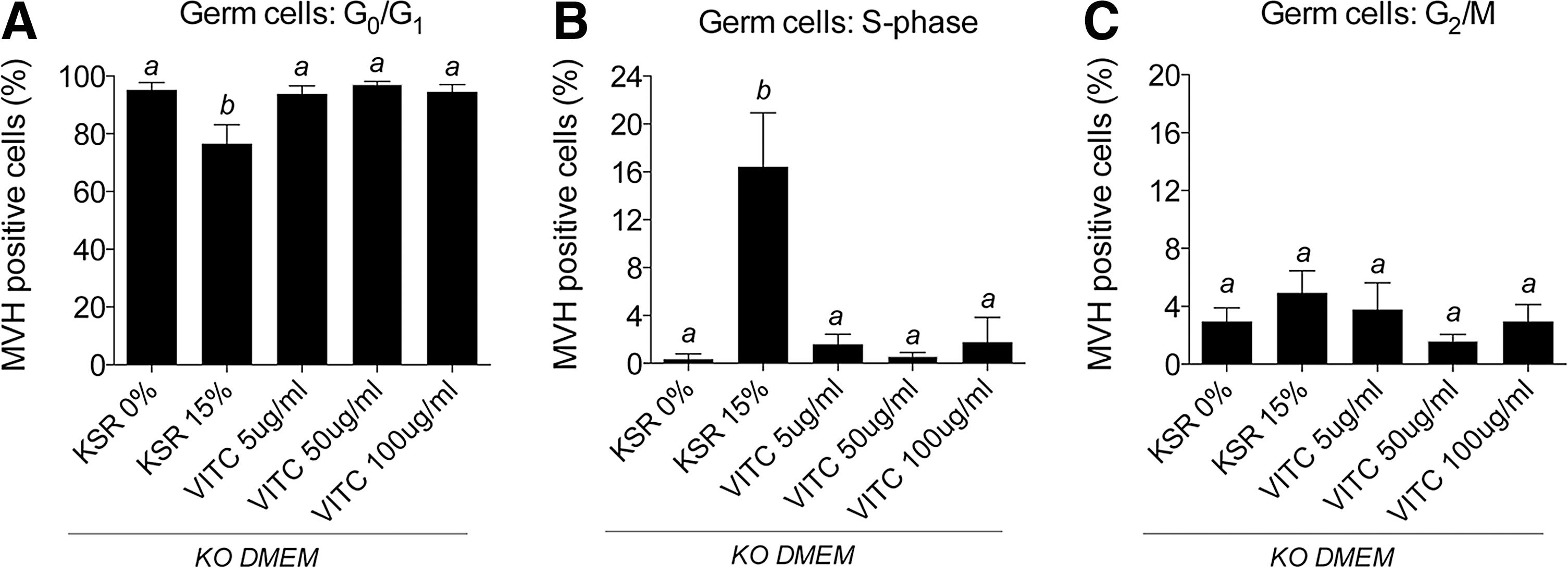
Vitamin C does not recapitulate the proliferative effect of KSR on germ cells in ex vivo fetal testis culture. Cell cycle was assessed in germ cells (MVH-labeled) isolated from E12.5 male gonads cultured for 72 h in KO DMEM supplemented with 0% or 15% KSR, or 5, 50 or 100 μg/mL vitamin C for
Discussion
In this study, we demonstrate that the serum replacement product, KSR, significantly affects cell cycle regulation and differentiation of germ cells and somatic cells in ex vivo fetal testis cultures. Rather than providing more defined conditions for ex vivo fetal gonad culture, we conclude that KSR has detrimental effects on normal gonad development, particularly when used in conjunction with KO DMEM.
Despite its status as a serum-free alternative to FBS, KSR does contain animal product [47]. Patenting information details the inclusion of a number of trace elements, vitamins, amino acids, antioxidants, transferrin, insulin substitutes, and collagen precursors [47]. In addition, ∼10% of KSR comprises AlbuMAX [47]. AlbuMAX is a lipid-rich formulation containing free fatty acids and cholesterol in addition to purified bovine serum albumin (BSA). The replacement of FBS with AlbuMAX in culture is known to enhance cell growth [62,63], strongly indicating that AlbuMAX contains undefined animal components that can impact cell biology.
Supporting our observations that KSR has substantial biological activity in gonad cultures, KSR is broadly used in the derivation and maintenance of reprogrammed or embryonically derived totipotent cell lineages. For example, KSR enhances the growth rate of both human and mouse-derived ES cells [64 –66] and induced pluripotent stem (iPS) cells [67 –69]. Of particular relevance to our study is the widespread use of KSR to enhance the derivation of EG cells from mouse PGCs before sex determination [48 –51]. Evidently, in these systems, KSR contains agents that promote self-renewal and the maintenance of pluripotency; however, the impacts of KSR on fetal gonad development or germ cell differentiation remain unknown. In this study, we demonstrate that KSR prevents normal entry of male germ cells into mitotic arrest during a critical window of gonad development (E12.5–E14.5). Remarkably, in the presence of increasing doses of KSR, a large proportion of germ cells (10%–17%) remained in S-phase at the end of the culture period. In contrast, the culture of E12.5 gonads for 72 h in DMEM:F12K with or without FBS produced cell cycle data most comparable with that of the in vivo situation [27], with complete exit of male germ cells from S-phase (<2%) and entry into the G0 stage of G0/G1 (>90%).
