
Abstract
Central nervous system (CNS) inflammation is primarily driven by microglial cells which secrete proinflammatory cytokines and undergo proliferation upon activation, as it occurs in neurodegenerative diseases. Uncontrolled or prolonged CNS inflammation is potentially harmful and can result in cellular damage. Recently, many studies have focused on human adipose tissue as an attractive source of cytokines with immunosuppressive properties that potentially modulate inflammation. Our study aimed to evaluate if different methods of human tissue collection could affect adipose mesenchymal stem cell (ADSC)-derived cytokine secretion and investigate the effects of ADSC secretome in modulating microglia activation and the possible implication of sphingosine-1-phosphate (S1P) in these effects. Our results demonstrate that the conditioned medium (CM) of ADSCs isolated by two different processing methods (lipoaspirate and Lipogems) significantly inhibited the lipopolysaccharide (LPS)-induced effects on microglia activation, including microglial expression of CD68, cytokine secretion, proliferation, and migration. Pulse studies with radiolabeled sphingosine demonstrated that LPS treatment of resting microglia induced a significant increase of both cellular and extracellular S1P. Moreover, and of relevance, FTY720, a functional antagonist of S1P receptor, inhibited the multiple LPS-induced proinflammatory effects on microglia, and S1P suppressed the anti-inflammatory effect of ADSC-CM. This suggests that LPS-mediated microglial activation is countered by ADSC-CM through the modulation of sphingosine kinase/S1P signalling.
Introduction
T
Among the multiple regulatory signals of microglia, sphingosine-1-phosphate (S1P) has emerged as a critical one, in both physiological and pathological conditions. Indeed, this bioactive sphingoid, derived from sphingolipid metabolism, is relevant to the neuroprotective effects of microglia, as well as to the etiology, course, and treatment of neuroinflammation and neurodegenerative disorders [6 –8]. S1P, synthesized from sphingosine (Sph) and ATP by sphingosine kinases (SphKs), exerts biological essential roles, including the regulation of cell proliferation, migration, fate, and immunity, predominantly through five specific G-protein-coupled receptors (S1PR1–5). As in other CNS cells, microglia can express a range of S1P receptors, including S1P1–3 and S1P5, and this expression is dynamically regulated upon activation [9 –12]. S1PR activation can be crucial to activating immune cells, including microglia [11], and this can contribute to neurogenesis regulation [13]. SphKs were reported to regulate the microglial production of inflammatory signals [9,11,14], and extracellular S1P has been shown to act as a powerful chemoattractant to the sites of injury [15] and increase the release of proinflammatory cytokines from activated microglia [11]. Furthermore, increased microglial activation in experimental autoimmune encephalomyelitis in CNS cell lineages was reduced by S1P1 deletion and administration of the S1P1 indirect antagonist FTY720 [16], indicating that S1P1 can control microglial activation and suggesting that microglial S1PRs are involved in the pathogenesis of neurodegenerative diseases. In agreement, FTY720 reduced microglial recruitment into lesions, activation, and cytokine production in rodent cerebral ischemic lesions, traumatic brain injury, and demyelination models [17 –20].
In the last decade, a big effort in different fields, including basic research, preclinical studies, and regenerative medicine, has been made to understand the properties and regenerative abilities of human adipose tissue that rely upon adipose mesenchymal stem cells (ADSCs). ADSCs possess not only multilineage differentiation capacity [21], ability to homing to areas of injury [22], and in vivo integration and regeneration [23] but are also recognized as crucial players in the modulation of the inflammatory state. Indeed, adipose tissue exhibits pleiotropic immune regulatory activities mediated by complex mechanisms. These activities lead to the functional inhibition of different immune cell subpopulations of the innate and adaptive immunity through the release of soluble paracrine factors as well as direct cell-to-cell interactions [24,25]. Moreover, fat tissue is an important source of secreted restorative growth factors [26] able to repair neural damage [27].
ADSCs can be isolated from adipose tissue biopsies by several protocols, different in time to digestion, enzyme type and concentration, and in culture media [28,29]. In addition, nonenzymatic procedures have been described [30 –32]. In this field, an innovative system, named Lipogems, was recently developed and provides a nonexpanded, ready-to-use fat product [33,34]. This system uses mild mechanical forces in a completely closed system, thus avoiding enzymes, additives, and other manipulations. This innovative enzyme-free technology was developed to process variable amounts of lipoaspirates, resulting in a nonexpanded adipose tissue product that contains viable and functional human ADSCs [33,35].
In the last years, safety and efficacy of implanted ADSCs have been investigated in different animal models, and promising preclinical studies with good perspectives for translational approaches are underway. Increasing evidence in animal models of stroke [36,37], traumatic brain injury [38] and spinal cord injury [39], demonstrates that ADSCs play a crucial role in the repair of CNS damage, by stimulating neurogenesis and exerting neuroprotective effects [40,41]. A growing body of evidence indicates the potential therapeutic capacity of ADSCs in a variety of diseases as amyotrophic lateral sclerosis, Parkinson's disease, autoimmune diseases, and multiple sclerosis, highlighting the ability of ADSCs to reduce inflammatory states and induce beneficial effects [42].
Despite this, little is known on whether ADSCs exert their beneficial roles by affecting the microglial cell activity, and the possible role of S1P in these effects remains uninvestigated. In this study, we tested the anti-inflammatory role of human ADSC secretome on the N9 microglial cell line and the possible involvement of S1P signalling in this effect. To this purpose, we triggered an inflammatory response in vitro by stimulating microglia with lipopolysaccharide (LPS) and investigated whether ADSC-conditioned media (CM) might have any influence on microglia activation, in terms of cytokine production, cell viability, proliferation, and chemotaxis. Finally, we evaluated the metabolism, extracellular release, and effects of S1P on LPS-induced microglia activation, in the absence or presence of ADSC-CM.
Materials and Methods
Patient eligibility
The protocol was reviewed and approved by the Internal Institute Board, and the study was performed according to the amended Declaration of Helsinki. Liposuction aspirates from subcutaneous adipose tissues were obtained from six subjects (Table 1), two males and four females, aged between 35 and 60 years, undergoing elective plastic surgical procedures at the Image Institute (Milan, Italy). Before sample collection, a written informed consent was obtained from all patients.
ADSC, adipose mesenchymal stem cells.
Adipose tissue samples
Specimens of fat from the abdomen region were collected by a lipoaspirate technique. All specimens were verified for the presence of human immunodeficiency virus 1 and 2, hepatitis C virus, hepatitis B virus, and cytomegalovirus. Fat tissues were serially processed to obtain two different aliquots: a first unprocessed lipoaspirate (ASP) specimen and a second processed sample, obtained by treating the ASP with the Lipogems® device (GEMS). Briefly, lipoaspirated tissues were introduced and shaken into the Lipogems device (LIPOGEMS International SRL, Milan, Italy), containing beads that allow the fragmentation of the fat tissue and the removal of red blood cells and oil residues [34,43]. An aliquot of both ASP and GEMS tissue samples (0.5 mL) was frozen dry at −80°C for enzyme-linked immunosorbent assay (ELISA), and an aliquot (0.5 mL) was processed for ADSC isolation by selective culture conditions as described below.
