
Abstract
Marrow stromal cells (MSCs) can be induced to differentiate into Schwann-like cells under classical induction conditions. However, cells derived from this method are unstable, exhibiting a low neurotrophin expression level after the induction conditions are removed. In Schwann cell (SC) culture, progesterone (PROG) enhances neurotrophic synthesis and myelination, specifically regulating the expression of the myelin protein zero (P0)- and peripheral myelin protein 22 (PMP22)-encoding genes by acting in concert or in synergy with insulin and glucocorticoids (GLUCs). In the present study, we investigated whether combined PROG, GLUC, and insulin therapy induced MSCs to differentiate into modified SC-like cells with phenotypes similar to those of mature SCs. After being cultured for 2 weeks in modified differentiation medium, the modified SC-like cells showed increased expression of P0 and PMP22. In addition, morphological and phenotypic characterizations were conducted over a period of over 2 weeks, and functional characteristics persisted for more than 3 weeks after the induction reagents were withdrawn. The transplantation of green fluorescent protein-labeled, modified SC-like cells into transected sciatic nerves with a 10-mm gap significantly increased the proliferation of the original SCs and improved axon regeneration and myelination compared with original BM-SCs. Immunostaining for P0 revealed that more of the transplanted modified SC-like cells retained the phenotypic characteristics of SCs. Taken together, these results reveal that the combined application of PROG, GLUC, and insulin induces MSCs to differentiate into cells with phenotypic, molecular, and functional properties of mature SCs.
Introduction
S
Marrow stromal cells (MSCs) have been proposed as a promising substitute for autologous SCs [8,9] based on their easy accessibility, rapidity of expansion, low immunogenicity, and multipotency [9,10]. Through exposure to classical chemical cocktails, MSCs adopt SC phenotypes, express cell-specific markers such as glial fibrillary acidic protein (GFAP) and S100 [11,12], and exhibit SC functional characteristics such as neurotrophin secretion [13 –15]. Nevertheless, these differentiated MSCs express very low levels of the mature SC markers, myelin protein zero (P0) and peripheral myelin protein 22 (PMP22) [16,17], which play indispensable roles in myelination and maintenance of myelin integrity [18]. Furthermore, the expression levels of SC markers and SC functional characteristics are transient after the induction reagents are withdrawn [4,13]. Therefore, it is necessary to optimize the differentiation culture conditions to produce cells with stable features of mature SCs for clinical application.
In cultured SCs, glucocorticoids (GLUCs) [19,20] and progesterone (PROG) [19,21,22] initiate myelination and increase the rate of myelin synthesis, particularly the stimulation of P0 and PMP22 promoter activities. Moreover, treatments using pairwise combinations of GLUC, PROG, and insulin result in the strong synergistic stimulation of neurotrophin synthesis and myelin protein expression [23,24]. However, to the best of our knowledge, the combined application of PROG, GLUC, and insulin for inducing MSCs into SC-like cells has not yet been reported. In the present study, we explored whether the combined application of PROG, GLUC, and insulin induced MSCs to differentiate into cells with more mature, stable SC-like phenotypes with functional characteristics in vitro and whether this treatment promoted axonal regeneration and remyelination in vivo.
Materials and Methods
Isolation and culture of rat MSCs
The isolation and culture procedures for MSCs were based on their ability to adhere to plastic surfaces and were performed according to previously reported protocols [8]. Briefly, under sterile conditions, the femur and tibia were excised from 6-week-old male Sprague–Dawley rats. The epiphyses were removed, and the bone marrow cavities were flushed thoroughly with cell culture medium using a syringe with a 22-gauge needle. The whole marrow plugs were dispersed homogenously and plated in Dulbecco's modified Eagle's medium-low glucose (Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS; Gibco) and 100 U/mL penicillin/streptomycin (Invitrogen, Carlsbad, CA). The MSCs (passage 4) were harvested and used in the subsequent experiments.
Characterization of cultured MSCs
To verify the multipotency of the rat MSCs, the cells were induced to differentiate into adipocytes and osteoblasts using differentiation medium following the supplier's instructions (Cyagen Biosciences, Guangzhou, China). MSCs from passage 4 were plated at a density of 4 × 103 cells/cm2 and were then incubated for 3 weeks in osteogenic differentiation medium or for 2 weeks in adipogenic differentiation medium. Osteogenesis and adipogenesis were evaluated using Alizarin Red and Oil Red O staining, respectively.
Transfection with lentivirus particles expressing green fluorescent protein
The transduction of MSCs with a lentivirus encoding green fluorescent protein (GFP, Catalog ID: LVCON145; GeneChem, Shanghai, China) was performed according to the manufacturer's instructions. At 6 days after transfection, the viral transfection efficiency was observed under a fluorescence microscope (Olympus BX51; Olympus, Center Valley, PA). Populations of more than 95% GFP+ cells were used for the differentiation and grafting experiments.
