
Abstract
DNA methylation, an epigenetic control mechanism in mammals, is widely present in the intestinal tract during the differentiation and proliferation of epithelial cells. Cells in stem cell pools or villi have different patterns of DNA methylation. The process of DNA methylation is dynamic and occurs at many relevant regulatory elements during the rapid transition of stem cells into fully mature, differentiated epithelial cells. Changes in DNA methylation patterns most often take place in enhancer and promoter regions and are associated with transcription factor binding. During differentiation, enhancer regions associated with genes important to enterocyte differentiation are demethylated, activating gene expression. Abnormal patterns of DNA methylation during differentiation and proliferation in the intestinal tract can lead to the formation of aberrant crypt foci and destroy the barrier and absorptive functions of the intestinal epithelium. Accumulation of these epigenetic changes may even result in tumorigenesis. In the current review, we discuss recent findings on the association between DNA methylation and cell differentiation and proliferation in the small intestine and highlight the possible links between dysregulation of this process and tumorigenesis.
Introduction
I
Although DNA methylation patterns have been identified and studied for many years, major aspects of the process, especially its dynamics and role during tumorigenesis, remain enigmatic. In recent years, the combination of bisulfite conversion and high-throughput sequencing (BS-Seq) has made it possible to fill in long-standing voids in our understanding of epigenetic phenomena at single-nucleotide resolution. In this study, we review the current understanding of the role of DNA methylation during the processes of differentiation and proliferation in the intestinal tract with a special focus on epigenetic dynamics and tumorigenesis.
Role and Profile of DNA Methylation in Enterocytes
DNA methylation plays an important role during differentiation and proliferation in normal intestinal epithelium and is thought to act as a safety mechanism to reinforce gene silencing [13 –17]. In general, the methylation of regulatory regions, such as promoters, enhancers, introns, and 3′ untranslated regions, precludes the binding of multiple factors required for intestinal epithelial differentiation, including HNF-4α, Onecut, Cdx2, Hnf1α, and GATA [12,18 –22]. This may initiate gene repression by leading to changes in chromatin organization that antagonize the binding of transcription factors to regulatory regions. Conversely, demethylation of regulatory regions may activate gene expression. For example, reducing DNA methylation causes intestinal crypt expansion in vivo and leads to increased expression of the Wnt-responsive IESC genes, Sox9 and Msi1, which are related to IESC proliferation [12]. Stem cells and highly specialized differentiated cells share the same genetic information. Epigenetic modifications, such as DNA methylation patterns in stem cells, are stable and heritable through mitosis, preserving an accurate differentiation process over many cell divisions. The barrier and absorptive functions of enterocytes can be destroyed during IESC differentiation and proliferation due to aberrant DNA methylation patterns.
Generally, in both IESCs and differentiated cells, DNA methylation is associated with CpG island (CGI)-associated promoters, which can be classified into three categories based on CpG density [14,16,23]. High CpG density promoters are rarely DNA methylated, which supports the traditional viewpoint that DNA methylation represses transcription and is correlated with lower expression levels of the associated genes. Intermediate CpG density promoters (ICPs) are inactive when methylated and active when unmethylated [16,17]. Interestingly, ICPs more frequently acquire differentiation-dependent hypermethylation at pluripotency gene promoters. Finally, low CpG density promoters are generally hypermethylated; however, they remain transcriptionally active regardless of methylation status [14,16]. CGIs that are associated with gene promoters, particularly those driving the expression of developmental or housekeeping genes, remain hypomethylated even if a locus is transcriptionally silent [24]. Consequently, CGIs across the genome are highly unmethylated in both Lgr5-positive IESCs and differentiated cells. In the intestinal genome, promoters are hypomethylated, while the remainder of the genome is hypermethylated [12,25].
Non-CpG methylation in the genomes of IESCs and differentiated cells is unusual. Conversely, nearly 25% of embryonic stem cell genome methylation is in a non-CpG context, suggesting that non-CpG methylation is much more common in embryonic stem cells than in other cell types [26].