ELISA on adipose tissues
The frozen adipose tissues were homogenized in ice-cold lysis buffer using a homogenizer. After a 10-min incubation at 4°C with constant rotation, the homogenate was centrifuged (300 g, 10 min) and the subnatant (whole tissue extract) below the lipid cake was aspirated and denatured. Total protein concentrations of the whole tissue extracts were measured using the Bradford method. Subsequently, both ASP and GEMS tissue samples were analyzed by Multianalyte Elisa Array-Human Inflammatory Cytokines (M0H-E004A; Sa Bioscience, a Qiagen company, Manchester, UK) following the manufacturer's protocols.
ADSC culture
ADSCs from both ASP and GEMS tissues were obtained as previously described [23,44]. Briefly, tissues were washed with saline D-PBS and centrifuged for 10 min at 920 g at room temperature (RT) to recover the stromal vascular fraction, which was immediately plated in T25 flasks in Dulbecco's modified Eagle's medium/Nutrient F-12 Ham (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) (Euroclone, Milan, Italy) and 1% penicillin and streptomycin solution (Sigma-Aldrich, Milan, Italy), optimized for the selective growth of ADSCs. The ASP- and GEMS-ADSCs were routinely seeded at 2 × 104 cells/cm2 in T75 flasks and passed weekly. Cell viability was assessed by the Trypan blue (Life Technologies, Carlsbad, CA) dye-exclusion assay. Cells were passaged at a density of 75%–90% for ∼14 days in culture and maintained in culture through passage 4. Cells were counted in three microscopic fields in each well and expressed as a percentage of the total number of cells.
FACS analysis
ASP- and GEMS-ADSCs were evaluated for the expression of mesenchymal, hematopoietic, endothelial, and immunological markers: anti-human CD14-PE, CD31-PE, CD34-PE, CD45-FITC, CD73-PE, HLA-DR-FITC (BD Pharmingen, San Jose, CA), and CD105-FITC (AbDSerotec, Raleigh, NC). Briefly, 5 × 104 cells/tube were stained with fluorochrome-conjugated monoclonal antibodies and incubated for 15 min at RT in the dark. Samples were centrifuged at 300 g for 5 min, washed twice with D-PBS and then fixed with 4% paraformaldehyde (PFA). Isotype-matched mouse immunoglobulins were used as control. At least 5 × 103 events were acquired for each sample. Nonviable cells were excluded by physical gating. For data acquisition, a FACS Canto II (BD Pharmingen) with DIVA software was used, whereas the analysis of the data was performed using the Cell Quest software (BD Pharmingen).
ADSC-CM collection and preparation
ASP- and GEMS-ADSCs were plated at density of 7 × 105 cells/cm2 for 7 days and supernatants were collected from both cultures, centrifuged for 10 min at 500 g, and stored at −20°C for the subsequent experiments. The final supernatant is referred as CM.
Microglial cell culture
The murine microglial cell line N9 was cultured in the IMDM medium supplemented with 4 mM glutamine, 25 mM HEPES (Invitrogen, Carlsbad, CA), 1% penicillin/streptomycin solution, and 5% FBS. This culture condition is referred in the text as the basal medium (BM). The cells were grown at 37°C in a humidified incubator in 95% air-5% CO2 and were split once every 3 days at 4 × 103 cells/cm2.
Microglial cell treatments
Microglial cells were challenged for 48 h in the BM with 3 μg/mL LPS (from Escherichia coli, serotype 055:B5; Sigma-Aldrich) alone, to induce their activation, or in the presence of ASP-CM or GEMS-CM (1:4, v/v). Therefore, microglial cells were divided into the following six experimental groups: (1) Control group (CTR): BM incubation; (2) LPS-treated group (LPS): incubation with 3 μg/mL LPS in BM; (3) ASP-CM group: incubation in ASP-CM; (4) ASP-CM+LPS group (LPS+ASP-CM): incubation in ASP-CM supplemented with 3 μg/mL LPS; (5) GEMS-CM group: incubation in GEMS-CM; (6) GEMS+LPS group: (LPS+GEMS-CM): incubation in GEMS-CM supplemented by 3 μg/mL LPS.
In the case of S1P treatment, a stock solution of S1P (100 mM; Enzo Life Sciences, Farmingdale, NY) was prepared in fatty acid-free bovine serum albumin (4 mg/mL in BM). For N,N-dimethylsphingosine (DMS) and FTY720 treatments, stock solutions of 5 mM DMS (Enzo Life Sciences) and 10 mM FTY720 (Cayman Chemicals, Ann Arbor, MI) were prepared in absolute ethanol. At the time of the experiments, stock solutions were administered to cells at the following final concentrations: 200 nM–1 μM for S1P, 100 nM for FTY720, and 1 μM for DMS, for the indicated periods of time. At the used concentrations, ethanol had no effect on cell growth and viability.
Assessment of ADSC and microglia viability and proliferation
The viability and proliferation of ASP- and GEMS-ADSCs were assessed by trypan blue staining. Briefly, cells were plated at a density of 2 × 104 cells/cm2 in 48-well culture plates (Corning Life Sciences, New York, NY). After 14 days of culture, cells were harvested, counted, and seeded at the same initial density for next passage count. The total number of dead and viable cells was counted at each passage by Trypan Blue dye-exclusion assay in a Fuchs-Rosenthal chamber, and the percentage of viability was assessed by the following formula: viable cell number/(viable cell number + dead cell number) × 100. Growth curves were obtained by counting the number of viable cells at each passage and calculating the cumulative population at each passage [45].
The effect of ADSC-CM and/or S1P modulators on microglial proliferation was determined by assessing 3H-thymidine (PerkinElmer, Boston, MA) incorporation into DNA [46]. Cells were plated (2 × 104/cm2) in 35-mm Falcon dishes in BM and grown for 24 h. Cells were then treated with ADSC-CM and/or S1P modulators (as described above) for 48 h. 3H-thymidine (1 μCi/mL) was added to each plate 12 h before cell harvesting. At the end, cells were harvested with phosphate-buffered saline, and the thymidine incorporation in trichloroacetic-insoluble material was determined by liquid scintillation counting.
Microglial cell immunofluorescence
To confirm microglial lineage and the effect of the different treatments on its activation, we assessed the expression of ionized calcium-binding adapter molecule 1 (IBA1) and CD68 by immunofluorescence analyses.