Differentiation of MSCs into SC-like cells
The induction of MSCs and GFP-labeled MSCs was performed as shown in Fig. 1. Briefly, cells were induced over 5 days using the standard method [4,11]. MSCs and GFP-labeled MSCs were incubated with α-MEM containing 1 mM β-mercaptoethanol (β-ME; Sigma-Aldrich, St. Louis, MO) for 24 h and then with α-MEM supplemented with 10% FBS and 35 ng/mL all-trans retinoic acid (ATRA; Sigma-Aldrich) for 3 days, followed by incubation in original differentiation medium (ODM) consisting of α-MEM, 10% FBS, and a mixture of 5 μM forskolin (FSK; Sigma-Aldrich), 10 ng/mL rat basic fibroblast growth factor (b-FGF; PeproTech, Inc., Rocky Hill, NJ), 5 ng/mL platelet-derived growth factor (PDGF; PeproTech), and 200 ng/mL heregulin-β1 (HRG-β1; R&D Systems, Minneapolis, MN). The cells were incubated with the ODM for another 9 days and were termed original bone marrow stromal cell-derived Schwann cells. After incubation for another 9 days with modified differentiation medium containing α-MEM supplemented with 10% FBS, 5 ng/mL PDGF, 10 ng/mL b-FGF, 5 μM FSK, 200 ng/mL HRG-β1, 20 nM PROG (Sigma-Aldrich), 0.5 μg/mL hydrocortisone (Sigma-Aldrich), and 5 μg/mL insulin–transferrin–selenium (ScienCell Research Laboratories, Carlsbad, CA), the cells were termed modified BM-SCs. MSCs without induction were used as the control. Fully induced MSCs were used in the subsequent in vitro experiments, and fully induced GFP-labeled MSCs were used in the subsequent in vivo experiments.
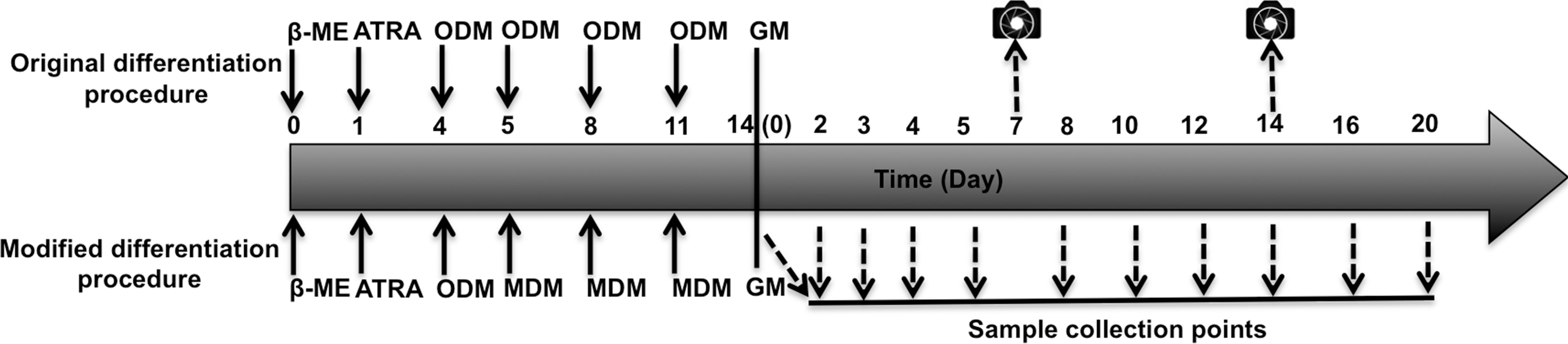
Schematic diagram illustrating the procedures for the differentiation of original BM-SCs and modified BM-SCs. Solid arrows represent the addition of factors at the indicated time points. Dashed arrows show the collection of samples for analyses. Camera icons represent the acquisition of photographs. β-ME, β-mercaptoethanol; ATRA, all-trans retinoic acid; ODM, original differentiation medium; MDM, modified differentiation medium; GM, growth medium; BM-SCs, bone marrow stromal cell-derived Schwann cells.
To compare the stability and functional characteristics of modified BM-SCs with original BM-SCs, the fully induced cells were cultured in growth medium (α-MEM supplemented with 10% FBS) for 3 weeks. The samples were collected at the indicated time points (Fig. 1).
Flow cytometry
The surface markers for undifferentiated MSCs, original BM-SCs, and modified BM-SCs were identified using flow cytometry (FCM). Briefly, four samples containing 5 × 105 single-cell suspensions were incubated with Alexa Fluor 488-, Alexa Fluor 647-, PE/Cy7-, or PE-conjugated antibodies against rat CD90, CD11b, CD29, or CD45 (Biolegend, San Diego, CA), respectively, in the dark for 30 min at 4°C. The remaining samples were subsequently incubated with all four antibodies. The data were acquired using FACSCalibur (BD Biosciences, San Jose, CA) and were analyzed using FlowJo 7.6.1 software (Tree Star, Inc., Ashland, OR).
