
Abstract
Human umbilical cord-derived mesenchymal stem cells (hUC-MSC) have been considered as promising candidates for cell-based regeneration medicine. However, the application was limited to its poor in vitro proliferation ability against the huge demand of cells. MicroRNA plays important roles in the regulation of cell proliferation, apoptosis, and differentiation. The objective of this study is to explore the roles of miRNAs in regulating the in vitro proliferation of hUC-MSC and unveil their possible mechanism. In this study, we found that miR-26b-3p was significantly upregulated during serial in vitro passage of hUC-MSC and was correlated with cellular senescence and cell cycle genes. The overexpression of miR-26b-3p greatly inhibited the proliferation of hUC-MSC in vitro, which is indicated by 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay, cell cycle, and cell growth curve analyses. miR-26b-3p suppression partly rescued this phenotype by maintaining its proliferation ability in vitro. For mechanism studies, we predicted and validated that miR-26b-3p suppresses estrogen receptor 1 (ESR1) expression by directly binding to the coding sequence (CDS) region of its message RNA (mRNA), thus subsequently changing the expression of its downstream effector Cyclin D1. In conclusion, we found that miR-26b-3p played an important role in the regulation of hUC-MSC proliferation in vitro by targeting the ESR-CCND1 pathway.
Introduction
M
In recent years, human umbilical cord-derived mesenchymal stem cells (hUC-MSC) attract attentions as a more promising source of MSCs [5]. Accumulating evidence showed that hUC-MSC may have therapeutic advantages to treat several diseases, especially autoimmune and neurodegenerative diseases. However, the ultimate requirement of cell-based therapy is that the functional cells reach their target in sufficient numbers to achieve a good clinical outcome. But as hUC-MSC gradually lost their therapeutic properties and proliferation abilities during in vitro amplification [6], there is a compelling need for finding a way to suppress hUC-MSC cellular senescence and maintain their proliferation abilities in vitro that enable more effective application for cell-based therapy in the future.
MicroRNAs (miRNAs) are small (20–22 nucleotides) noncoding RNAs that cause translational repression and/or message RNA (mRNA) destabilization by binding to the target mRNAs [7]. MiRNAs are reported to play important roles in the regulation of cell proliferation, apoptosis, and differentiation [8,9]. Herein, we screened a set of miRNAs that may have strong influence on hUC-MSC. Among which, miR-26b-3p, a reported miRNA that inhibits the growth in many cancer cells [10] that significantly upregulates during serial in vitro passage, inhibits the proliferation of hUC-MSC by regulating the mRNA and protein level of estrogen receptor 1 (ESR1), thus influencing its downstream target CCND1. To this end, we identified that serial in vitro passage upregulated miR-26b-3p could inhibit hUC-MSC proliferation through ESR1-CCND1 pathway, thus providing new insights into in vitro proliferation control of hUC-MSC.
Materials and Methods
Cell culture
Human umbilical cords were obtained from the Changhai Hospital affiliated to the Second Military Medical University, all umbilical cords were obtained with patients' informed written consents. hUC-MSC were then isolated and cultured as follows. Briefly, fresh umbilical cords and skin were firstly washed with 70% ethanol and Dulbecco's modified Eagle's medium (DMEM; Gibco-Invitrogen) to remove excess blood. The tissues were then minced and cultured in DMEM containing 10% fetal bovine serum (FBS) at 37°C. When the cells reached 70% confluence, they were trypsinized for subculture with 0.05% trypsin and 0.025% ethylenediamine tetraacetic acid (EDTA). Daily cultures were done using DMEM with 10% FBS, 100 IU/mL streptomycin and penicillin, and 2 mM
Characterization of hUC-MSC by flow cytometry
The isolated hUC-MSC were detached from the culture flask, washed, and suspended in 1% bovine serum albumin (BSA) containing relevant antibodies including CD29, CD90, CD44, CD34, CD14, CD45, and HLA-DR (BD Biosciences), and incubated at room temperature for 30 min. The isotype antibody was used as control. The cells were then washed with the BSA and analyzed using BD FACSCalibur flow cytometry analyzer (BD Biosciences).
RNA isolation and real-time polymerase chain reaction analysis
Total RNA was extracted using TRIzol (Invitrogen). The miRNA was reverse transcribed with specific primer (Supplementary Table S1; Supplementary Data are available online at
Plasmids construction
For screening which part of ESR1 mRNA is effective to miR-26b-3p binding, we first cloned the coding sequence (CDS) region and 3′ untranslated region (UTR) region into the pmiR-Report vector (Promega). The 3′UTR region was separated into two parts and together with the CDS region we generated three reporter vectors using the primers listed in the Supplementary Table S1. The ESR1 CDS containing putative miR-26b-3p binding site, which we identified later, was amplified from hUC-MSC cDNA and cloned into pmiR-Report vector (the vector contains flanking sequence ∼200 bp up- and downstream of the target site; Promega) downstream of the luciferase coding gene to generate wild-type (wt) ESR1-CDS. ESR1-CDS vector with mutated bases within the putative miR-26b-3p binding site is also cloned and named variant ESR1-CDS. The primers for PCR or quantitative real-time PCR used in this study were listed in Supplementary Table S1.