One of the most obvious cell biological markers of male germ cell differentiation is their entry into mitotic arrest [33]. Given the pronounced ability of KSR to block mitotic arrest in male germ cells, we examined the transcriptional profile of cell cycle regulators and well-established markers of male germline differentiation in FACS-purified germ cells. In normal gonad development, male germ cells repress the cell cycle regulators, Ccne1 and Ccne2, between E12.5 and E15.5, concomitant with germ cell entry into mitotic arrest [27]. In line with KSR delaying male germ cell entry into G0/G1 arrest, KSR promoted continued expression of Ccne1 and Ccne2. In addition, the S-phase marker, Pcna, remained highly expressed in KSR-treated conditions, while Cdkn2b, which encodes the G1-S inhibitor p15INK4b [70], remained at low levels. Significantly, Cdkn2b is robustly upregulated in germ cells entering mitotic arrest in vivo [27,29]. Combined, these data clearly demonstrate that KSR acts as a potent mitogen on fetal male germ cells, blocking their entry into mitotic arrest and maintaining their proliferation. In addition to this mitogenic affect, KSR significantly impacted the molecular profile of male fetal germ cell differentiation. This included a failure to upregulate a number of male germ cell markers, including Dppa4, Piwil1, Piwil4, Tdrd9, Map2k7, and Dnmt3L, demonstrating a broad repressive effect on male germ cell differentiation.
Germ cell development is strongly regulated by factors produced by the somatic cells of the developing testis [3,33]. In view of the positive effect of KSR on pre-Sertoli cell proliferation and our findings that germ cell differentiation was impaired, we examined the expression of genes that regulate somatic cell development in the testis and signaling to the germ cells. KSR reduced levels of Amh, Fgf9, Ptgds, and Cyp26b1 in the somatic cell population, indicating that the differentiation of Sertoli cells was inhibited. Since male germ cells require growth factors such as FGF9 for differentiation and survival [6,71], KSR may prevent germ cell differentiation by blocking normal Sertoli–germ cell interactions. However, other studies that have used KSR to promote EG cell derivation indicate that KSR acts directly on germ cells as these cells are commonly derived in the absence of Sertoli cells [48,50,51].
One gene that was not properly upregulated in developing testes cultured with KSR was Cyp26b1, which encodes an enzyme that metabolizes retinoic acid in XY germ cells [16,17]. Insufficient expression of Cyp26b1 could lead to precocious meiotic entry in the male germline; however, KSR did not increase the proportion of male germ cells residing in G2/M or the expression levels of the γH2AX, which are definitive markers of germ cell entry into meiosis [72,73]. Moreover, expression levels of the ovarian marker, Rspo1, remained unaltered in KSR cultures, indicating that female gonad differentiation is not activated in the presence of KSR. Consistent with this, the essential male sex-determination gene, Sox9, was expressed at normal levels in XY gonads cultured with KSR.
DMRT1 is robustly expressed in both Sertoli and germ cells after E11.5 [54,74] and is required to maintain male Sertoli cell identity at postnatal stages [75]. Deletion of Dmrt1 in male germ cells results in a greater susceptibility to teratomas due to an inability of germ cells to enter mitotic arrest and repress pluripotency markers, Sox2 and Nanog [54]. Although, the significant increase in Dmrt1 expression in response to KSR appears inconsistent with the downregulation of other markers of male germ cell differentiation observed in this study, it may explain why pluripotency markers, Sox2 and Nanog, remain efficiently repressed in the presence of KSR.
Observations that KSR enhances EG and iPS cell derivation suggest a role for KSR in promoting germ cell pluripotency [48,50,51,67 –69]. This is of potential concern in an ex vivo gonad development model, whereby pluripotency genes should be transcriptionally downregulated during male germ cell development [28,34]. However, despite the ability of KSR to block germ cell differentiation and promote germ cell proliferation in the fetal gonad, germ cell transcription of Oct4, Sox2, and Nanog and protein expression of OCT4 and the reprogramming marker E-cadherin were not increased by KSR. Interestingly, like germ cells, somatic cells also proliferated more rapidly and failed to differentiate properly. Combined, these observations indicate that KSR promotes proliferation and reduces differentiation in both germ and somatic cells of the developing gonad, but does not directly maintain germline pluripotency. It is therefore possible that the ability of KSR to enhance pluripotent cell derivation may be mediated through its capacity to reduce cell differentiation and promote proliferation rather than directly promoting pluripotency.