Real-time polymerase chain reaction
The extraction of total RNA and the synthesis of first-strand cDNA templates were performed using TRIzol reagent and a SuperScript II First-strand Synthesis Kit (Invitrogen) according to the manufacturer's instructions. Eight sets of primers (Table 1) were designed with Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA) and synthesized by Sangon Biotechnology Co., Ltd. (Shanghai, China). Real-time quantitative polymerase chain reaction (RT-PCR) was performed using the StepOnePlus System (Applied Biosystems, Foster City, CA). All samples were repeated in triplicate and normalized to β-actin amplification as the endogenous internal control. The relative gene expression was calculated using the comparative CT (2−ΔΔCT) method, and the results are indicated as relative ratios of MSCs. RT-PCR data were analyzed using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems).
BDNF, brain-derived neurotrophic factor; GDNF, glial cell line-derived neurotrophic factor; GFAP, glial fibrillary acidic protein; NGF, nerve growth factor; P0, myelin protein zero; PMP22, peripheral myelin protein 22.
Western blot
Immunoblotting analysis was performed as previously described [25] with few modifications. Briefly, 30 μg of each protein sample was separated with 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) at 90 V and blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA) at 300 mA for 60 min. The membranes were blocked with 5% skim milk and then incubated at 4°C overnight with the following primary antibodies: mouse anti-S100 (1:500; Abcam, Cambridge, United Kingdom), rabbit anti-GFAP (1:5,000; Abcam), rabbit anti-P0 (1:2,000; Abcam), and rabbit anti-β-actin (1:2,000; Santa Cruz Biotechnology, Santa Cruz, CA). The blots were incubated with horseradish peroxidase-conjugated secondary antibodies (1:3,000; Cell Signaling Technology, Beverly, MA) for 1 h and visualized with an ECL chemiluminescence system (Amersham, Arlington Heights, IL). The band intensity of the primary antibody binding was analyzed using Quantity One 4.6.2 software (Bio-Rad Laboratories, Hercules, CA) and normalized to β-actin.
Enzyme-linked immunosorbent assay
Fully induced cells, after culture for 0, 2, 4, 8, 10, 12, 16, or 20 days in growth medium, were seeded at 2 × 105 cells per well (800 μL of growth medium) in 12-well plates for 48 h. Culture supernatants were measured with the rat β-nerve growth factor (NGF) enzyme-linked immunosorbent assay (ELISA) Kit (Sigma-Aldrich) against NGF and the Emax Immunoassay System (Promega, Madison, WI) against brain-derived neurotrophic factor (BDNF) according to the manufacturers' protocols. Sample absorbance was recorded with a plate reader at 450 nm in triplicate within 30 min after the addition of the Stop reagent. All samples were tested at least thrice, and the growth medium was used as the negative control.
Surgical procedures and transplantation of cells to the sciatic nerve
All animal study protocols were approved by the animal experimental committee of Huazhong University of Science and Technology. Surgery and animal care were performed in compliance with institutional guidelines and ethical standards (IACUC No. 410). Before transplantation, GFP-labeled cells were harvested and homogenously suspended at a final concentration of 1–2 × 108 cells/mL in Matrigel (BD Biosciences) containing 0.1% Fast Green (Sigma-Aldrich) to monitor the injection process and to ensure that no significant leakage occurred.
A total of 60 adult (14 ± 2 weeks old) male Sprague–Dawley rats weighing 300–350 g were used, and the experiments were performed as detailed in Table 2. All surgical procedures were performed using aseptic techniques under general anesthesia and according to Cui et al. [26], with slight modifications. The right sciatic nerves were completely exposed through a 3-cm skin incision at the middle of the thigh. Two tiny cuts, 2 and 12 mm distal to the internal obturator tendon and each 10 mm wide, were made in the epineurium. One end of the sciatic nerve was transected underneath the cuts, and the isolated nerve segment was then removed by the other end through the cuts. The cuts in the epineurium were anastomosed using 10-0 nylon sutures under a surgical microscope, and a total of 5–10 × 105 cells were then injected into the nerve gap through the epineurium using a 10-μL Hamilton syringe (Hamilton Co., Reno, NV). Matrigel alone was injected as a control. All surgical procedures were carefully performed to ensure the integrity of the epineurium. The surgical wounds were sutured after cell transplantation.
BM-SC, bone marrow stromal cell-derived Schwann cell; GFP, green fluorescent protein; MSC, marrow stromal cells.
Immunocytochemistry and immunohistochemistry
Immunocytochemistry and immunohistochemistry (IHC) analyses were performed as previously described [6]. For the immunocytochemistry of differentiated cells, primary antibodies against Nestin (1:300; Santa Cruz Biotechnology), P0 (1:400; Abcam), GFAP (1:1,500; Abcam), and S100 (1:300; Abcam) were used. The experiments assessing these markers were performed using MSCs as a negative control and the rat SC line, RSC96 (Cell Bank; Chinese Academy of Sciences, Shanghai, China), as a positive control. Alexa Fluor 488-conjugated anti-mouse IgG and anti-rabbit IgG (1:300; Invitrogen) were used as secondary antibodies.