Luciferase reporter gene assays
HEK293 cells were plated into 96-well. When 70% confluent, the cells were co-transfected with the pRL-TK plasmid, the indicated reporter construct containing either wild-type or mutant binding site of miR-26b-3p and miR-26b-3p mimics/inhibitor. At 48 h post-transfection, dual luciferase reporter assay was performed according to the manufacturer's instruction. The results were normalized using pRL-TK. All experiments were performed with four replicates.
Western blot
The protein concentration was determined using a BCA Protein Assay Kit (Beyotime). The protein extracts were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to poly-vinylidene difluoride membranes (Millipore). Nonspecific protein was blocked with 5% dried skim milk for 1 h, and the membranes were incubated overnight with antibodies directed against ESR1 (1:500; Proteintech) and GAPDH (1:1,000; ABclonal). Horseradish peroxidase (HRP)-conjugated secondary antibodies (Abgent) were diluted at 1:5,000. The signals were assessed using Chemiluminescent HRP Substrate (Millipore) and a Bioshine ChemiQ imaging scanner (Bioshine).
5-ethynyl-2′-deoxyuridine incorporation assays
Five-ethynyl-2′-deoxyuridine (EdU) incorporation assay was carried out according to the following procedure. hUC-MSC were plated into 48-well plates. When cells reach 70% confluent, the medium was changed containing 50 μM EdU and incubated for 4 h at 37°C. After the incubation, cells were washed twice with phosphate-buffered saline (PBS) and fixed in 4% poly formaldehyde for 30 min. Then, the cells were stained with Apollo 567 and Hoechst 33342 solution according to the manufacturer's instructions.
Cell cycle analysis
Cell cycle distribution was analyzed using flow cytometry. hUC-MSC treated with miR-26b-3p mimics or inhibitor were harvested using trypsin/EDTA and washed twice with PBS. Aliquots of at least 2 × 106 cells were centrifuged, fixed in 70% ethanol, and stained with 500 μL propidium iodide solution. Labeled cells were analyzed using a BD FACSCalibur (BD Biosciences) system and the data were analyzed using CellQuest software.
Growth curve
hUC-MSC transfected by miR-26b-3p were seeded to 96-well plates with three replicates for each group. Cells were counted by cytometry, and the mean value was calculated for 7 consecutive days. The medium was changed at a 3-day interval for uncounted cells.
Cell differentiation assay
hUC-MSC were induced with DMEM supplemented with 10% FBS, 50 mg/mL ascorbate-2 phosphate, 10−8 M dexamethasone, and 10 mM β-glycerophosphate to differentiate into osteoblasts. DMEM supplemented with 10% FBS, 50 mg/mL ascorbate-2 phosphate, 10−7 M dexamethasone, 50 mM indomethacin, 0.45 mM 3-isobutyl-1-methylxanthine, and 10 mg/mL insulin to differentiate into adipocytes, respectively. After 3 weeks of induction, cells treated with osteoblast induction medium were stained with alizarin red S, and cells treated with adipocyte induction medium were stained with oil red O. Samples were run in triplicates.
Statistical analysis
Each experiment was repeated at least three times independently. Statistical significance was assessed by comparing mean values ± standard deviation using a Student's t-test for independent groups and was assumed for *P < 0.05.
Results
The expression level of miR-26b-3p was associated with hUC-MSC proliferation and cellular senescence in vitro
The surface markers of the primary hUC-MSC were first detected by flow cytometry. The cells isolated and cultured were of hUC-MSCs traits, which were positive for CD29, CD44, and CD90 while negative for CD14, CD34, and CD45 (Fig. 1A). Serial passage of the primary hUC-MSC resulted in poor morphology (a more flattened and spiked morphology) and delayed doubling time as we assessed (Fig. 1B). This phenotype is accompanied by a reduction in expression of cell cycle promoting genes like CCND1, CCNE1, CDK2, and CDK4 (Fig. 1C), while a relative upregulation of negative factors (Fig. 1D).
