
Abstract
Andersen's syndrome (AS) is a rare disorder characterized by a triad of symptoms: periodic paralysis, cardiac arrhythmia, and bone developmental defects. Most of the patients carry mutations on the inward rectifier potassium channel Kir2.1 encoded by the KCNJ2 gene. kcnj2 knockout mice are lethal at birth preventing, hence, thorough investigations of the physiological and pathophysiological events. We have generated induced pluripotent stem (iPS) cells from healthy as well as from AS patient muscular biopsies using the four-gene cassette required for cellular reprogramming (Oct4, Sox2, Klf4, and c-Myc). The generated AS-iPS cells exhibited the gold standard requirement for iPS cells: expression of genetics and surface pluripotent markers, strong alkaline phosphatase activity, self-renewal, and could be differentiated by the formation of embryoid bodies (EBs) into the three germ layers. Sequencing of the entire coding sequence of the KCNJ2 gene, in AS-iPS cells, revealed that the reprogramming process did not revert the Andersen's syndrome-associated mutation. Moreover, no difference was observed between control and AS-iPS cells in terms of pluripotent markers' expression, self-renewal, and three germ layer differentiation. Interestingly, expression of osteogenic markers are lower in EB-differentiated AS-iPS compared to control iPS cells. Our results showed that the Kir2.1 channel is not important for the reprogramming process and the early step of the development in vitro. However, the osteogenic machinery appears to be hastened in AS-iPS cells, strongly indicating that the generated AS-iPS cells could be a good model to better understand the AS pathophysiology.
Introduction
A
Two genes have been associated with AS: KCNJ2 and KCNJ5 [2,3]. Over 30 mutations were identified on the KCNJ2 gene, and led to a loss of function of the Kir2.1 channel with a dominant negative effect [2,4], and one mutation on the KCNJ5 gene that was postulated to inhibit Kir2.1 channel function [3]. The role of the channel and the functional consequence of its loss of function have been investigated in vitro, in transfected cells or ex vivo in patient cells [4,5]. Myoblasts from vastus lateralis muscle biopsies of AS patients or control individuals were isolated and the electrical properties of the channel were studied. These data confirmed those found in heterologously transfected cells.
Mutations in Kir2.1 led to a complete loss of the corresponding current in human myotubes [5]. However, in vivo investigations of the channel were difficult, because Kir2.1 knockout mice died few hours after birth because of a cleft of the secondary palate [6]. In a recent study, kcnj2 knockout newborn mice showed that the anterior and posterior palatine process and vomer bones were reduced in size; they also suffered from digit defects, and apparent decreased ossification in the pups [7]. The use of induced pluripotent stem (iPS) cells may turn out to be a good alternative to these animal models [8,9], in particular, to investigate the role of the dominant loss of Kir2.1 function associated with the mutations reported in humans.
In this study, we report for the first time the generation of functional iPS cells obtained from AS patients. This work shows that the functional Kir2.1 channel is not important for the reprogramming and the early steps of the development in vitro, but it is crucial during osteogenesis.
Materials and Methods
Standard protocol approvals, registrations, and patient consents
The human samples were derived from a declared collection of samples (# DC-2008-391). This work was approved by the French Ethics Committee and all the patients signed an informed consent.
Cell culture and iPS cell generation
Control and AS myoblasts used to generate iPS cells were obtained from vastus lateralis muscle biopsies, and were cultured as previously described [5]. Patient myoblasts, carrying the C154Y mutation, were characterized phenotypically and showed functionally the absence of the inward rectifier current.
Control and patient iPS cells were obtained after lentiviral infection. Lentiviral preparation was made at the Plateforme de Vectorologie with the human STEMCCA plasmid [10]. Control and patient myoblasts were plated at 1 × 105 cells per dish and infected at one multiplicity of infection with lentiviruses carrying the STEMCCA reprogramming cassette [octamer-binding transcription factor 4 (Oct4), sex-determining region Y-box 2 (Sox2), Kruppel-like factor 4 (Klf4), and c-Myc], in myoblasts' culture medium containing Ham's-F10, 20% fetal bovine serum (FBS), 2 mM Glutamine, 1 mM penicillin/streptomycin, and 10 ng/mL basic fibroblast growth factor.