To determine the morphological changes in response to new induction medium, the MSCs, original BM-SCs, modified BM-SCs, and RSC96 cells were immunostained for p75 neurotrophin receptor (p75NTR) (1:500; Abcam). Then, the cells were quantitatively evaluated based on their aspect ratios (cell length vs. cell width) as previously described [25].
At 1, 3, and 4 weeks after surgery, the animals were sacrificed, and regenerating sciatic nerve samples for the GFP+ modified BM-SC group (n = 5), the GFP+ original BM-SC group (n = 5), the GFP+ MSC group (n = 5), and the Matrigel group (n = 5) were harvested (Table 2). All sections were cut at a thickness of 15 μm, and the distribution and migration of transplanted GFP+ cells were immediately observed under a fluorescence microscope at three locations: 3 mm toward the distal end from the proximal epineurium cut (Prox), 3 mm toward the proximal end from the distal epineurium cut (Dis), and the remaining end (Med). Proximal nerve sections harvested at either 1 or 3 weeks were processed for P0 staining (1:300; Abcam). The distal nerve sections harvested at 4 weeks and without GFP signals as confirmed by fluorescence microscopy were stained for the axon marker NF-200 (1:400; Sigma-Aldrich) or for the myelin marker P0 (1:300; Abcam). Alexa Fluor 594-labeled donkey anti-rabbit IgG, Alexa Fluor 488-labeled donkey anti-rabbit IgG, and Alexa Fluor 488-labeled donkey anti-mouse IgG (1:300; Invitrogen) were used as secondary antibodies.
All slides were incubated with primary antibodies at 4°C overnight and subsequently incubated with secondary antibodies at 37°C for 60 min. All slides were stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for nuclear staining, and the omission of primary or secondary antibodies was used as the negative control for the staining process. At least five specimens from each treated animal were quantitatively examined, and the effect of cell implantation on nerve regeneration was quantified based on the following parameters: (1) GFP−/P0+-SC proliferation in the proximal sections at 1 week; (2) the percentage of implanted GFP cells colocalized with P0 in the proximal sections at 3 weeks; and (3) the areas of P0-myelin and the numbers of NF-200-axons in the distal sections at 4 weeks. The images were captured under a confocal laser scanning microscope (TCS-SP5; Leica, Mannheim, Germany) at consistent laser intensities and detection sensitivities in a randomized and double-blinded method. The data were quantified using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD). The number of GFP−/P0+-SCs and the percentage of implanted GFP+/P0+ cells were counted in five random fields of each specimen (using a 400× objective). The number of axons and area of myelin were counted in five different fields at a magnification of 200× and then extrapolated by using the area algorithm to estimate the total number of axons and total area of myelin for each nerve.
Statistical analyses
Data from the experiments are expressed as the mean ± standard error of the mean. Data were analyzed using one-way or repeated-measures analysis of variance, followed, if warranted, by multiple mean comparisons, using SPSS software (version 12.0; SPSS, Chicago, IL). P < 0.05 was considered statistically significant.
Results
Morphology and phenotypic characterization of rat MSCs
The MSCs at passage 4 displayed a high degree of morphologic consistency, showing large, flattened, and fibroblast-like morphologies (Fig. 3A). FCM revealed that these MSCs expressed CD29 (98.4%) and CD90 (90.8%), although a few CD11b- and CD45-positive cells were present (0.93% and 4.82%). In addition, 89.9% of MSCs were positive for both CD29 and CD90. Further analyses of CD29+/CD90+ MSCs revealed that 95.3% of these cells were negative for CD11b and CD45 (Fig. 2A). The adipogenic and osteogenic potentials were determined based on the production of lipid-rich vacuoles (Fig. 2B) and mineralized nodules (Fig. 2C).