Microglial cells cultured for 1 day on permanox chamber slide at the concentration of 2.5 × 104 cell/cm2 were treated with LPS alone or in combination with ADSC-CM. After 48 h at 37°C, the cells were fixed with 4% PFA (15 min, RT). To avoid quenching, microglia were treated with 0.1 M glycine (10 min, RT). Subsequently, the samples were permeabilized with 0.1% Triton X-100 for 15 min. The following primary antibodies were applied overnight at 4°C: mouse anti-IBA1 (1:500) and mouse anti-CD68 (1:400), (both purchased from Santa Cruz Biotechnology, Inc., Heidelberg, Germany). Cells were then incubated with Alexa488 conjugated secondary antibody (1:1,000; Life Technologies) at RT for 45 min, washed, and the nuclei were counterstained with DAPI (Life Technologies). The sample was mounted with ProLong® Gold Antifade Mountant (Life Technologies) and images were captured using a Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). Cells were counted in three microscopic fields in each well (three wells/treatment) and expressed as a percentage of the total number of cells. Each treatment was performed in triplicate.
Chemotaxis of microglial cells in presence of ASP- and GEMS-CM
Chemotaxis of microglia was performed using the modified Boyden chemotaxis chamber assay (8.0 μm pore size; Falcon, Corning, New York, NY). Briefly, microglia (5 × 104 cells/mL) were suspended in BM, and 200 μL of the cell suspension was added to the top compartment of the chamber. LPS was added to the lower chamber, in the absence or presence of ASP- or GEMS-CM and/or FTY720 or S1P. The chambers (set up in triplicate) were incubated for 48 h. At the end, nonmigrated cells on the upper side of the filter membrane were gently wiped off with a cotton swab. Subsequently, the filters were removed, and cells were fixed in methanol, stained with hematoxylin/eosin, and then scanned with the EVOS® FL Auto Imaging System, using a 4 × objective. Multiple images were then tiled and stitched together using the ImageJ software by the plugin Stitching. Microglia cells migrated in the lower compartment of the chamber and were stained with Calcein AM (Life Technologies). The fluorescent dye (1:1,000) was administered directly into each well and the cells were incubated at 37°C for 30 min. At the end of the incubation, the chambers were washed, acquired by using an inverted Eclipse Ti-E microscope (Nikon Instruments, Florence, Italy), and the number of migrated microglial cells was determined in the whole well by ImageJ software.
ELISA on microglia medium after coincubation with ASP-CM or GEMS-CM
Cells were seeded into 24-well plates at the density of 5 × 104 cells/well. After 48 h in the presence of different treatments (see above), the supernatants were collected, centrifuged for 10 min at 500 g, and stored at −20°C. The supernatants were subjected to the evaluation of the expression levels of interleukin IL-10 and IL-17 by specific ELISA kits (Affymetrix eBioscence, San Diego, CA) according to the manufacturer's instructions. The absorbance at 450 nm was recorded using a microplate reader. Data analysis was performed by SP2000 Manager software.
S1P metabolism and extracellular release in microglial cells
To evaluate S1P metabolism and extracellular release, pulse studies with [C3-3H]-sphingosine (3H-Sph; PerkinElmer, Boston, MA) were performed as previously described [47,48]. Briefly, cells were incubated with 3H-Sph (20 nM, 0.4 μCi/mL) in the S1P trapping medium for 1 h. At the end, media were collected, and cells were harvested. Total lipids were extracted from cells at 4°C and partitioned as previously reported [49]. After centrifugation, the upper alkaline aqueous phase, containing S1P, was evaporated under a nitrogen stream and counted for radioactivity by liquid scintillation. Extracellular S1P was extracted from the pulse medium and partially purified by two-step partitioning, at first in alkaline conditions. Then a back extraction of the aqueous phase was carried out in acidic conditions [49]. The final organic phase, containing S1P, was evaporated under a nitrogen stream. The fractions containing cellular and extracellular S1P were submitted to high-performance thin layer chromatography (HPTLC) on silica gel plates, using n-butanol/acetic acid/water (3:1:1, v/v/v) as solvent system. Standard 3H-S1P was chromatographed on the same plate and used as internal standard. At the end of the chromatography, HPTLC plates were submitted to digital autoradiography (Beta-Imager 2000; Biospace, Paris, France), and S1P was quantified by the beta vision analysis software provided with the instrument.
Statistical analysis
Results are expressed as the means ± SD of three independent assays. The comparisons between two groups were undertaken by using the unpaired Student's t-test. A P value <0.05 was considered statistically significant.
Results
ASP and GEMS tissues express different levels of inflammatory cytokines
As shown in Fig. 1A and B, GEMS tissue color was clearer than the ASP one. The ELISAs of a focused panel of 12 proteins related to inflammation (Fig. 1C) revealed that the levels of several cytokines were significantly different in GEMS and ASP (Fig. 1C). In particular, the content of some inflammatory cytokines (including interleukin [IL-]1β, IL-6, IL-8, and tumor necrosis factor-alpha [TNF-α]) in the GEMS tissue was significantly lower than that in ASP.