A second round of infection was performed at day 1. At day 3, the medium was changed to Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS, 2 mM glutamine, and 1 mM penicillin/streptomycin. At day 4, infected cells were plated on mitotically inactivated mouse embryonic fibroblasts (MEF). At day 6, the medium was changed to classical iPS cell medium containing 80% DMEM-F12, 20% knockout serum replacer (KSR; Invitrogen), 1 mM nonessential amino acids, 1 mM penicillin/streptomycin, 100 μM 2-mercaptoethanol, and 10 ng/mL FGF2, and changed every day.
From day 8 to 20, medium was supplemented with 2 μM SB431542, 3 μM CHIR99021, 5 μM PS48, and 250 μM sodium butyrate. These molecules are histone deacytylase (sodium butyrate), TGFβ signaling (SB431542), GSK3 inhibitor (CHIR99021), and PDK activator (PS48).
At day 24, colonies with embryonic stem (ES) cell morphology were individually picked, and transferred into a 48-well plate, with inactivated MEF. iPS cells were maintained in classical iPS cell medium, as described above, with inactivated MEF feeder and passaged twice a week. See Fig. 1A for detailed protocol.
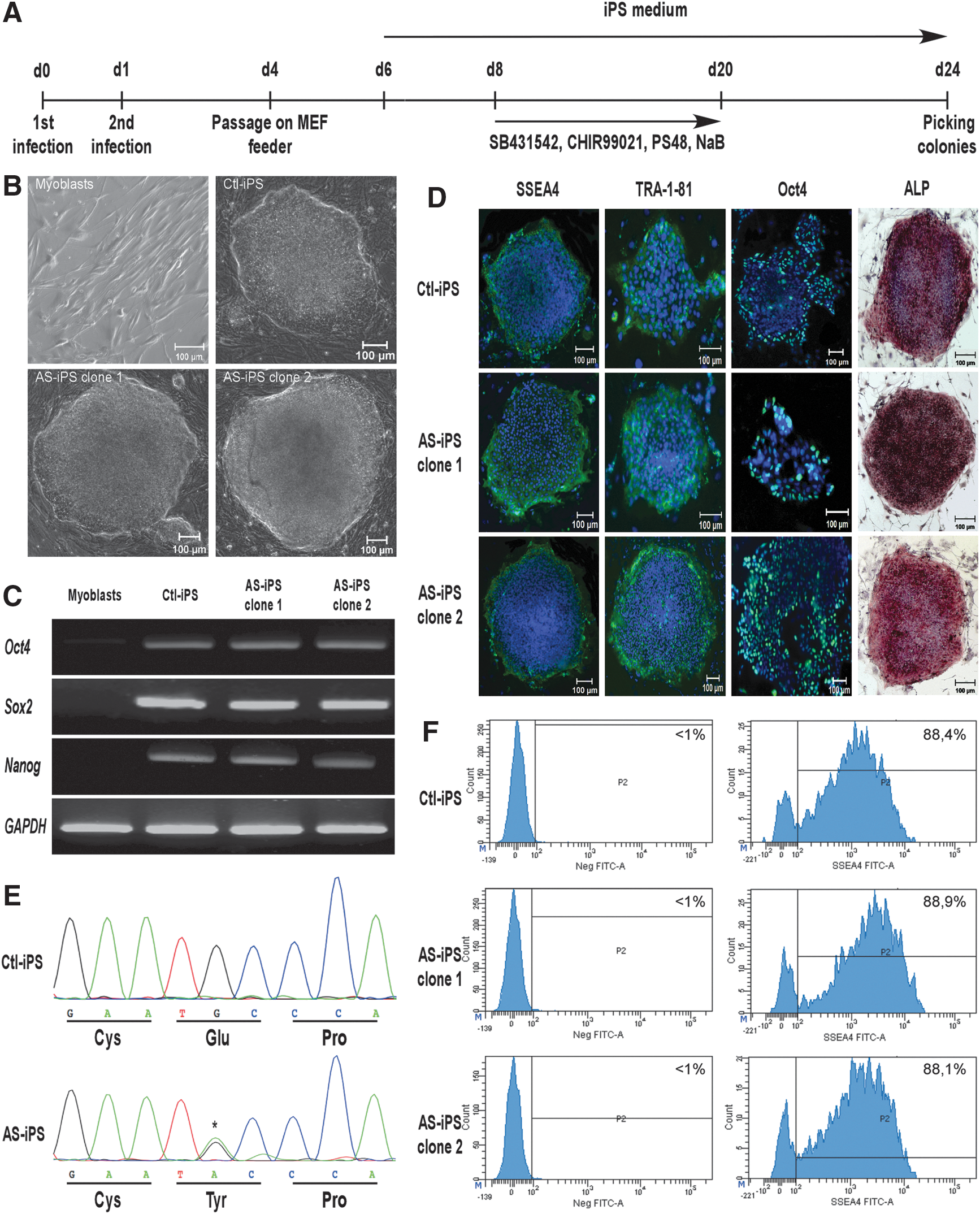
Generation and characterization of control and AS-iPS cells.