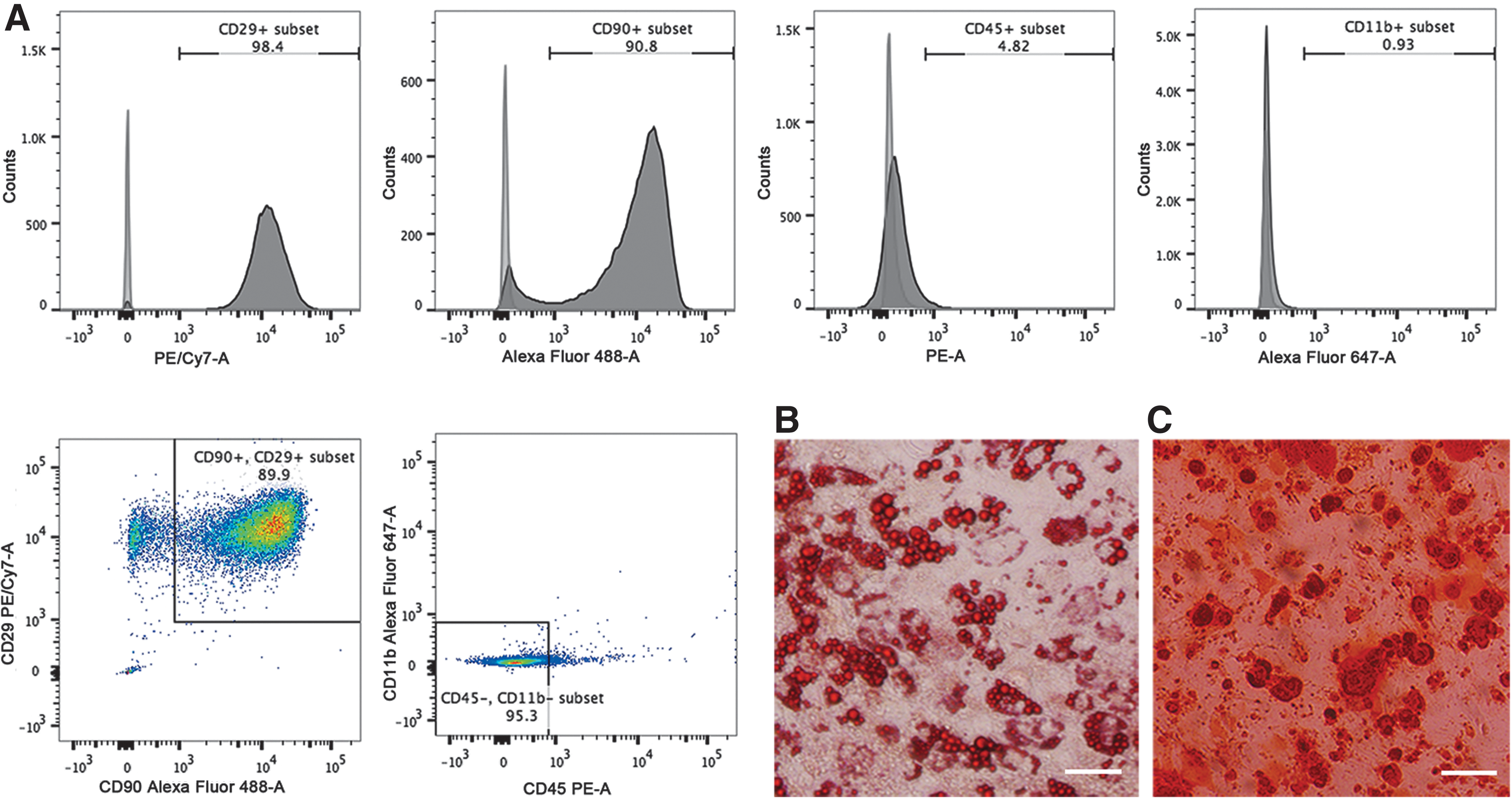
FCM analysis and multilineage potential of MSCs.

Morphological and phenotypic characterization of modified BM-SCs.
Morphological and phenotypic characterization of modified BM-SCs and original BM-SCs
Phase-contrast microscopy showed progressive morphological changes of MSCs during the SC induction process. After treatment with β-ME for 1 day, MSCs exhibited very rapid morphological changes, showing a refractive pyramidal or spherical cell body and fine, multiple, or bipolar processes, and they also completely stopped proliferating (data not shown). These cells gradually shifted their morphology to bipolar, small, and spindle-like shapes at 14 days after the induction of differentiation (Fig. 3A). Compared with original BM-SCs, modified BM-SCs displayed longer finer cellular processes and smaller bodies (Fig. 3A). Morphological measurements were obtained based on p75NTR expression as modified BM-SCs, original BM-SCs, MSCs, and RSC96 cells were all immunopositive for this protein (Fig. 3B). The aspect ratios of modified BM-SCs, original BM-SCs, and MSCs were 15.4 ± 2.6, 9.3 ± 2.64, and 3.2 ± 0.8, respectively, while that of the RSC96 cells was 15 ± 0.9 (Fig. 3C). However, FCM revealed that a few CD11b- and CD45-positive cells were present between both original BM-SCs (5.89% and 6.67%) and modified BM-SCs (0.81% and 1.70%). Moreover, most original BM-SCs and modified BM-SCs expressed the MSC markers, CD29 (99.6% and 97.6%) and CD90 (96.0% and 93.9%) (Supplementary Fig. S1; Supplementary Data are available online at
To further investigate whether these morphological changes were accompanied by phenotypic changes in the gene and protein expression levels of SC markers, RT-PCR, IHC, and western blot analyses were performed. Figure 3E shows that modified BM-SCs exhibited significantly upregulated mRNA expression of S100b, P0, and PMP22 (P < 0.01 for each), whereas the GFAP mRNA expression was downregulated compared with that of original BM-SCs (P < 0.01). The IHC analysis revealed that nearly all original BM-SCs were positive for Nestin and GFAP and weakly positive for S100 and P0 (Fig. 3D). Modified BM-SCs showed significantly increased immunoreactivity for S100 and P0 and decreased immunoreactivity for GFAP and Nestin (Fig. 3D). The western blot results further confirmed that modified BM-SCs showed upregulated levels of P0 and S100 and downregulated expression of GFAP (Fig. 3F). The morphology of the modified BM-SCs was elongated and slender, more closely resembling SCs than original BM-SCs. Moreover, modified BM-SCs exhibited increased gene and protein expression of mature SC markers.