DNA Methylation Is a Dynamic Process in the Small Intestine
DNA methylation in enterocytes is a dynamic process during IESC differentiation into mature cells. To study the dynamics of DNA methylation during this process, IESCs and differentiated cells must be effectively separated from the intestinal epithelium. Lgr5 has been identified as an IESC marker. Using an Lgr5-green fluorescent protein (GFP) knock-in model, the epithelial cells of the small intestine can be divided into three types based on GFP intensity: stem cells (GFP-High), their close descendent cells (GFP-Low), and terminally differentiated cells (Villus) [27 –29]. Either the gain or loss of DNA methylation at certain genes during differentiation is dependent on a complex mechanism. Generally, most unmethylated regions gain methylation during differentiation, whereas low-methylated regions show both gains and losses of methylation, and the number of low-methylated regions in differentiated cells is higher than that in IESCs. In a study by Sheaffer et al., in which 1,093 progenitor genes were evaluated, 140 were found to gain methylation during differentiation. Several of these genes are important for directing differentiation, including Bmp7, Ephb2, Olfm4, and Hes1, which are hypomethylated in stem and progenitor cells [12,30]. The 140 progenitor genes identified to possess increased methylation after differentiation were found to be enriched in functional categories such as translation initiation and Notch signaling, a pathway known to be critical for IESC proliferation and differentiation [12,31 –33]. A total of 523 of 3,758 differentiation-induced genes that are highly expressed in intestinal villus showed decreased methylation after differentiation, including enterocyte-specific genes such as Lct, Alpi, and Krt20 [12]. Similar findings were reported in a study by Forn et al., in which ∼1/3 of genes located near transcriptional start sites were found to be hypermethylated during differentiation [34]. Consequently, DNA methylation contributes to the shutting down of IESC-specific pathways during differentiation. However, cell fate decisions in differentiating cells are largely unaffected by a loss of methylation as proportions of differentiated cell types, including enteroendocrine, goblet, and Paneth cells, remain constant over time [12].
Regardless of whether DNA methylation is gained or lost during differentiation, these epigenetic alterations remain concentrated in regulatory regions, while other regions of the genome exhibit stable methylation patterns. These regulatory regions primarily act as enhancers or promoters and are often associated with genes that are important for both the maintenance and differentiation of stem cells. Promoters involved in stem cell gene expression frequently become more methylated as cells differentiate [35 –37]. Hypomethylation of promoter CGIs is associated with transcription factor binding, which prevents the DNA methylation machinery from targeting these regions [25,38 –40].
In fact, DNA methylation dynamics not only affect promoters but also enhancers. A study by Sheaffer et al. demonstrated that a loss of DNA methylation is associated with the activation of specific enhancers during differentiation, while increased DNA methylation at IESC enhancers, such as those found in the Olfm4 and Hes1 genes, is correlated with the repression of the corresponding genes. A proportion of IESC-associated genes that gain DNA methylation during differentiation lose H3K27ac, an active enhancer mark, while losses of methylation during differentiation lead to the acquisition of H3K27ac. Therefore, DNA methylation plays an active role in regulating gene expression by controlling enhancer accessibility during intestinal epithelial differentiation. As with promoters, enhancer hypomethylation is also associated with inappropriate binding of transcription factors such as Cdx2 and Hnf4α [12]. After intestinal differentiation, genomic regions that have reduced DNA methylation are occupied by multiple transcription factors. Transcription factor binding is important in maintaining DNA hypomethylation and chromatin accessibility at enhancers in both mouse and human cells [25,41,42]. Consequently, a gain or loss of methylation during differentiation is not a genome-wide phenomenon, but rather is targeted to regulatory regions, primarily enhancers, and promoters (Fig. 1).