Embryoid bodies formation
To form embryoid bodies (EBs), iPS cells were detached from the feeder layer with a dissociation buffer containing 0.25% trypsin without EDTA, 1 mg/mL collagenase type IV, 1 mM CaCl2, 20% KSR, and phosphate-buffered saline (PBS). The clumps of the iPS cells were maintained 1 h in suspension and the floating EBs were cultured in 1 g/mL poly(2-hydroxyl methacrylate) (polyHema)-coated low-attachment dishes in FGF2-depleted iPS cell medium. The medium was changed every other day. After 5 days as floating culture, EBs were transferred in 0.1% gelatin-coated dishes and cultured in the same medium for 3 days, to carry out RNA analysis of ectoderm, endoderm, and mesoderm markers' expression (8 days of differentiation in total).
For osteogenic marker analysis, iPS cells were differentiated as floating EB for 20 days, to carry out RNA analysis for RUNX2, osterix (OSX), alkaline phosphatase (ALP), collagen 1A1 (Coll1A1), osteocalcin (OCN), and KCNJ2 expression.
Genomic DNA extraction and sequencing
Genomic DNA was extracted with the REDExtract-N-Amp Tissue Polymerase Chain Reaction (PCR) Kit (Sigma) following the manufacturer's instructions. Specific KCNJ2 oligonucleotides were used to directly amplify the second KCNJ2 exon (encoding the entire open reading frame) [11]. PCR product was purified with the PCR Purification Kit (Qiagen) and DNA was analyzed by direct sequencing to ensure KCNJ2 sequence integrity.
Immunohistochemistry
Cells were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, and permeabilized in 0.2% Triton in PBS. Cells were then blocked in 5% horse serum in PBS for 1 h at room temperature. The following primary antibodies and dilution were used: mouse anti-stage specific embryonic antigen 4 (SSEA4) 1:1,000 (R&D system), mouse anti-TRA1-81 1:500 (Santa Cruz), and mouse anti-Oct4 1:500 (Santa Cruz).
Cells were incubated overnight at 4°C with the primary antibody. Secondary antibody was Alexa 488 donkey anti-mouse (Invitrogen), used at 1:1,000 dilution. Images were acquired using a fluorescence microscope (Zeiss Axio Observer D1) and pictures were taken with a fluorescence camera (Orca Flash 4.0 LT) using the ZEN software.
ALP activity
ALP activity was measured using the Leukocyte ALP Staining Kit (Sigma), following the manufacturer's instruction. Briefly, cells were first fixed in a citrate/acetone solution for 30 s. After several washes, cells were incubated 30 min in a Fast Violet solution, until coloration appeared. After several washes, cells were stained with Mayer's Hematoxylin, and images were taken using the Zeiss Axio Observer D1 microscope, with a color camera (Zeiss).
Gene expression analysis
mRNA were isolated using the Nucleo Spin RNA Kit (Macherey-Nagel), following the manufacturer's recommendations. Reverse transcription was performed using the RETROscript Kit (Ambion) following the manufacturer's instructions. PCR reactions were performed using the GoTaq DNA polymerase (Promega). Real-time PCR assays were run on a StepOnePlus real-time PCR system (Applied Biosystem), using the SensiFAST SYBR Hi-ROX Kit (Bioline). Transcript expression levels were evaluated using a comparative CT method (delta delta CT). Human 36B4 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as reference genes, for quantitative polymerase chain reaction (qPCR) and PCR, respectively. Specific primers were used for amplification (PCR in Table 1; qPCR in Table 2).
qPCR, quantitative polymerase chain reaction.