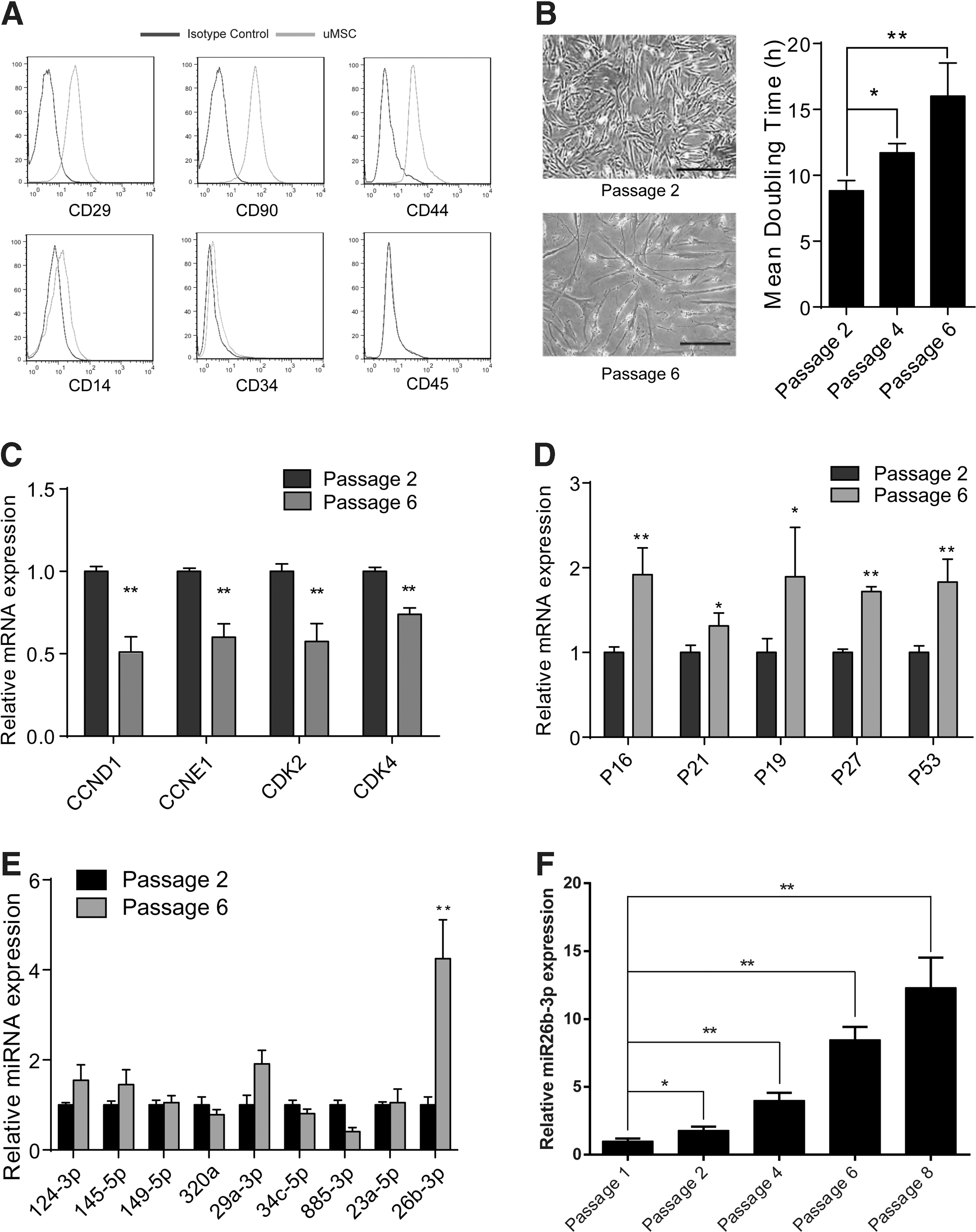
The expression level of miR-26b-3p is associated with lowered proliferation ability of human umbilical cord-derived mesenchymal stem cells (hUC-MSC).
To unveil the roles of miRNAs in such phenotypic change of hUC-MSC during serial cultivation in vitro, we screened a set of miRNAs that were reported to have strong influences in MSC lineage commitment [11 –16]. By assessing their expression change in early-passage (passage 2) and late-passage (passage 6) hUC-MSC, we found the expression level of miR-26b-3p was significantly increased in late-passage hUC-MSC (Fig. 1E), and further assessment showed that miR-26b-3p expression significantly increased after passage 4 (Fig. 1F), contrary to the proliferation ability of hUC-MSC with passage. These results indicated the proliferation ability of hUC-MSC was in inverse association with the expression level of miR-26b-3p. It made us suppose that potential relationship may exist between miR-26b-3p and the proliferation ability of hUC-MSC, which is worth further investigation.
miR-26b-3p regulated hUC-MSC proliferation and cellular senescence in vitro
To analysis the effect of miR-26b-3p on hUC-MSC proliferation, we synthesized miR-26b-3p mimics and inhibitors. Their efficacy was tested by qPCR (Fig. 2A, C). The proliferation ability, cell cycle, and cell growth curve were tested by EdU assay, flow cytometry, and growth curve analysis, respectively. The results showed EdU-positive cells were significantly lowered in 26b-3pOE group than negative control (NCOE) group (Fig. 2B), while the miR-26b-3p inhibitors transfected group has significantly more EdU-positive cells than negative control (NCInh) group (Fig. 2D). Flow cytometry showed the 26b-3pOE group has lower percentage of G2/M phase cells, while the 26b-3pInh group has more percentage of G2/M phase cells compared with relative control groups (Fig. 2E). The cell growth curve showed that the number of cells were significantly lower in the 26b-3pOE group than its control group (Fig. 2F, upper panel), while they significantly increased in the 26b-3pInh group (Fig. 2F, lower panel) at 72 h. The RNA levels of cell senescence and cell cycle-related genes were significantly increased in the 26b-3pOE group compared with the control group (Fig. 2G), while it significantly decreased in the 26b-3pInh group. These findings confirm that miR-26b-3p could inhibit hUC-MSC proliferation, while miR-26b-3p inhibitors promoted the proliferation of hUC-MSC in vitro.