PROG, GLUC, and insulin increased the stability of modified BM-SCs
To determine whether changes in PMP22 and P0 expression in modified BM-SCs increased the stability of the morphological and phenotypic characteristics, fully induced cells were cultured in growth medium for 3 weeks. After 7 days of culture, nearly all original BM-SCs reverted to the MSC morphology (cell length/cell width <4) (Fig. 4A). In contrast, after 2 weeks, ∼70% of modified BM-SCs maintained a spindle-like elongated morphology (cell length/cell width >9) (Fig. 4B). Moreover, even after 3 weeks, many of the modified BM-SCs maintained the spindle-like morphology (Supplementary Fig. S2).
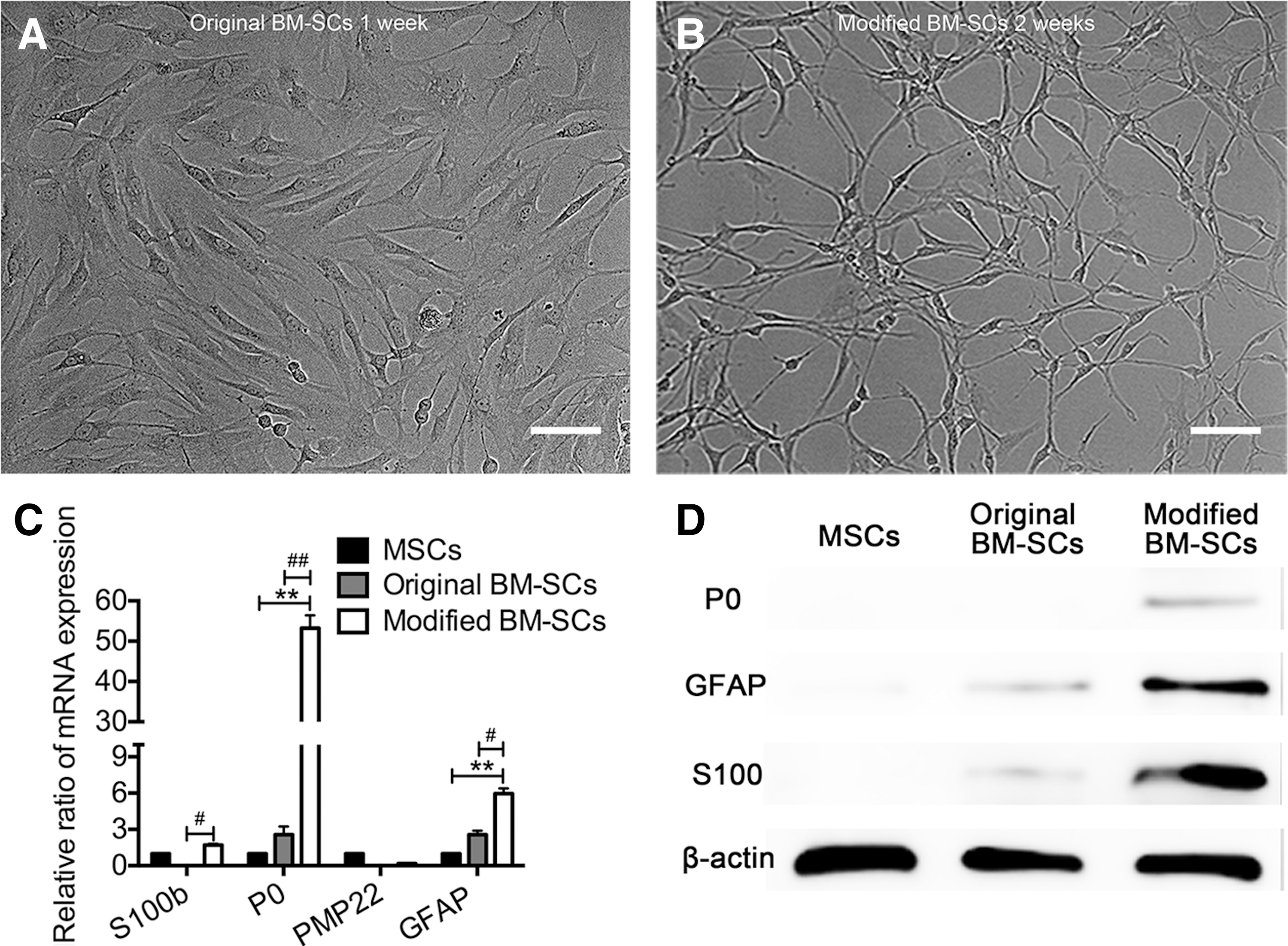
Morphological and phenotypic characterization of original BM-SCs and modified BM-SCs in growth medium.