Flow cytometric analysis
Cell suspensions of undifferentiated iPS cells were prepared by a 0.25%trypsin/EDTA treatment, and suspended in a solution containing 1% FBS, 2 mM EDTA in PBS. Approximately 2 × 105 cells were incubated with specific cell surface marker antibodies or isotype control, at 4°C for 15 min. A specific antibody for SSEA4 (clone MC813-70; BD Biosciences) was used for the labeling. Antibodies were diluted in PBS containing 1% FBS, 2 mM EDTA. After three washes in PBS/FBS/EDTA, samples were fixed in 0.4% paraformaldehyde, and analyzed on a FACS Canto II (BD), using the BD FACSDiva software.
Statistical analysis
To test for statistical significance, the Student's t-test for paired data was used. P value <0.05 was considered as statistically significant.
Results
Control and AS-iPS cell generation and characterization
Control and AS-iPS cells were generated from myoblasts obtained from vastus lateralis muscle biopsies. Around day 24 post infection, colonies with typical ES cell morphology were found and were individually picked and amplified to obtain pure iPS clones, derived from the same colony. We have successfully isolated two AS-iPS clones, derived from one AS patient and five control iPS clones. To minimize epigenetic or transcriptional pattern specific to the nonreprogrammed cells, we selected colonies at passage 10 or more to establish the control and AS-iPS cell lines. These iPS cell lines were cultured for several passages, and still displayed ES cell morphology (Fig. 1B).
We then investigated the expression of pluripotent markers of the generated cells by reverse transcription-polymerase chain reaction (RT-PCR) and immunohistochemistry. AS cells expressed genetic (Oct4, Sox2, and Nanog) as well as membrane (SSEA4, TRA1-81) pluripotent markers, at the same level than control iPS cells, whereas nonreprogrammed myoblasts did not (Fig. 1C, D). The generated control and AS-iPS cells expressed also Oct4, shown by PCR and immunohistochemistry. Moreover, control and AS-iPS cells have strong ALP activity (Fig. 1D). We have also tested the pluripotent markers expression on the whole iPS cell population. As shown in Fig. 1F, around 90% (P < 0.05) of control and AS-iPS clones 1 and 2 expressed high level of SSEA4.
The Kir2.1 channel is mainly involved in maintaining the resting membrane potential in excitable cells, but its precise role during the development is still unknown. We have tested the expression of KCNJ2 gene in undifferentiated iPS cells by RT-PCR. Control and AS-iPS cells did not express any KCNJ2 transcript (Fig. 1C).
To ensure that the integrity of the KCNJ2 gene sequence in the generated iPS was preserved, the entire coding region (exon 2), which contains the ORF of the KCNJ2 gene, was sequenced from the iPS genomic DNA for all the generated control and AS-iPS clone. Our data show that AS-iPS genome harbored the G461A transition that leads to the C154Y mutation on the Kir2.1 channel, whereas control iPS cells showed a wild-type KCNJ2 gene sequence (Fig. 1E, one control and one AS-iPS clone represented).
Three germ layer differentiation of control and AS-iPS cell types
One of the characteristics of iPS cells is the ability to differentiate into cells of the three germ layers. We have tested this property by the formation of EBs. EBs were formed from control as well as from AS-iPS cells, by floating culture of iPS cells for 5 days; followed by the adhering culture of EBs for an extra 3 days. After 8 days of culture, cells obtained from AS and control EBs displayed a completely different morphology compared to nondifferentiated iPS cells. After differentiation, we obtained a heterogeneous population of cells, containing epithelial cells, fibroblast-like cells, and other cell types, in both EBs differentiated control and AS-iPS cells (Fig. 2A).

Control and AS-iPs cells are able to differentiate into the three germ layers by EB formation.