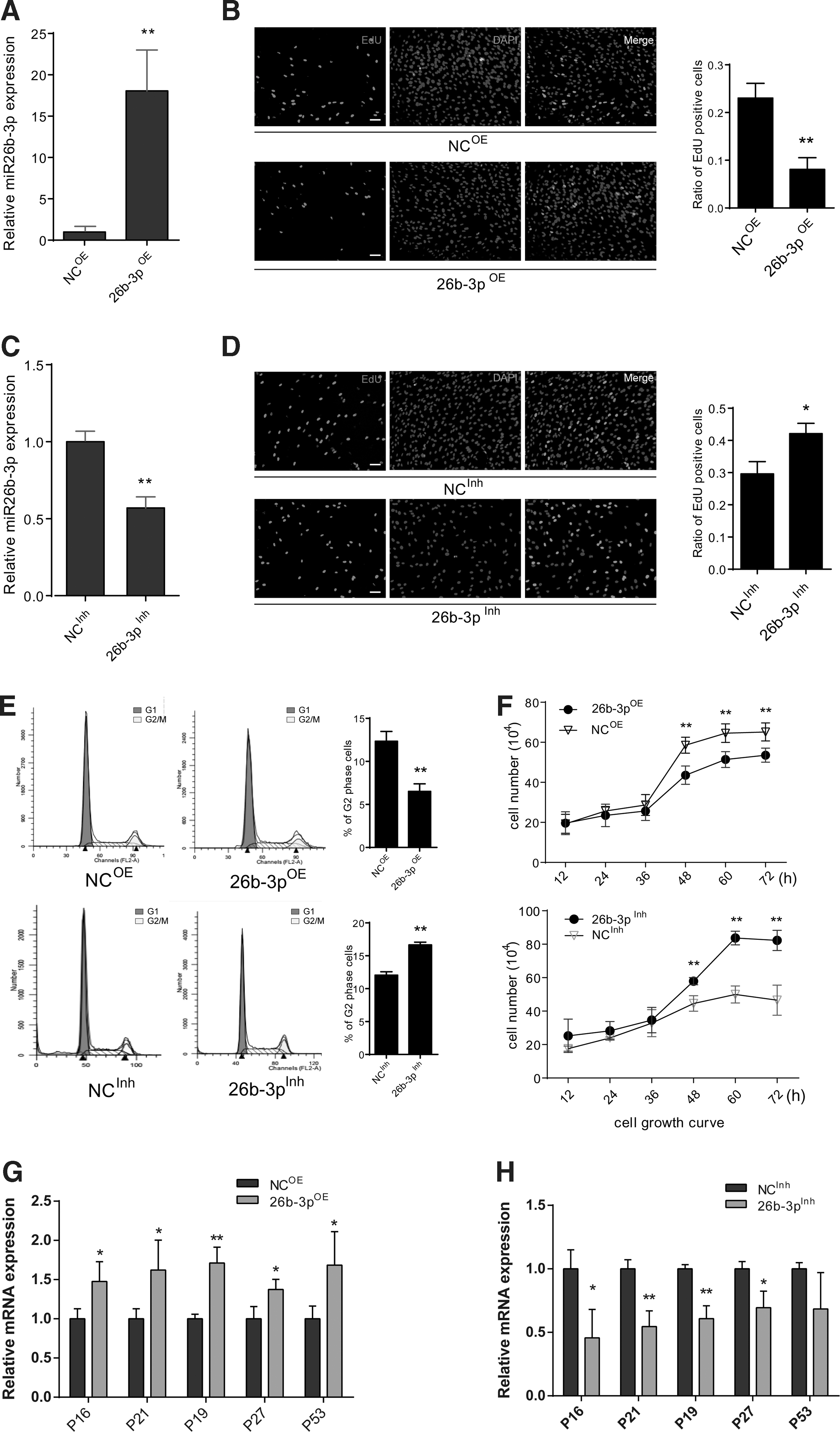
miR-26b-3p regulated hUC-MSC proliferation and cellular senescence.
miR-26b-3p directly targets the CDS region of ESR1 mRNA
To find out the target of miR-26b-3p, which may play an important role in mediating hUC-MSCs proliferation, we used TargetMiner database to search for putative targets of miR-26b-3p. To increase the probability of the prediction, we chose proliferation-related genes with more than 10 putative target sites as effective candidates (Table 1). The mRNA level of these candidates were analyzed using qPCR (Fig. 3A), among which the RNA level of ESR1 was found significantly decreased in the 26b-3pOE group. To further confirm the effect of miR-26b-3p on ESR1, we analyzed ESR1 protein and mRNA expression using western blot and qPCR respectively in both 26b-3pOE and 26b-3pInh groups. The ESR1 protein and mRNA expression in the 26b-3pInh group were significantly higher than that in the 26b-3pOE group (Fig. 3B). These results confirmed that miR-26b-3p indeed affects ESR1 expression.

miR-26b-3p directly inhibit estrogen receptor 1 (ESR1) message RNA (mRNA) by targeting its coding sequence (CDS) region.
5′, forward primers; 3′, reverse primers; ESR1, estrogen receptor 1; qRT-PCR, quantitative real-time polymerase chain reaction.
Since miR-26b has been implicated to take vital roles in cell proliferation control in several tumor cell lines [17 –22], we again evaluated the gene expressions of other reported targets in early and late passaged hUC-MSC. Results showed that none has significant changes in expression between passages as strengthened by analyzing the high-throughput profiles of their transcriptome expressions of passage 9 and passage 18 human MSCs (Supplementary Fig. S1A, B). However, the levels of ESR1 mRNA and protein during serial cultivation in vitro were decreased significantly (Fig. 3C), this change is also confirmed by the high-throughput profiles (Supplementary Fig. S1A). These results showed that among the reported targets and the newly tested targets of miR-26b-3p, only ESR1 showed a negative correlation with miR-26b-3p during serial passage of hUC-MSC.