The RT-PCR data showed that at 2 weeks in growth medium, modified BM-SCs exhibited upregulated GFAP mRNA expression (Fig. 4C). However, the mRNA levels of S100b decreased during the second week in culture, but remained higher than those in original BM-SCs (P < 0.05). The mRNA levels in original BM-SCs were approximately equal to or lower than those in MSCs (P > 0.05) (Fig. 4C). These results were further confirmed by western blots showing that modified BM-SCs still expressed S100 and P0 proteins and exhibited slightly increased levels of GFAP. However, these SC markers were nearly undetectable in original BM-SCs (Fig. 4D). These results demonstrated that modified BM-SCs were more morphologically and phenotypically stable than original BM-SCs after the removal of the induction reagents.
Modified BM-SCs enhanced the expression of neurotrophins, GDNF, NGF, and BDNF, for longer periods in growth medium
Although the above findings showed differences between modified BM-SCs and original BM-SCs in terms of the morphological and phenotypic characteristics, the functional properties of these cells remained unknown. Thus, the mRNA and protein expression of neurotrophic factor in modified BM-SCs was evaluated.
After 2 weeks of induction, modified BM-SCs did not show increased levels of glial cell line-derived neurotrophic factor (GDNF) mRNA (Fig. 5A). Moreover, decreased mRNA expression of NGF (P < 0.05) and BDNF (P < 0.01) was observed in modified BM-SCs compared with original BM-SCs (Fig. 5A). To determine whether neurotrophic factor mRNA levels changed after withdrawing the induction reagents, the original BM-SCs and modified BM-SCs were cultured in growth medium for 14 days. After 3 days, NGF, GDNF, and BDNF transcripts were highly expressed in original BM-SCs, and this expression decreased to basal levels after 5 days (Fig. 5A). In contrast, the mRNA expression levels of BDNF, NGF, and GDNF in modified BM-SCs increased after 5 days and remained significantly elevated for at least 2 weeks in growth medium (Fig. 5A).
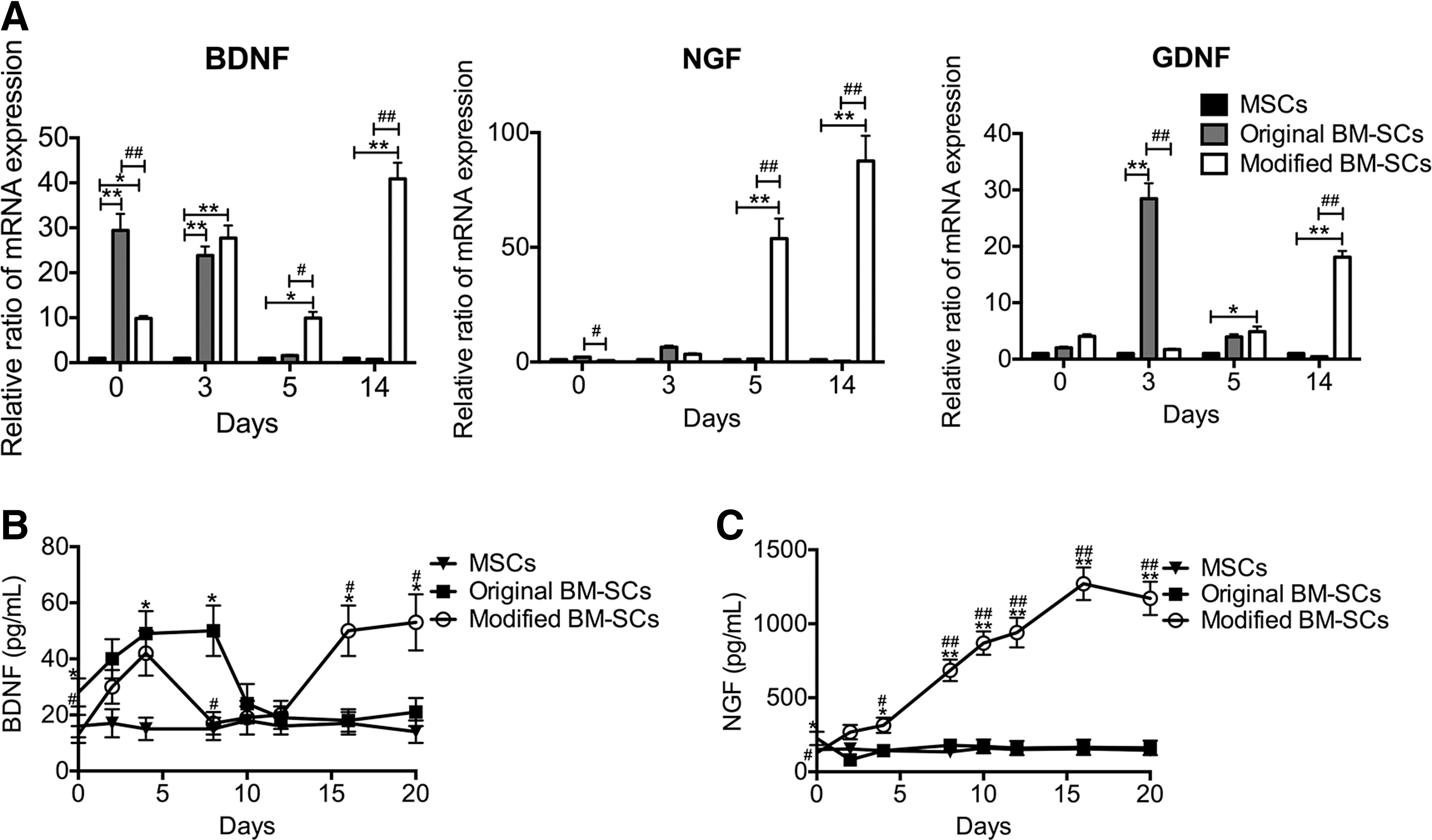
Neurotrophin analysis of cultured original BM-SCs and modified BM-SCs.