To test the three germ layer differentiation, EBs were collected and their RNA were analyzed for specific germ layer markers. As shown in Fig. 2B, expression of the pluripotent markers Oct4 and Nanog significantly decreased in differentiated cells from control and AS-iPS-derived EB. Moreover, expression of alpha-fetoprotein (AFP), insulin promoter factor (PDX1), and hepatocyte nuclear factor-3-beta (HNF-3B, FOXA2) for the endoderm; cluster differentiation 34 (CD34), Brachyury, runt-related transcription factor 1 (RUNX1) for the mesoderm; paired box gene 6 (Pax6), glial fibrillary acidic protein (GFAP), and sex determining region Y-box 1 (Sox1) for the ectoderm, significantly increased after the differentiation process. No significant difference was observed in the level of expression of the three germ layer markers between control and AS-differentiated iPS cells (Fig. 2B).
EB-differentiated AS-iPS cells present a lower expression of early osteogenic markers compared to control iPS cells
To investigate the role of the channel during the differentiation process, we have tested its expression after 20 days of differentiation, by EB formation. KCNJ2 expression increased after differentiation for control and AS-iPS cells, but no difference was found between EB differentiated from control and from AS-iPS cells (Fig. 3F).

Expression of osteoblastic markers in control and AS-iPS cells, after EB differentiation. Expression of osteoblast-associated genes RUNX2
We have then tested the expression of the main osteoblastic markers in EB-differentiated control and AS-iPS cells, after 20 days of differentiation. As shown in Fig. 3, differentiated cells from control and AS-iPS-derived EB showed an increased expression of osteoblastic differentiation master genes RUNX2 (Fig. 3A), OSX (Fig. 3B), and early osteoblast markers ALP (Fig. 3C), Coll1A1 (Fig. 3D), and OCN (Fig. 3E) compared to undifferentiated iPS cells. Significant increase in the marker expression was observed between undifferentiated and differentiated control and AS-iPS cells (P < 0.05), except for the ALP expression, but the extent of this increase is three to five times less compared to control EB.
Discussion
AS is associated with a loss of function of the KCNJ2 gene, encoding the strong inward rectifier K+ channel Kir2.1 channel [2]. This loss of function was demonstrated in vitro and ex vivo [4,5]. This disorder revealed that this channel was not only involved in maintaining the resting membrane potential in excitable tissues, but may also play an unexpected role in bone development.
In this study, we have generated iPS cells from control and AS myoblasts from vastus lateralis muscular biopsies [5,12]. The patient's DNA harbored a missense mutation on the KCNJ2 gene that led to a loss of the Kir2.1 channel function in vitro. To perform myoblast reprogramming, cells were infected with a lentivirus carrying the four reprogramming genes (Oct4, Sox2, Klf4, and c-Myc). These genes are known as the Yamanaka's factors, and are widely used to generate human iPS cells [9,13,14].
Few studies reported the generation of iPS cells from skeletal muscle cells [15 –17]. The difficulty to obtain iPS cells from myoblasts could arise from the MyoD-mediated Oct4 inhibition. This inhibition may prevent the reprogramming of myoblasts into iPS cells [17]. To improve the reprogramming efficiency, we added a cocktail of small molecules between days 8 and 20. These molecules were described to enhance the reprogramming process in myoblasts [16].
iPS cells have the same characteristics of ES cells. At early passages, the generated iPS cells can lose their stem cell properties because of epigenetic factors or spontaneous differentiation [13]. To encounter such problems, we have selected colonies at passage 10 at least to establish control and AS-iPS cell lines. These iPS cell lines might represent fully reprogrammed cells. Control and AS-iPS cells still displayed ES cell morphology after several passages in culture, showing that the cells are capable of self-renewal. Moreover, control and AS-iPS cells were characterized at the molecular and genetic levels and both cell types showed expression of multiple undifferentiating markers (SSEA4, TRA1-81, ALP, Oct4, Sox2, Nanog), demonstrating the successful reprogramming of the myoblasts of both genotypes.
KCNJ2 gene is located on the long arm of the chromosome 17 (17q24.3). As chromosome 17 is known as a hotspot for genetic anomalies in iPS cells [18], we sequenced the entire coding region of the KCNJ2 gene. Control and AS-iPS cells still displayed a wild-type and a mutant KCNJ2 gene, respectively, showing that the reprogramming process did not revert the mutation and no genetic anomalies were introduced in the open reading frame of the KCNJ2 gene during reprogramming. Interestingly, control and AS-iPS cells did not express the KCNJ2 mRNA, suggesting that the Ik1 current, carried by the Kir2.1 channel, does not play an important role in the undifferentiated cell stages, and may act later during the development process.