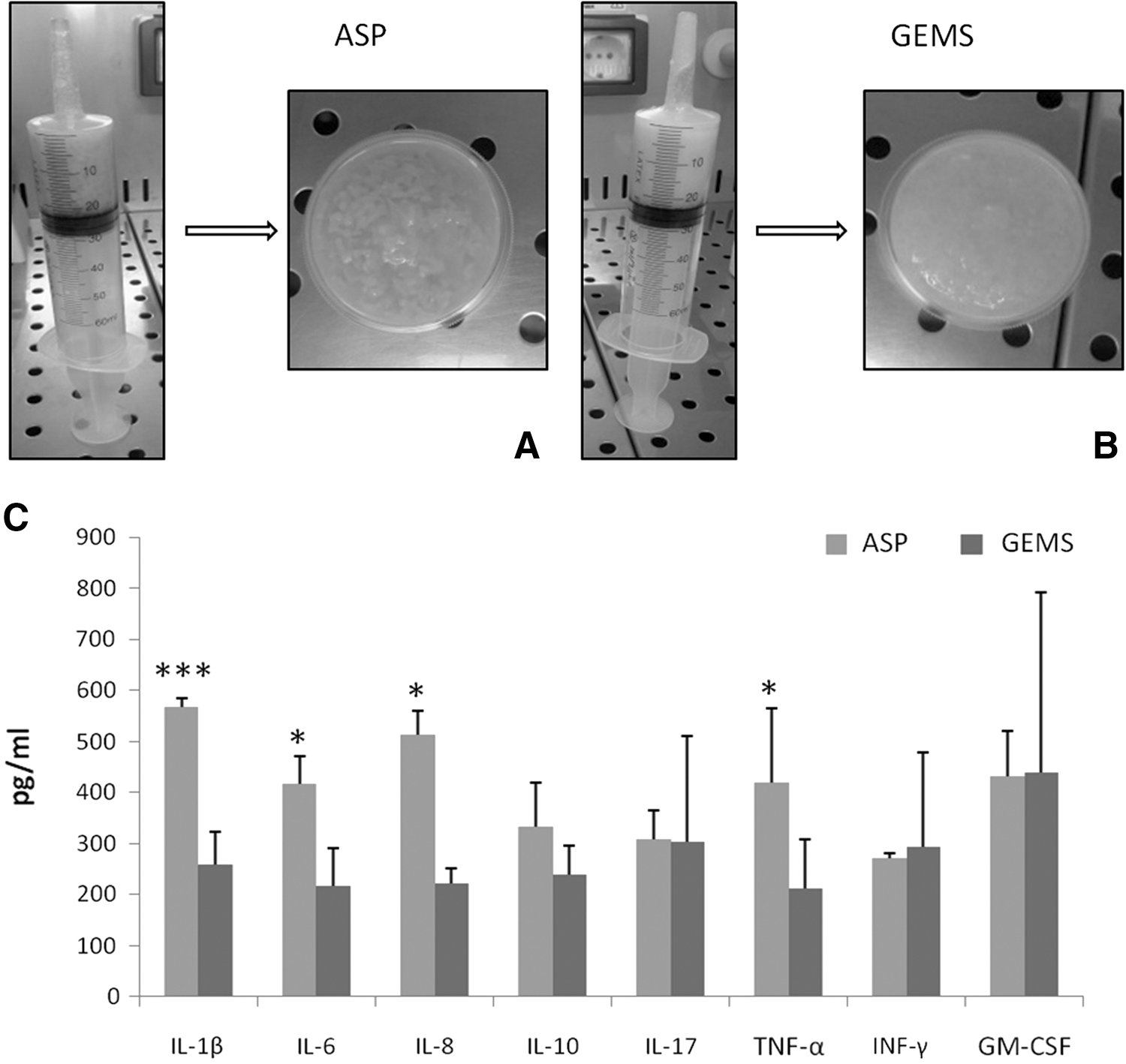
Characterization of ASP and GEMS adipose tissue. Photomicrographs of ASP
Human ADSCs isolated from ASP and GEMS tissues exhibit a mesenchymal stem cell phenotype and proliferative properties
To evaluate if the phenotype and growth properties of ASP- and GEMS-ADSCs were different, we analyzed the morphology and growth curve during first passages.
Once seeded in BM, both ADSCs grew adherent to plastic, and exhibited a fibroblast-like morphology (Fig. 2A, B). Cell growth analyses demonstrated that both ASP- and GEMS-ADSCs started to proliferate 7–10 days after plating, and continued to propagate along with passages (Fig. 2C). Of note, GEMS-ADSCs showed a higher proliferation rate than ASP-ADSCs (Fig. 2C), and the number of viable cells from ASP was found to be significantly lower than that from GEMS (Fig. 2D).
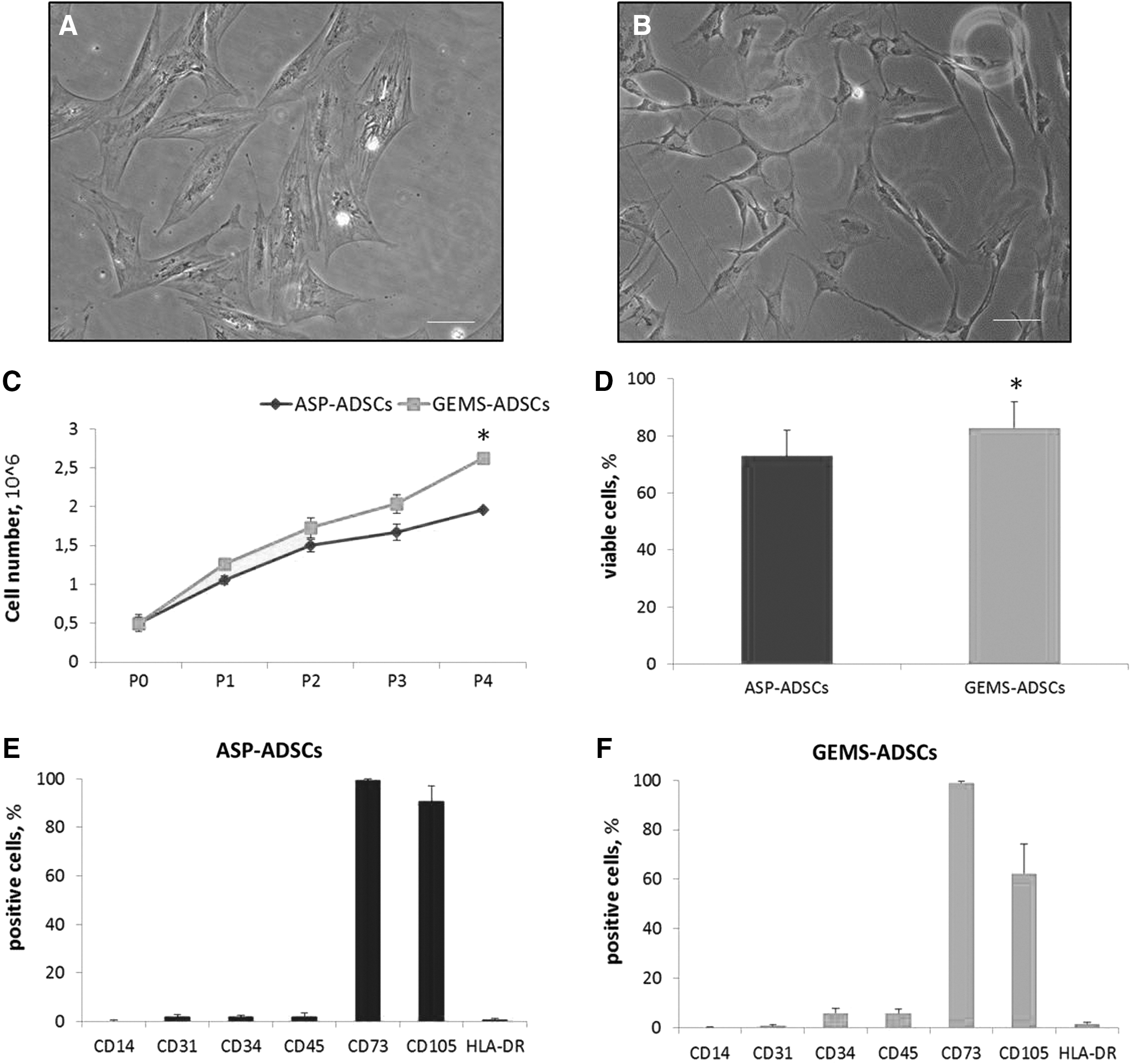
Characterization of ASP- and GEMS-ADSCs. Photomicrographs of ASP-ADSCs
To characterize the immunological phenotype of ASP- and GEMS-ADSCs, we next analyzed the expression of different surface markers. Our results show that both ASP- and GEMS-ADSCs highly expressed the mesenchymal stem cell (MSC) markers CD73 and CD105 (Fig. 2E, F), but did not express hematopoietic (CD34 and CD45), leukocyte (CD14 and HLA-DR), and endothelial (CD31) markers, thus excluding the contamination by different cell types, including lymphocytes and monocytes.