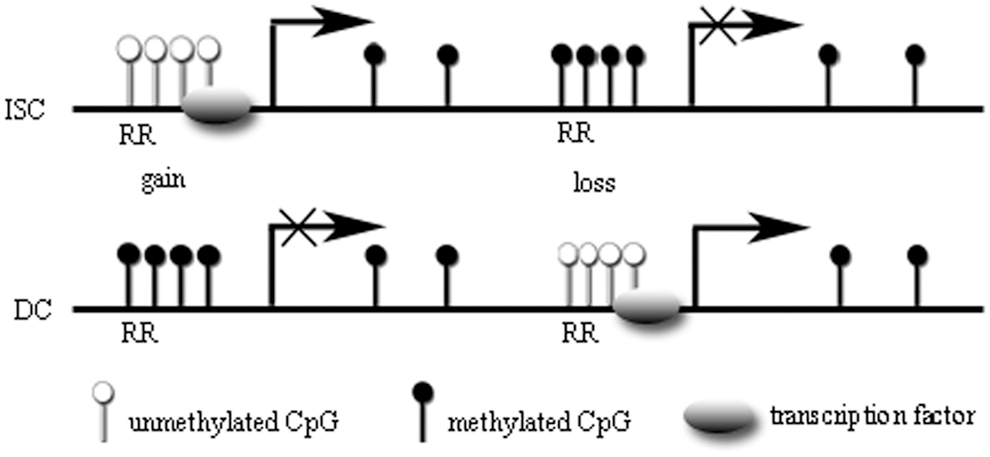
DNA methylation dynamics upon cellular differentiation. In both intestinal stem cells (ISCs) and differentiated cells (DCs), the presence of unmethylated CpG islands (CGIs) in regulatory regions (RRs) that often bind to transcription factors is related to active transcription, whereas methylated CGIs prevent transcription. During differentiation, genes important for directing differentiation gain methylation, whereas differentiation-induced genes, which are highly expressed in the intestinal villus, exhibit decreased methylation after differentiation.
A contradictory viewpoint to the above was noted in a study by Kaaij et al., in which DNA methylation patterns in Lgr5-positive IESCs underwent little change at transcriptional start sites and differentiation-related genes were found to be already primed for expression in stem cells [29]. Previous genome-wide studies have identified numerous differentiated methylation regions (DMRs) upon stem cell differentiation. Many of these regions are associated with transcriptional start sites and regulatory domains. In the study by Kaaij et al., however, only 50 DMRs were identified genome-wide. Almost all of these loci were associated with driving enhancer gene expression in the differentiated portion of the small intestine. Tcf4 was one transcription factor identified as contributing to the formation of DMRs that were hypomethylated. Therefore, it was concluded that limited DNA methylation dynamics occurred during IESC differentiation and that transcription factor binding significantly shapes the DNA methylation landscape during stem cell differentiation in vivo [29,43]. Indeed, transcription factor binding sites are often mildly methylated [25,26]. These limited DNA methylation dynamics were also observed in a study by Bock et al. that focused on stem cell differentiation into blood and skin lineage cells and demonstrated that changes in the genomic distribution of DNA methylation were small, but indicative of adult stem cell differentiation events in vivo [44].
DNA Methylation in Tumorigenesis
Although genetic mutations have been implicated in the initiation of many cancers, epigenetic and genetic alterations are likely to act synergistically during tumorigenesis. Disorders in proliferation, differentiation, and apoptosis within intestinal crypts caused by epigenetic changes can lead to the formation of aberrant crypt foci, which are thought to later progress into adenomas or cancer [45 –48]. Aberrant DNA methylation is the most common cause of epigenetic alterations in this context. Relevant studies of these phenomena have more frequently focused on the large intestine because it is a common site of intestinal tumors. There is compelling evidence of an association between colorectal carcinogenesis and aberrant DNA methylation [49]. The first epigenetic abnormality discovered in a number of cancers has been the loss of DNA methylation at CGIs genome-wide [50 –52]. This DNA hypomethylation corresponds with increased gene expression, loss of heterozygosity, and global chromosomal stability [53 –55]. However, tumorigenesis is not only associated with global hypomethylation but also with site-specific gene hypermethylation. Gene hypermethylation leads to the inactivation of important tumor suppressor genes by inducing transcriptional silencing of genes, such as the gene encoding the mismatch repair enzyme, MLH1, which has been observed to be hypermethylated in colon cancer [56]. The silencing of tumor suppressors and tumor-related genes through hypermethylation at promoter CGIs is a major event during tumorigenesis. Hypermethylation of gene promoters in colorectal carcinoma is extensive and affects several key tumor suppressors, including retinoblastoma, P16, RARB, SFRP, CDX1, and PRC2 [34,57 –62]. Site-specific hypermethylation of genes encoding signaling pathway elements has also been observed. For instance, hypermethylation of the gene encoding epidermal growth factor receptor pathway substrate 8 leads to a loss of its expression and plays an important role in the formation of colorectal adenomas and carcinomas [63]. Another example is the hypermethylation of suppressor of cytokine signaling 3, which plays a tumor-suppressive role during tumorigenesis of ulcerative colitis (UC)-related colorectal cancers [64]. It is notable that epigenetic alterations are also evident in normal aged colonic tissue [65 –67]. Interestingly, most methylation changes that have been observed in cancers occur in CGI shores rather than in promoter regions [68]. In addition, cancer-specific differentially methylated loci vary between normal tissues and colon, lung, breast, thyroid, and Wilms' tumor subtypes [69]. These variable cancer loci are often differentially methylated or misregulated when comparing embryonic stem cells and induced pluripotent stem cells [70]. Like promoters, enhancers have characteristic DNA methylation patterns, and hypomethylation is correlated with active gene expression [25,71,72]. Remarkably, such enhancer methylation has been shown to be more closely associated with gene expression changes in cancer cells than promoter methylation itself [73].