It is unclear, however, whether miR-26b-3p directly target ESR1. Through TargetMiner prediction, we found 13 putative binding sites, among which ten 6mer and one 7mer-m8 sites were located in the 3′UTR region, and the 8mer site was located in the coding sequence (CDS) region of ESR1 mRNA. We thus constructed one CDS region and two separated 3′UTR region luciferase reporters as indicated in the Fig. 3D. Through dual-luciferase assay, we found only the luciferase activities of CDS region reporter were downregulated upon miR-26b-3p transfection (Fig. 3D). These results indicated that the predicted 8mer site may contribute most to the targeting of miR-26b-3p. To validate this binding site, we cloned the CDS of ESR1, which contained the wild-type or the mutated miR-26b-3p binding site (including its flanking sequences) into a new luciferase reporter plasmid (Fig. 3E). Luciferase activities of the wild-type reporter were significantly decreased in 26b-3pOE group while increased in 26b-3pInh group compared with control. However, no significant difference of the luciferase activities were observed among the mutated ESR1 CDS groups (Fig. 3F). The results validated that miR-26b-3p regulates the expression of ESR1 via directly binding to the CDS region of ESR1 mRNA.
ESR1 is essential for the function of miR-26b-3p
ESR1 is an important nuclear receptor and transcript factor that plays an important role in various cellular processes. To further validate miR-26b-3p exerts its function of proliferation regulation through ESR1, we analyzed the EdU incorporation rate of hUC-MSC treated by 17β-estradiol (E2), an ESR1 agonist, or ESR1 antagonist fulvestrant (F). The results showed that the EdU incorporation rate was increased significantly in E2 (0.1 nM) treated hUC-MSC, while ESR1 antagonist fulvestrant (1 nM) decreased the EdU incorporation rate (Fig. 4A). To further analyze ESR1 perturbation affected the function of miR-26b-3p, we added F to the 26b-3pInh transfected cells and reduced the EdU incorporation rate, which partially reversed the effect of miR-26b-3p inhibition to a normal level (Fig. 4B). As expected, the lowered EdU incorporation rate was significantly reversed by adding E2 to the 26b-3pOE transfected cells (Fig. 4C). These results indicate that miR-26b-3p inhibits hUC-MSC proliferation at least partially through inhibiting ESR1 expression.
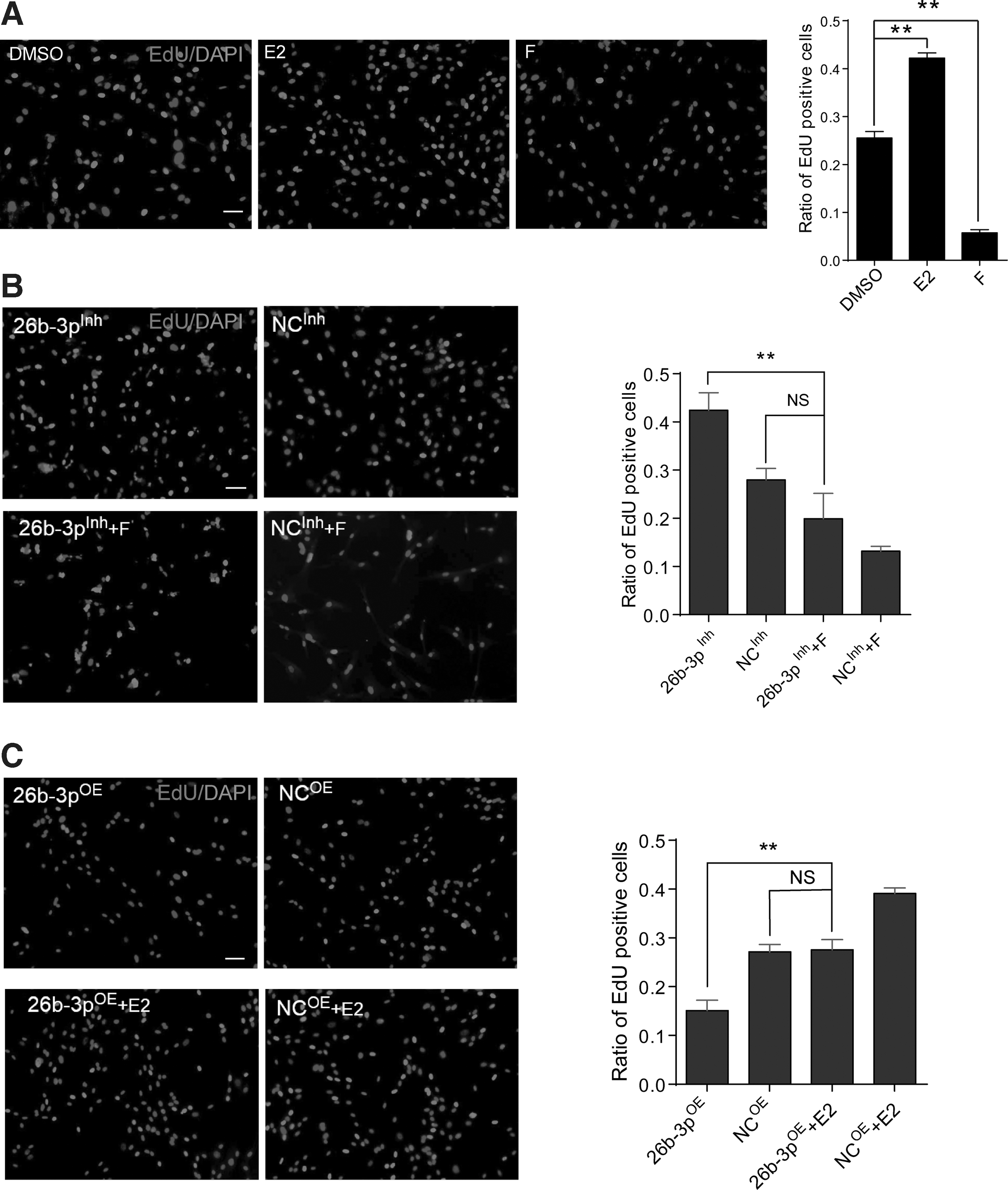
miR-26b-3p exert proliferation regulatory function through ESR1.