ADSC-CM inhibit LPS-induced activation of microglial cells
We then investigated whether ADSC-CM were able to affect microglial activation induced by LPS, a well-recognized inflammatory stimulus. In the presence of LPS, morphological analyses of microglial cells showed that the ramified morphology of cell resting state (Fig. 3A) changed into an amoeboid morphology, the typical morphological state of activated microglial cells (Fig. 3B). After treatment with ASP-CM or GEMS-CM alone, microglial cells maintained their resting morphology (Fig. 3C, D). In contrast and of interest, the morphology of microglial cells stimulated with LPS combined with either ASP-CM or GEMS-CM showed the presence of both amoeboid and ramified cells, with the predominance of the ramified morphology (Fig. 3E, F). Thus, both ASP- and GEMS-CM were able to reduce/inhibit LPS-induced changes in microglial morphology.
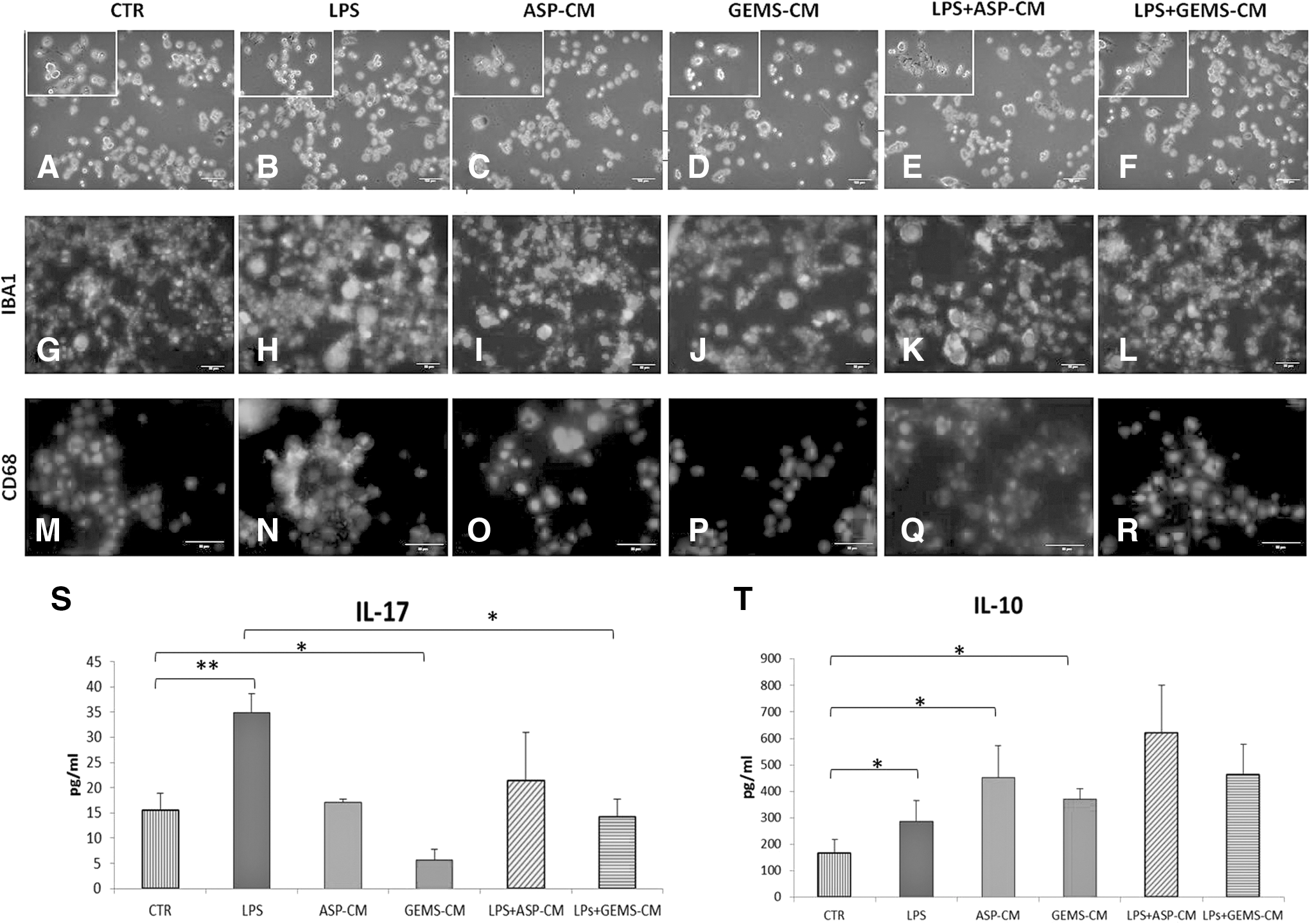
ADSC-CM treatment inhibits LPS-induced activation in microglial cells.
Then, to assess the expression markers of resting/activated microglia, we performed immunofluorescent analyses. In all experimental conditions, DAPI counterstaining showed that microglial cells exhibited IBA1-immunoreactivity in a similar manner (Fig. 3G–L). On the contrary, the expression of CD68, a recognized marker for activated phagocytic microglia, was different among the tested treatments. Indeed, CD68 was expressed at very low (if any) levels before microglial LPS activation (Fig. 3M), as well as after ASP- or GEMS-CM treatment (Fig. 3O, P), but prominently increased after LPS treatment (Fig. 3N). Of relevance, treatments with both types of ADSC-CM were able to suppress the LPS-induced expression of CD68 (Fig. 3Q, R). This indicates that ADSC-CM were both able to potently inhibit the upregulation of functional phagosomes in LPS-treated microglia.
Our investigation proceeded with the aim of assessing if ADSCs were able to affect the microglia production of inflammation-related ILs. As shown in Fig. 3S, T, after 48 h of culture, resting microglia (CTR) released both IL-10 and IL-17 in the medium. As expected, LPS exposure increased the extracellular level of both cytokines, with a high significance for the IL-17 one. Of note, both ADSC-CM alone affected the basal release of IL-17 and IL-10 in LPS-treated microglia. Indeed, in the presence of LPS and ASP- or GEMS-CM, a significant decrease of the proinflammatory IL-17 and a significant upregulation of the anti-inflammatory IL-10 occurred (Fig. 3S, T).