Aberrant DNA methylation is a major event during carcinogenesis, primarily occurring during the early stages of this process. A number of studies have suggested that a loss of cell fate hierarchies precedes the dysregulation of proliferation that is stimulated by signaling pathways, such as the Wnt/β-catenin pathway. Changes in the intestinal cell epigenome may precede the activation of oncogenes, such as Wnt, RAS, and p53, which are needed for neoplastic progression [11,74 –81]. Cadherin family member, CDH13, is frequently silenced by aberrant methylation in colorectal cancers and adenomas and is methylated at an early stage during colorectal tumorigenesis [82]. A similar example is found regarding estrogen receptor methylation during colorectal tumorigenesis in patients with UC [83].
It is well known that tumorigenesis is influenced by age, diet, metabolic status, and other environmental factors. Notably, DNA methylation patterns are also affected by these factors [84,85]. This DNA methylation plasticity, however, leads to a risk of misregulation and may underlie a number of hereditary disorders, developmental defects, and cancers. The micronutrients, folate and selenium, may modulate DNA methylation patterns by affecting intracellular levels of the methyl donor, S-adenosylmethionine (SAM), and/or of S-adenosylhomocysteine (SAH), a product of methylation reactions. Exposure of normal human cells to the supraphysiological folic acid concentrations that are present in commercial cell culture media perturbs intracellular SAM/SAH ratios and induces aberrant DNA methylation. Media containing supraphysiological concentrations of folic acid induce CGI hypermethylation and decrease intracellular SAM/SAH ratios in fibroblasts and colon epithelial cells [86]. Deficient dietary intake of selenium and folate increases tumorigenesis by decreasing DNA methylation [87,88]. A low selenium status affects DNA promoter region methylation and results in the downregulation of tumor suppressor genes in von Hippel–Lindau tumors [89]. Maternal folic acid supplementation significantly reduces the odds of colorectal adenocarcinoma. This protective effect may be mediated, in part, by increased global DNA methylation [90]. In addition to selenium and folate, chronic inflammation is associated with high levels of methylation, perhaps as a result of increased cell turnover. This can be demonstrated in UC, a condition associated with a markedly increased risk of colon cancer [91]. In summary, disorders affecting the complex environment of the gut may lead to changes in DNA methylation patterns and accumulation of aberrant DNA methylation, which may or may not act in cooperation with genetic alterations during the development of adenomas or cancer.
Concluding Remarks
Changes in DNA methylation patterns during the processes of differentiation, proliferation, and tumorigenesis in intestinal tract cells are widespread. These patterns are heritable and stable in normal intestinal epithelium, and the fidelity of this process requires precise signals from the local tissue environment. Disorders such as the development of adenomas or the formation of cancer occur in response to disruptions in DNA methylation dynamics. In certain tumors without genetic mutations, accumulations of aberrant DNA methylation have been found. Significantly, the cumulative acquisition of aberrant DNA methylation patterns proceeds in an orderly way that is influenced at every stage by dietary nutrient intake, metabolic status, chronic inflammation, and other environmental factors. Therefore, further studies into the mechanisms that underlie the accumulation of aberrant DNA methylation may provide significant understanding of intestinal epithelial cell epigenetics.
Footnotes
Acknowledgments
The authors thank the anonymous reviewers for their valuable comments and suggestions to improve the quality of the article. This study was supported by the National Natural Science Foundation of China (no.81270442 and no. 81370475).
Author Disclosure Statement
No competing financial interests exist.