CCND1 is mainly regulated by miR-26b-3p-ESR1 axis
To further explore the downstream target of miR-26b-3p, we examined the expression of ESR1 regulated genes, among which CCND1 showed strong correlation with miR-26b-3p perturbation (Fig. 5A). It is known that CCND1 plays an important role in regulating proliferation and cell cycle, and the RNA level of CCND1 was significantly increased in E2 group, which is consistent with the previous findings that CCND1 is regulated by ESR1 (Fig. 5B). When adding E2 to miR-26b-3pOE transfected cells, the lowered expression of CCND1 caused by miR-26b-3p overexpression was significantly reversed (Fig. 5C). And opposite change was observed in 26b-3pInh group treated with F compared with miR-26b-3p inhibition alone (Fig. 5D, E). In conclusion, miR-26b-3p could inhibit hUC-MSC proliferation via ESR1 by changing the expression level of ESR1 downstream effector CCND1.
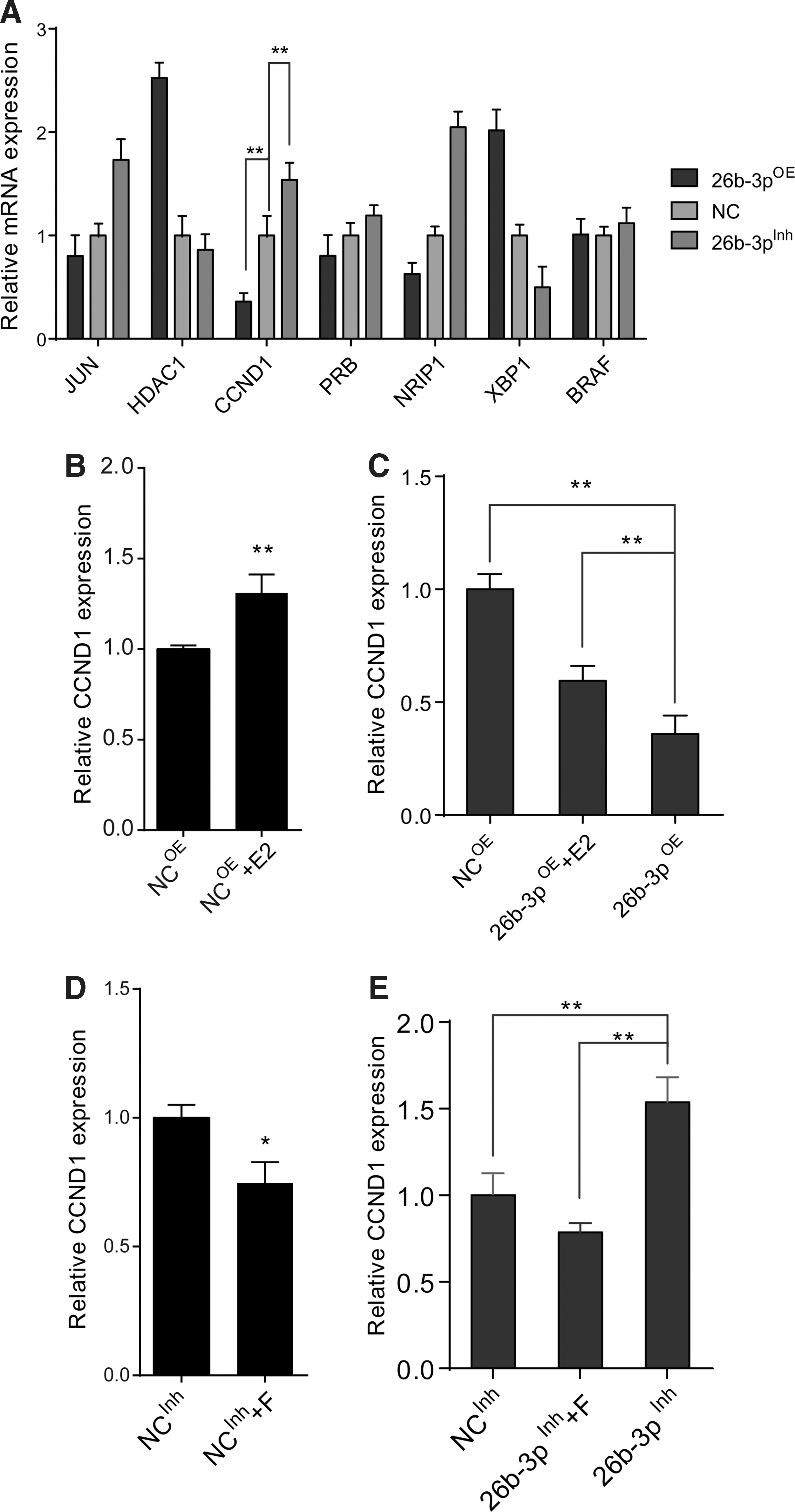
CCND1 was regulated by miR-26b-3p-ESR1 axis.