ADSC-CM inhibit LPS-induced proliferation and chemotaxis of microglia
We next investigated the effect of CM on microglia viability, growth, and chemotaxis prior and after LPS treatment (Fig. 4). In the used experimental conditions, LPS treatment, either alone or in combination with ASP- or GEMS-CM had no effect on microglia survival. Indeed, after 48 h of different treatments, microglia viability was higher than 95% in all cases (Fig. 4A). The results of the proliferation analyses demonstrated that microglial proliferation was significantly enhanced by LPS (Fig. 4B), and the treatment with either ASP- or GEMS-CM was able to counteract the LPS-induced proliferative effect. Indeed, the proliferation of microglial cells cotreated with LPS and ASP-/GEMS-CM did not differ from untreated cells (Fig. 4B).
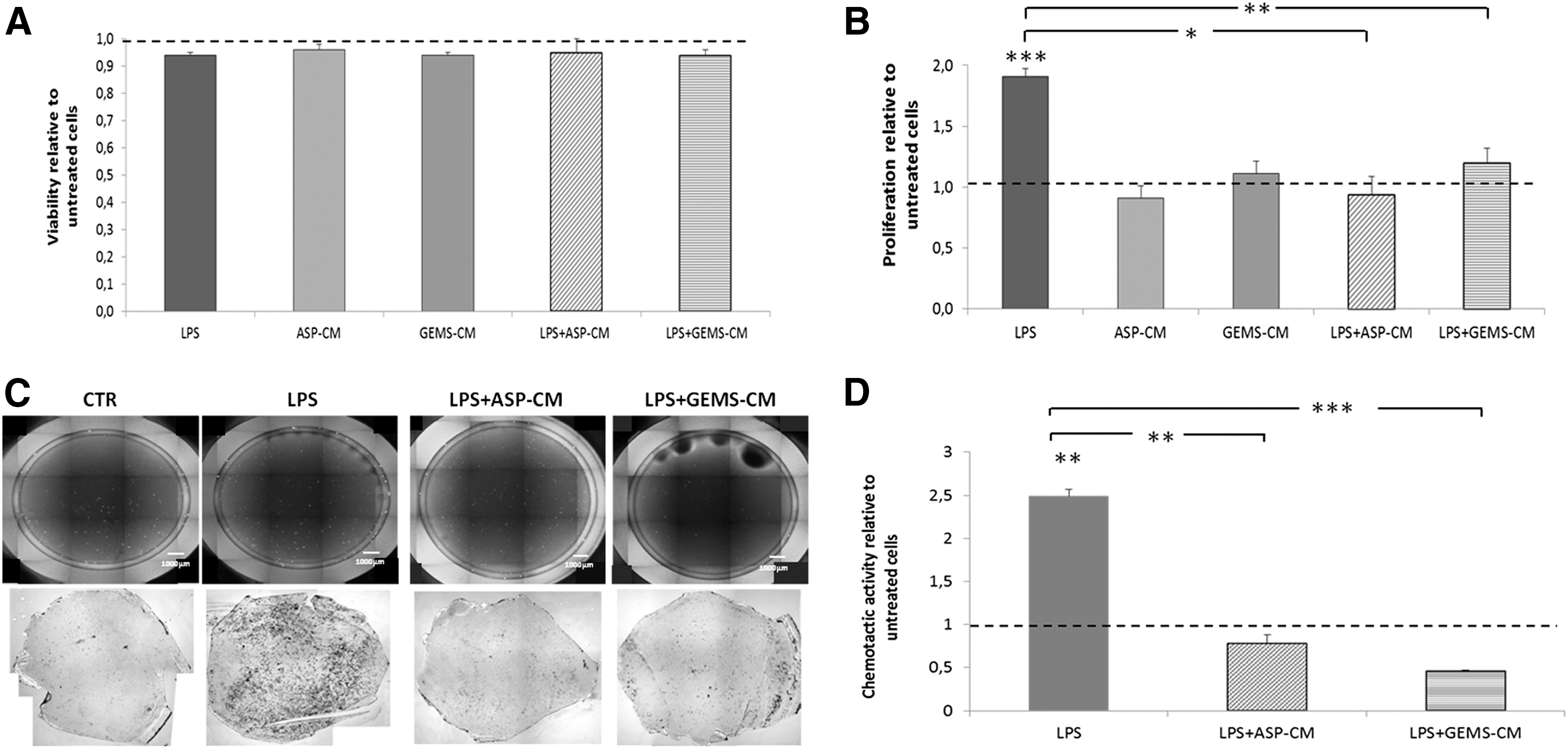
ADSC-CM inhibits microglial proliferation and chemotaxis. Cell viability
In further studies, microglial chemotaxis was investigated. We first found that, in resting conditions, N9 microglial cells were able to spontaneously migrate, and this migration was significantly promoted by LPS (Fig. 4C, D). Both ASP- and GEMS-CM treatments exerted a significant antichemotactic effect on LPS-induced microglia migration. In fact, the migration of microglial cells treated with LPS together with ADSC-CM was significantly decreased when compared with that of cells treated with LPS alone (Fig. 4C). Hematoxylin/eosin staining confirmed these trends, in that event microglial migration was lower in CTR condition than after LPS stimulation (Fig. 4C). The quantification of migrated microglial cells revealed that the LPS-induced chemotactic activity was significantly reduced by both ADSC-CM (Fig. 4D).
S1P is involved in the proliferative behavior of LPS-stimulated microglia
To investigate whether LPS-induced proliferation depends on the SphK activity, microglial cells were exposed to LPS in the absence or presence of DMS, a potent inhibitor of SphKs. As shown in Fig. 5A, the administration of DMS significantly decreased the LPS-induced incorporation of tritiated thymidine into microglial cells, suggesting that the SphK activity and thus S1P formation are required by LPS to exert its growth promoting effect on microglia. In support, cell exposure to S1P induced a potent proliferative effect in both the absence and presence of ADSC-CM (Fig. 5B). This indicates that S1P is able to stimulate microglial proliferation, even in the presence of ADSC-CM. Notably, similar results were obtained at 200 nM (Fig. 5B) and 1 μM (data not shown) concentration of S1P, suggesting that S1P stimulates microglial proliferation through a receptor-mediated mechanism.

Effects of DMS and S1P on microglial cell proliferation. Incorporation of 3H-thymidine into microglial cells after 48-h incubation in BM in the absence or presence of LPS, GEMS-CM, 1 μM DMS
ADSC-CM inhibits S1P synthesis and secretion in LPS-stimulated microglia
Aiming to evaluate the possible role of cellular and/or extracellular S1P in ADSC-CM anti-inflammatory effects on microglial cells, we investigated whether the administration of ADSC-CM, in the absence or presence of LPS, results in any variation of intracellular and/or extracellular S1P production by microglia. Since both metabolism and release of S1P can be evaluated after administration of radiolabeled Sph [49], microglial cells were pulsed with radiolabeled Sph, in the absence or presence of the different treatments, and radiolabeled S1P was evaluated in both cells and media. As shown in Fig. 6A, after a 2-h LPS treatment, radiolabeled S1P was significantly increased, whereas ADSC-CM alone did not affect the basal S1P value. Of relevance, ADSC-CM in combination with LPS treatment were able to significantly counteract the LPS-induced S1P production by microglia.