To confirm the presence of other ES cell characteristics, we have formed EBs from both control and AS-iPS cells. EBs are three-dimensional structures made of aggregated iPS cells, in which the cells could spontaneously differentiate into the three germ layers, reproducing in vitro the in vivo early steps of the development [19]. Control and AS-iPS could differentiate into the three germ layers (ectoderm, mesoderm, and endoderm) in such EB structures. Pluripotent marker expression decreased, and typical three germ layer marker expression increased upon the differentiation as expected. Interestingly, control and AS-iPS cells exhibited the same differentiation ability by having the same expression pattern in the three germ layers. These results suggest that the channel activity was not important for the early steps of the development, but rather later in the embryogenesis process.
Although all the iPS cell lines generated around the world are morphologically indistinguishable, they can present a variable pluripotency marker expression and behavior upon differentiation. This could be due to the reprogramming process, culture conditions, or batch effect [13,20]. Moreover, the different methodology used to generate iPS cells, in different laboratories, and the use of those cells for comparison can improve this differential expression [20]. In this study, the same cell type, that is myoblasts, was used to generate control and AS-iPS cells. The common origin of these cells could explain the similar pluripotent marker expression, and behavior upon the differentiation process, in the EBs.
AS is characterized by bone developmental defects. Several iPS cell differentiation protocols are based on EB formation [21 –23]. Based on this, we have investigated the osteoblastic differentiation process in control and AS-iPS cells, through the EB formation. Both control and AS-iPS cells showed an increased expression level of the main osteogenic markers, but this increase was hastened in AS-iPS cells. RUNX2 and OSX are the two master genes of the osteoblastic differentiation. Their expression is required for the expression of all the genes involved in the differentiation process and the bone matrix production [24,25]. RUNX2 or OSX knockout mice present a complete lack of bone [26,27]. Their low expression, in AS-iPS cells, should indeed have major consequences on the ability of these cells to differentiate into the osteoblastic lineage.
Bone mineralization is a two-step process where osteoblasts synthesize first a collagen type 1 extracellular matrix and then hydroxyapatite is deposited in the extracellular matrix [28]. ALP is a key enzyme for this process, as it provides inorganic phosphate to promote mineralization [28]. Interestingly, the level of expression of ALP is not different between nondifferentiated and EB-differentiated AS-iPS cells, whereas all the other early osteogenic markers present an increased expression.
KCNJ2 expression is not different between control and patient cells meaning that only the function of the channel is important for osteogenesis. These results are in agreement with previous findings showing that in vivo models presented developmental features. Indeed, kcnj2 knockout mice had an abnormal skeleton development [7]. Drosophila invalidated for the three kir2.1 orthologs (Irk channels) exhibited a wing patterning defect [7]. In a Zebra fish model, transient expression of human mutated kcnj2 gene and fish wild-type kcnj2-12 also displayed dysmorphic features [29]. Further investigations are needed to better understand the precise role of the Kir2.1 channel in the osteoblastic differentiation process. Despite the fact that a KCNJ2 mutation can produce different clinical phenotypes among the same family members, generating iPS cells carrying other KCNJ2 mutations may be helpful in investigating the complex AS pathophysiology.
In this study we report for the first time the generation of iPS cells from AS patient. The lower expression of osteoblastic genes in EB differentiated from AS-iPS cells that recapitulate one of the three AS clinical phenotypes, indicates that AS-iPS cells could be a valuable cell model to further investigating the developmental events requiring Kir2.1 channel function.
Footnotes
Acknowledgments
The authors would like to thank the patient and her family for their participation and the members of the RESOCANAUX. The authors thank G. Mostoslavsky (Boston University School of Medicine) for providing the STEMCCA vector. This work was supported by CNRS, Association Française contre les Myopathies (AFM)–Téléthon (research grant to S.B.), and Labex ICST (fellowship to J.P.).
Author Disclosure Statement
No competing financial interests exist.