miR-26b-3p promotes adipogenic differentiation in hUC-MSC by modulating estrogen signaling
To further explore the effect of miR-26b-3p in hUC-MSC differentiation, and to clarify the function of miR-26b-3p regulated estrogen signaling during differentiation, we setup osteogenic and adipogenic differentiation model by using commercially available induction medium. By overexpressing miR-26b-3p, we found that the adipogenic differentiation-related gene expression upregulated significantly, while osteogenic-related genes downregulated to some extent (Fig. 6A, B). The relative staining of the adipogenic and osteogenic induction cells also supported this change. However, inhibiting miR-26b-3p showed inversed effect to that of miR-26b-3p overexpression (Fig. 6C, D). These results indicated that miR-26b-3p may play a role in promoting adipogenic differentiation of hUC-MSCs.
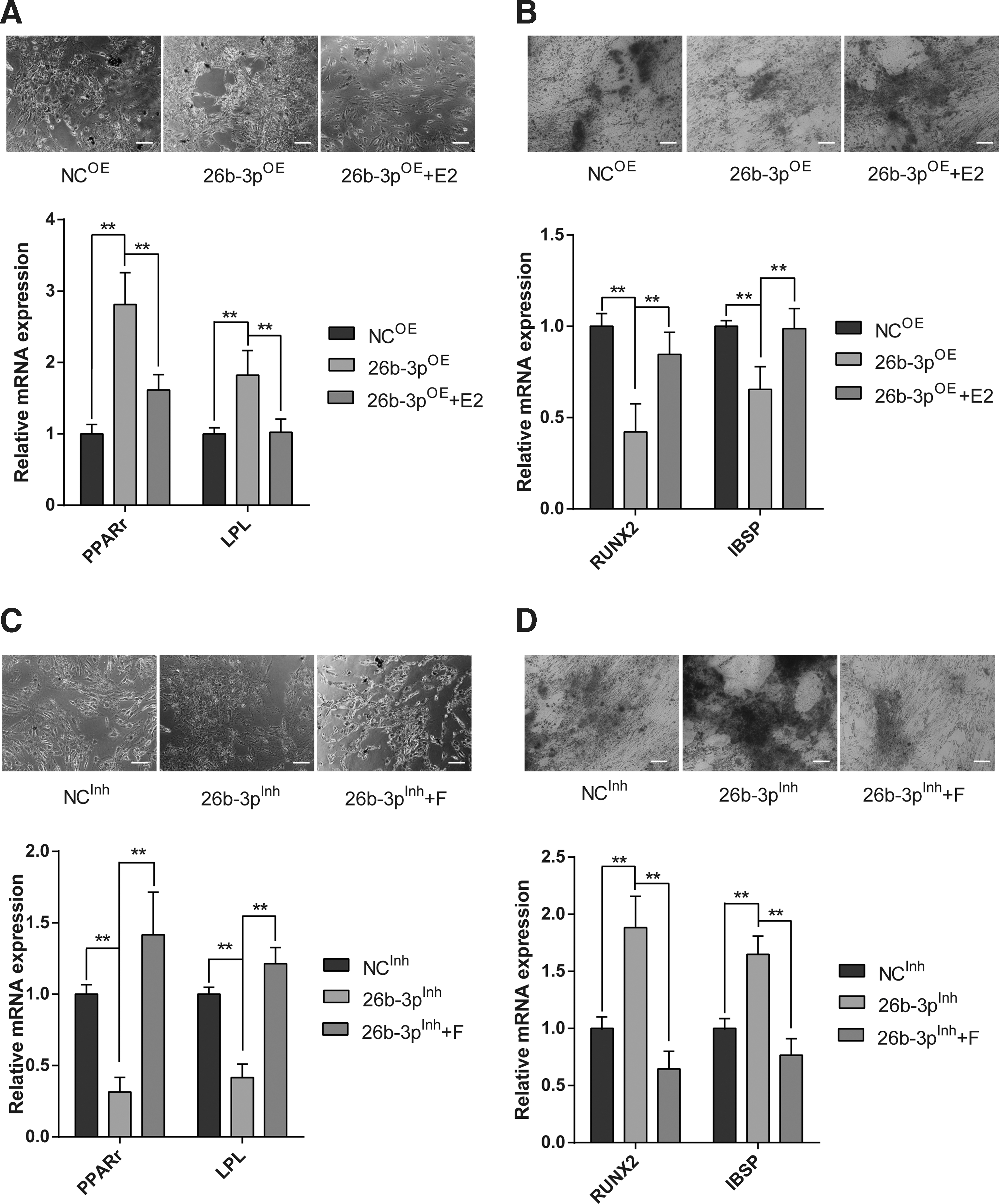
miR-26b-3p promotes adipogenic differentiation by modulating estrogen signaling.
To test whether modulation of estrogen signaling is required for this effect, we added E2 to the miR-26b-3p overexpression group, and found that the elevated PPARγ and lipoprotein lipase decreased upon treatment and the osteogenic factors increased compared with miR-26b-3p overexpression group (Fig. 6A, B). While adding F to the miR-26b-3p inhibiting group showed significant induction of adipogenic genes' expression and decreased osteogenic genes' expression (Fig. 6C, D). These results together implied that miR-26b-3p promoted adipogenic differentiation through modulating estrogen signaling, and probably by direct modulation of ESR1.