Effects of LPS and ADSC-CM on S1P metabolism and extracellular release in microglia. Microglial cells were fed with 20 nM 3H-Sph for 1 h in the absence (CTR) or presence of LPS and/or ADSC-CM. At the end, cells and media were collected and processed as described in Materials and Methods. Radioactivity incorporation into cellular S1P
Of interest, after medium extraction and partitioning, radiolabeled S1P was found as a medium component in both untreated and treated microglial cells. The radioactivity associated to extracellular S1P was found similar in untreated and ADSC-CM-treated cells (Fig. 6B). We also found that LPS stimulation significantly increased S1P secretion by microglia, and, most importantly, both types of ADSC-CM were able to nullify the LPS-induced microglia release of S1P (Fig. 6B).
ADSC-CM downregulate the induction of extracellular S1P pathway upon LPS-stimulated microglia
On the basis of the above results, it was of interest to investigate whether the modulation of extracellular S1P pathway might influence ADSC-CM-induced inhibition of LPS microglial activation. To this purpose, we first tested the effect of FTY720, an S1PR functional antagonist [50,51]. We found that LPS stimulation of cell growth (as thymidine incorporation into DNA), chemotactic activity, or CD68 expression was significantly reduced by FTY720 (Fig. 7A–C). These results support that extracellular S1P, after binding to S1PRs, acts as an extracellular mediator of LPS-induced microglial activation, and FTY720 was able to inhibit this, in a similar manner to what it occurs with ADSC-CM treatments. In agreement, when LPS- and ADSC-CM-treated cells were incubated with exogenous S1P, a significant stimulation of microglial cell proliferation, chemotactic behavior, and CD68 expression was found (Fig. 7A–C).
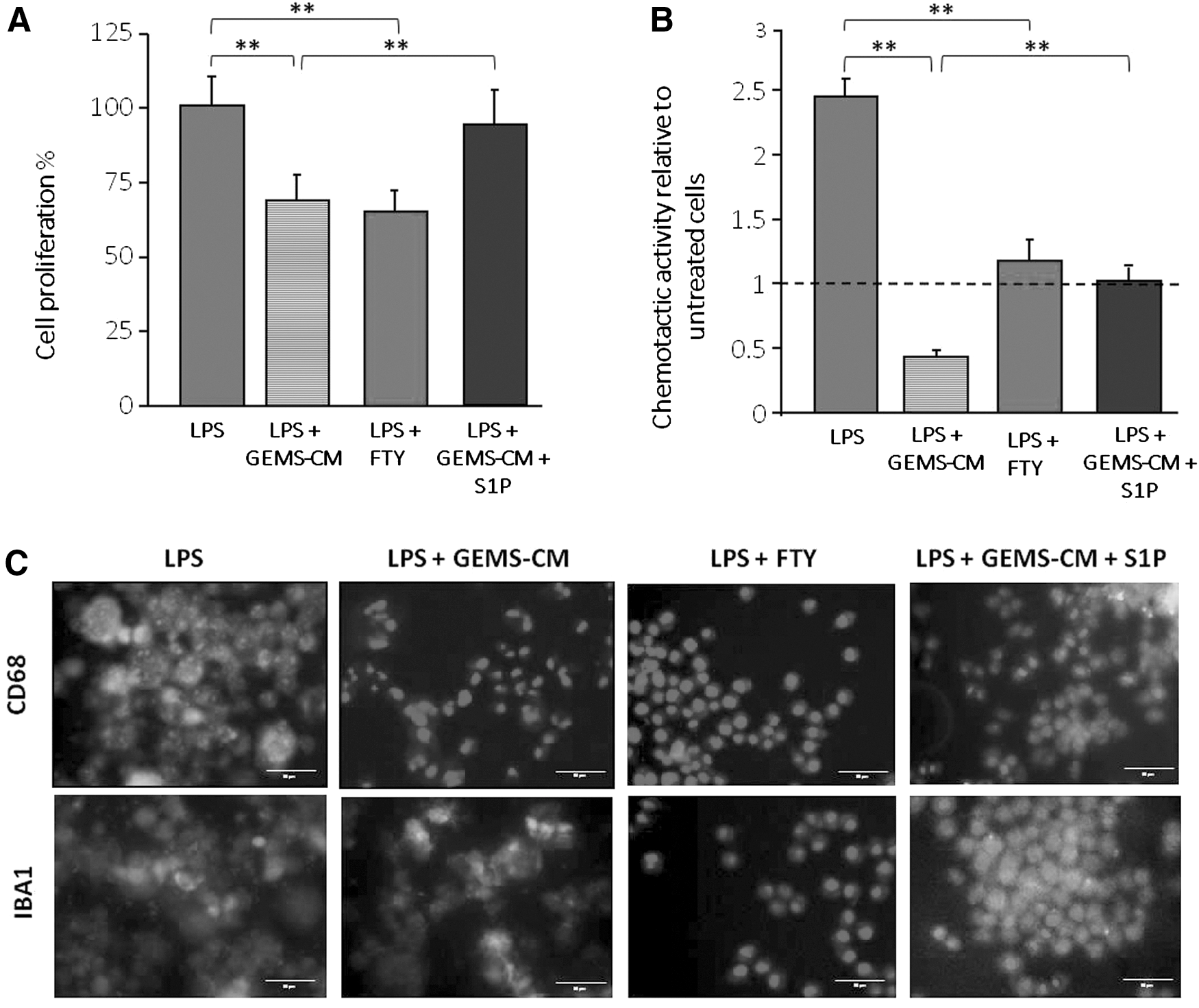
Effects of FTY720 and S1P on microglia proliferation, migration, and activation. Microglial cells were incubated 48 h in BM without or with LPS, GEMS-CM, 100 nM FTY720, and 200 nM S1P. The effects of the different treatments on cell proliferation
Discussion
Adipose tissue and ADSCs represent one of the major research topics in regenerative medicine. A great benefit is represented by the minimally invasive accessibility to adipose tissue and its ready availability [52]. To this purpose, in our study, we investigated the inflammatory profile of tissue isolated from fast, safe, and readily available source of fat explants [35] by lipoaspirate procedure and Lipogems device. This latter new technique, derived from lipofilling surgical method [43], represents a new field of regenerative surgery, whose therapeutic criteria are based on the possibility of transferring the MSCs [53] in those damaged areas of the human body that require repair and immunomodulation. We found that adipose tissues secreted several inflammation-related cytokines with different expression levels in two adipose sources. In particular, GEMS tissue presented a significantly lower content of some inflammatory cytokines, including IL-1β, IL-6, IL-8, and TNF-α, than the ASP tissue. Recent studies have shown that adipose tissue is a potent source of inflammatory cytokines by two types of cells: fat cells (mainly adipocytes) and non-fat cells [54]. As GEMS tissues were prepared by fragmentation of the fat ASP tissue, yielding a stromal vascular fraction highly enriched in pericytes and MSCs from human lipoaspirates, it is likely that the depletion of nonfat cells in GEMS-CM contributes to its low profile of inflammatory cytokines. The expression level of inflammation-related cytokines in both ADSC-CM was lower than that expressed from other human mesenchymal tissues, such as bone marrow [55], anyway.