A number of compounds contained in KSR may promote the observed alterations to germ cell differentiation, cell cycle, and gonad biology in culture, and other studies have attempted to identify the component/s responsible for the KSR self-renewal effects. In spermatocyte cultures, Sato et al. found that KSR promoted spermatocyte progression through meiosis and maturation to a greater extent than FBS and generated sperm that produced fertile offspring. These authors concluded that the AlbuMAX component of KSR was most critical in improving their culture system since replacement of KSR with AlbuMAX could recapitulate the apparent benefits of KSR in culture [76]. Furthermore, Aoshima et al. reported that KSR supported the long-term survival of mouse spermatogonial stem cells and stem cell activity in vitro, which was augmented by the addition of BSA to the culture medium [53]. However, since AlbuMAX is poorly defined and potentially contains a number of biologically active ingredients, we did not analyze its activity here.
Vitamin C (ascorbate) is also a promising candidate responsible for the stimulatory effects of KSR in gonad culture. Chung et al. identified that vitamin C is an ingredient of KSR that acts directly on human ES cells to promote cell survival and reduce spontaneous apoptosis through epigenetic modulation of CD30 [60]. Indeed, the same authors found that vitamin C led to extensive human ES cell DNA demethylation at genes affecting pluripotency/differentiation and reprogramming [59]. Moreover, Blaschke et al. demonstrated that vitamin C promoted genome-wide DNA demethylation and the altered expression of germline genes in mouse ES cells [61].
The reported effect of KSR influencing epigenetic marks in human and mouse ES cells [60,61] is of particular interest in the fetal gonad given the extensive epigenetic reprogramming that takes place in germ cells during development [56]. Our finding that KSR prevented upregulation of the DNA methyltransferase cofactor gene, Dnmt3L, in male germ cells indicates a significant block on de novo methylation capacity with potential for disrupting the establishment of paternal imprints and allowing aberrant reactivation of retrotransposable genomic elements [32,77,78].
We tested the hypothesis that vitamin C might mediate the inhibition of germ cell development observed in gonads cultured with KSR in this study. However, exposing fetal testis to similar doses of vitamin C present in KSR (according to patent information [47]) did not enhance male germ cell proliferation ex vivo, indicating that vitamin C did not mediate similar effects to KSR. Despite this, it is possible that the lack of effects on gonadal germ cells in the current study may be explained by the very low DNA methylation levels in germ cells at this stage of development [56,79], and it remains possible that vitamin C could impact gene expression changes observed in this study.
From approximately E8.0 to E12.5, concurrent with genome-wide DNA demethylation, there is extensive remodeling of nuclear chromatin, whereby the repressive histone mark, H3K9me2, is replaced by the more plastic repressive mark, H3K27me3 [80,81]. This epigenetic remodeling is considered to facilitate repression of genes promoting somatic differentiation, the reactivation and maintenance of pluripotency, and activation of transcriptional networks appropriate for germ cell development [82]. In our study, we observed KSR-mediated dysregulation of specific histone methyltransferases in male germ cells. Ehmt2, which encodes the H3K9-specific methyltransferase, G9a, was significantly repressed in the presence of KSR. Conversely, Ehz2, a subunit of the polycomb repressive complex 2 and required for H3K27me3 methylation, was significantly upregulated in the presence of KSR. The biological relevance of potential histone variant changes in germ cells at this slightly later stage in development (approximately E14.5–E15.5 postculture) is unclear. However the ability of KSR to alter these epigenetic modifiers is significant given the widespread use of KSR in biological systems where correct epigenetic programming is critical for cell function, including studies of ES and iPS cells.
We were surprised to find that KSR was less effective at promoting germ cell proliferation when present in DMEM:F12K medium than in KO DMEM, despite the absence of significant changes in cell cycle parameters when the two media were directly compared. Although combining KO DMEM with KSR seems to produce an additive effect on cell proliferation compared with other basal media, the KO DMEM formula is proprietary and the factors that might mediate these effects remain unknown.
In summary, using ex vivo fetal gonad culture, we found that supplementing the culture medium with KSR significantly altered male gonad developmental milestones. In the fetal testis, KSR exposure resulted in significant dysregulation of male-specific germ and somatic cell differentiation markers and disruption of normal progression of germ cells into mitotic arrest. Consistent with a number of other studies, our data reinforce the observed ability of KSR to promote self-renewal and block differentiation/promote dedifferentiation. Moreover, our data question the use of KSR as a serum replacement in situations where the aim is to maintain physiological conditions and normal development. Clearly, in the context of mouse fetal gonad development, our findings indicate that inclusion of KSR in culture medium is detrimental for establishing serum-free conditions that recapitulate normal development. By contrast, our data indicate that the use of basal DMEM/F12 media or DMEM/F12 containing FBS supports relatively normal germ and supporting cell differentiation and robust fetal testis gonad development.
Footnotes
Acknowledgments
This work was supported by the NHMRC Project Grant, 1043939, awarded to P.S.W. and the Victorian Government Operational Infrastructure Support Program. The authors thank the staff at the Monash Medical Centre Animal Facility for animal care, in addition to staff at the MHTP Medical Genomics Facility and the FlowCore Flow Cytometry Unit at Monash University, for processing samples. The authors also thank Lexie Prokopuk for manuscript review and comments.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.