Discussion
MSC-based therapy is a fast-growing field that has proven safe and effective in the treatment of various degenerative diseases and tissue injuries [23]. Self-renewal and multipotency are the key hallmarks of MSC, which features establish MSC as the most promising tool for regenerative medicine [24,25]. However, it remains difficult to culture sufficient numbers of effective MSC for treatment. On the one hand, the large number of MSC needed for clinical application limited its usage. On the other hand, after cells passage cultivation, MSCs become large and flatten, and may lose therapeutic properties gradually. And in general, administered MSCs were limited in passage 1–5 in reported clinical trials [26,27]. In this study, we observed cell morphology become longer and larger, proliferation become slower, and proliferation relative gene was decreased in late passages of hUC-MSC. This is consistent with the results of the published report. The mechanism for this phenomenon remains elusive.
MicroRNAs (miRNAs) participate in the regulation of a large variety of cellular processes including proliferation, differentiation, migration, metabolism, and apoptosis [27,28]. We screened a set of reported functional miRNAs in MSCs and found miR-26b is significantly upregulated during continued passages in hUC-MSC, and correlates negatively with cell cycle promoting genes.
miR-26b encoded in an intron of the CTDSP2 [17], was indicated recently a downregulation in many cancer cells. miR-26b is downregulated in epithelial ovarian carcinoma, and directly targets KPNA2. miR-26b/KPNA2 axis suppresses tumor proliferation and metastasis through decreasing OCT4 expression [18]. Overexpression of miR-26b dramatically inhibited proliferation, invasion, and migration of HCC cells by targeting EphA2 [19]. miR-26b regulates β-catenin and cyclin D1 levels by inhibiting GSK-3β expression, which in turn alters the Wnt/GSK-3β/β-catenin pathway to lower rheumatoid arthritis fibroblast-like synoviocyte proliferation and elevate cell apoptosis and the secretion of TNF-α, IL-1β, and IL-6 cytokines [20]. miR-26b exerts a tumor suppressive role in breast cancer and the miR-26b-mediated growth inhibition is achieved through suppression of a new target gene CDK8 [21]. miR-26b promoted differentiation and, at least partly by targeting cyclin D2, attenuated cell proliferation via arresting the G1/S transition [22]. Cancer cells share some characteristics with stem cells, including self-renewal and differentiation. One more important feature of stem cell is that it may accumulate genetic changes during prolonged culturing in vitro. Some of those changes resemble the ones found in tumors. Therefore, for safety issues, there is also a need to confirm their stem cell genetic integrity [29]. It has been reported that human germline stem cell (SSC) line was able to colonize and proliferate in vivo in the recipient mice. Neither Y chromosome microdeletions of numerous genes nor tumor formation was observed in human line although there was abnormal karyotype in this cell line [30]. Although these studies have demonstrated the effects of miR-26b on inhibiting proliferation of various cancer cells, its effects on hUC-MSC were unknown. In this study, we found miR-26b-3p inhibits cell proliferation and miR-26b-3p could inversely regulate the mRNA and protein level of ESR1 through directly binding to CDS of ESR1.
ESR1 is transcript factors and mediate the growth effect of estrogen. E2 treatment stimulates proliferation in cultured bovine satellite cells (BSCs) through interaction with ESR1 as E2-stimulated BSC proliferation completely suppresses estrogen receptor (ER) blocker and E2 does not stimulate proliferation in BSC cultures once ESR1 expression has been silenced, which establish that ESR1 is required for E2 to stimulate proliferation of cultured BSCs [31,32]. Studies have identified the proliferation promotion of estrogen on MSC in rats and human [33 –35]. Estrogen stimulates MSC proliferation and ESR1 expression [36]. In this study, we also observed that E2 could stimulate hUC-MSC proliferation. Estrogen exerts its effects on cell growth and function via intracellular ERs and eventually by activating particular gene [37 –39], ESR1 could interact with special DNA sequence referred as estrogen-responsive elements located within the promoter of genes, thus regulating the expression of target genes [40], such as NRIP-1 [41]. We also detect signal molecule expression of ESR1 pathway, we found the level of CCND1 and NRIP1 decreased in 26b-3pOE group and increased in 26b-3pInh group, which suggested that miR-26b-3p could change biology of ESR1. The association of CCND and CDKs can promote the progression of cell cycle by accelerating the transition of G1/S phase [42], thus promoting cell proliferation. In the study, we found that miR-26b-3p can regulate the expression of ESR1 and influence the level of CCND1 to inhibit the hUC-MSC proliferation.
In this study, we identified that miR-26b-3p regulate MSC proliferation through ESR1-CCND1 pathway. It suggests the inhibition of miR-26b-3p could be a potential, novel tool for increasing the growth of MSC and have benefits for clinical application of MSC. We also believe that there are other miRNAs or miRNA/mRNA pairs involved in MSC proliferation. More intensive studies are required to further illustrate the cellular and molecular mechanisms that regulate the proliferation of hUC-MSC.
Footnotes
Acknowledgments
This study was supported by the National Key Basic Research and Development Project (Beijing, China) (no. 2011CB965101), National Natural Science Foundation of China (no. 31371507), and the Foundation of Science and Technology Commission of Shanghai Municipality (no. 13JC1407101).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.