In an effort to characterize resident ADSCs in fat tissue derivatives, it was observed that GEMS and ASP fat explants showed similar tissue architecture. In this study, we demonstrate that a subpopulation of stem cells, with mesenchymal phenotype, can be obtained from adipose tissue by both lipoaspirate and Lipogems procedures. Indeed, no difference in phenotypic profile in ADSCs obtained from GEMS or ASP tissues was observed [43]. In line with this, Bianchi and collaborators demonstrated that the in vitro culture of GEMS-ADSCs allows the isolation of a population of stem cells with a high degree of purity. These cells fulfilled the definition of MSCs, being able to differentiate into different cells of mesoderm type, including osteogenic, chondrogenic, and adipogenic ones (such as ASP cells) [33]. Moreover, we found that the Lipogems procedure was associated to a higher expression of different antigens and cell multipotency with respect to the ASP one, in agreement with recent reports [33,43]. This supports that Lipogems represents a valid and efficient procedure for obtaining ADSCs with a high degree of purity and low expression of proinflammatory cytokines.
In CNS, microglia are activated by various stresses and brain injury to provide supportive signals that lead to neuronal recovery. However, the persistence of the inflammatory processes in the CNS can cause serious damages to the neural complex and compromise its functional integrity, resulting in neuronal injury and neurological diseases, such as multiple sclerosis, trauma, Parkinson's disease, and Alzheimer's disease [56,57]. There is growing evidence that the modulation of microglia activation and the brain inflammatory state in general is able to improve the symptoms of many pathological conditions and decrease the phenomenon of neurodegeneration [58,59]. Based on these observations, microglial activation represents a potential pharmacological target to treat neurodegenerative and inflammatory diseases [60,61].
In this study, to evaluate the potential effects of ADSCs on microglia, we administered the supernatant of cultured ADSCs to resting and activated cells. Our results clearly show that ADSC-CM does not affect cell viability, but exhibits a potent anti-inflammatory effect on microglia, in a similar manner for both ASP- and GEMS-CM. Indeed, the LPS-induced inflammatory activation of microglia was counteracted by the ADSC secretome, by inhibiting multiple processes of activated microglia, including cytokine secretion, expression of molecular activation markers, cell proliferation rate, and chemotaxis. Thus, our data demonstrate that ADSC-CM exhibit potent inhibitory effect on the LPS-induced activation of microglia and prompt the maintenance of a resting phenotype.
Several studies have shown that MSCs from bone marrow are able to exert inhibitory effects toward microglia cells [41,55,62 –64]. Indeed, it was shown that the CM from rat bone marrow MSC was able to significantly inhibit the LPS-induced microglia proliferation, maintain the resting phenotype, and control markedly by suppressing major microglia functions, such as the phagocytic activity and cytokine production [55]. In addition, it was demonstrated that bone marrow MSC-CM exerts modulation on microglia in a cell contact-independent manner, through paracrine effects [41]. Thus, despite the evidence that human MSCs from different sources diverge in their expression of cell surface proteins and display distinct differentiation patterns [65], it appears that ADSCs share with MSCs from bone marrow the capacity to produce a secretome with anti-inflammatory properties against microglia. In agreement, a recent report showed that murine AMSCs, or their CM, induced changes in microglia morphology, decrease of proinflammatory cytokines, and increase in the phagocytic activity [66]. As human adipose tissue can easily be obtained in large quantities and represents a good source of ADSCs, our results underscore the potential of ADSC secretome for the development of acellular therapeutic interventions for diseases involving excessive microglial activation.
A major finding of this study is the evidence that the anti-inflammatory effects of ADSC-CM require microglial SphK/S1P as a target. First, we found that LPS-induced proliferation of microglia was strongly inhibited by the SphK inhibitor DMS. Moreover, exogenously administered S1P was able to mimic the proliferative effect of LPS, suggesting S1P as mediator of LPS effect. It is well recognized that SphK1 plays a key role in inflammatory responses, such as production of cytokines in immune cells (see Aoki et al. [67] for a recent review). Our findings are in agreement with previous reports, showing that SphK1 microglial expression is greatly and rapidly enhanced by LPS [11,14], and the evidence that blockade of SphK1 activity suppresses inflammatory responses in microglial cells [11,14,68].
Further pulse studies with radiolabeled Sph demonstrated that LPS treatment of resting microglia induced not only an increase in the cellular S1P but also, most importantly, a significant elevation of S1P in the cell medium. To our knowledge, this is the first report demonstrating that microglial cells are able to extracellularly release S1P and this secretion is potentiated by LPS. This finding may be of relevance for the understanding of not only microglia inflammatory properties but also of the pathogenesis of neurodegenerative diseases.
Final experiments of this investigation show that FTY720, a functional antagonist of S1PRs [51], inhibited multiple LPS-induced proinflammatory effects in microglia and provided the novel evidence that S1P suppressed the anti-inflammatory effect of ADSC-CM, suggesting that LPS-mediated microglial activation is countered by ADSC-CM through the modulation of SphK/S1P.
Conclusions
Overall, this study demonstrates that the proinflammatory cytokine expression level is different in adipose tissue obtained by lipoaspirate and Lipogems techniques, with Lipogems exhibiting markedly lower levels. Notwithstanding, both ADSC-CM potently inhibited LPS-induced microglial activation, by reducing or halting multiple inflammation-associated microglial properties. The main novelty of this study is to show, for the first time, the SphK/S1P axis as a target of ADSC-CM in exerting its anti-inflammatory action on microglial cells. Further in vitro and in vivo studies are needed to dissect the molecular mechanisms underlying ADSC secretome modulation of S1P signalling, and to confirm this finding.
Footnotes
Acknowledgments
This work was supported by IRCCS Foundation Ca’ Granda Ospedale Policlinico Milan and Università degli Studi di Milano, Italy. The authors thank Dr. Clara Di Vito for performing ELISAs, Dr. Letizia Venturini for assessing immunofluorescent analyses, and Prof. Giovanni Ricevuti for critical reviewing of the article.
Author Disclosure Statement
Carlo Tremolada is the inventor and pct patent holder of Lipogems. All other authors declare no competing financial interests.