These results were further confirmed by an ELISA showing that original BM-SCs produced slightly higher amounts of BDNF and NGF than modified BM-SCs. NGF levels were 138 ± 15 and 76 ± 8 pg/mL in original and modified BM-SCs, respectively, and BDNF levels were 21 ± 4 and 10 ± 2 pg/mL (P < 0.05 for each) (Fig. 5B). In addition, the levels of BDNF and NGF secretion were significantly increased in these cells compared with the negative control (with medium only) (data not shown), and increased protein expression levels were observed in MSCs, 11 ± 2 and 68 ± 10 pg/mL for BDNF and NGF, respectively. After the first 7 days in growth medium, BDNF secretion increased from both original BM-SCs and modified BM-SCs. This secretion then significantly decreased. However, after 16 days, BDNF production increased again in modified BM-SCs, but not in original BM-SCs. At 20 days in growth medium, 53 ± 8 pg/mL of BDNF was expressed in modified BM-SCs, nearly 2.5 times more than in original BM-SCs (21 ± 3 pg/mL) (P < 0.05) (Fig. 5B). After 2 days in growth medium, the NGF secretion from original BM-SCs significantly decreased. However, NGF secretion from modified BM-SCs increased significantly and remained elevated, even after 20 days (Fig. 5C). At 20 days in growth medium, the ELISA quantification showed an average of 1,564 ± 160 pg/mL NGF in modified BM-SCs, 20 times the expression in original BM-SCs (70 ± 10 pg/mL) (P < 0.01) (Fig. 5C). This result indicated that modified BM-SCs showed enhanced secretion of neurotrophic factors for longer durations under induction agent-free conditions than did original BM-SCs.
Modified BM-SCs increased nerve regeneration through the promotion of host-derived SC proliferation and axon regeneration
The evaluation of the efficacy of transplanted cell stimulation on host-derived SC proliferation and axon regeneration is essential for future clinical applications. Consistent with previous reports [27 –30], transduction with GFP lentiviral vector has no adverse effects on MSC morphology, phenotypic characteristics, or differentiation potential (Supplementary Fig. S3A, B). Therefore, GFP-labeled cells were transplanted into the defects of transected sciatic nerves. After 1 week, the IHC analysis of the transverse sections stained for P0 revealed that the mean numbers of GFP−/P0+ cells (original SC) per field of vision increased nearly twofold in the GFP+ MSC group (19 ± 7), sevenfold in the GFP+ original BM-SC group (65 ± 10), and eightfold in the GFP+ modified BM-SC group (73 ± 12) compared with the control group (10 ± 4) (Fig. 6A, B). The continuous observations of longitudinal sections obtained at 1, 3, and 4 weeks after implantation revealed that the GFP-expressing cells (considered as viable implanted cells) populated these nerve defects within 3 weeks (Fig. 6D) and that there were no significant differences in the total numbers of surviving GFP+ cells among the three groups. However, low GFP signal was detected at 4 weeks in all GFP-labeled cell groups (Fig. 6D). These results indicated that implanted cells survived ∼3 weeks and may have facilitated the proliferation of host-derived SCs. In addition, the GFP+ modified BM-SCs displayed the most significant effects on host-derived SC proliferation. To quantify the GFP-labeled implanted cells expressing P0, the GFP+ and GFP+/P0+ cells were counted in transplanted nerves at 3 weeks after surgery (Fig. 6E). Quantitative analysis showed that at least 30% of the GFP+ modified BM-SCs colocalized with P0, while <5% of the GFP+ original BM-SCs and MSCs colocalized with P0 (Fig. 6C), suggesting that more modified BM-SCs maintained the phenotypic characteristics of SCs.
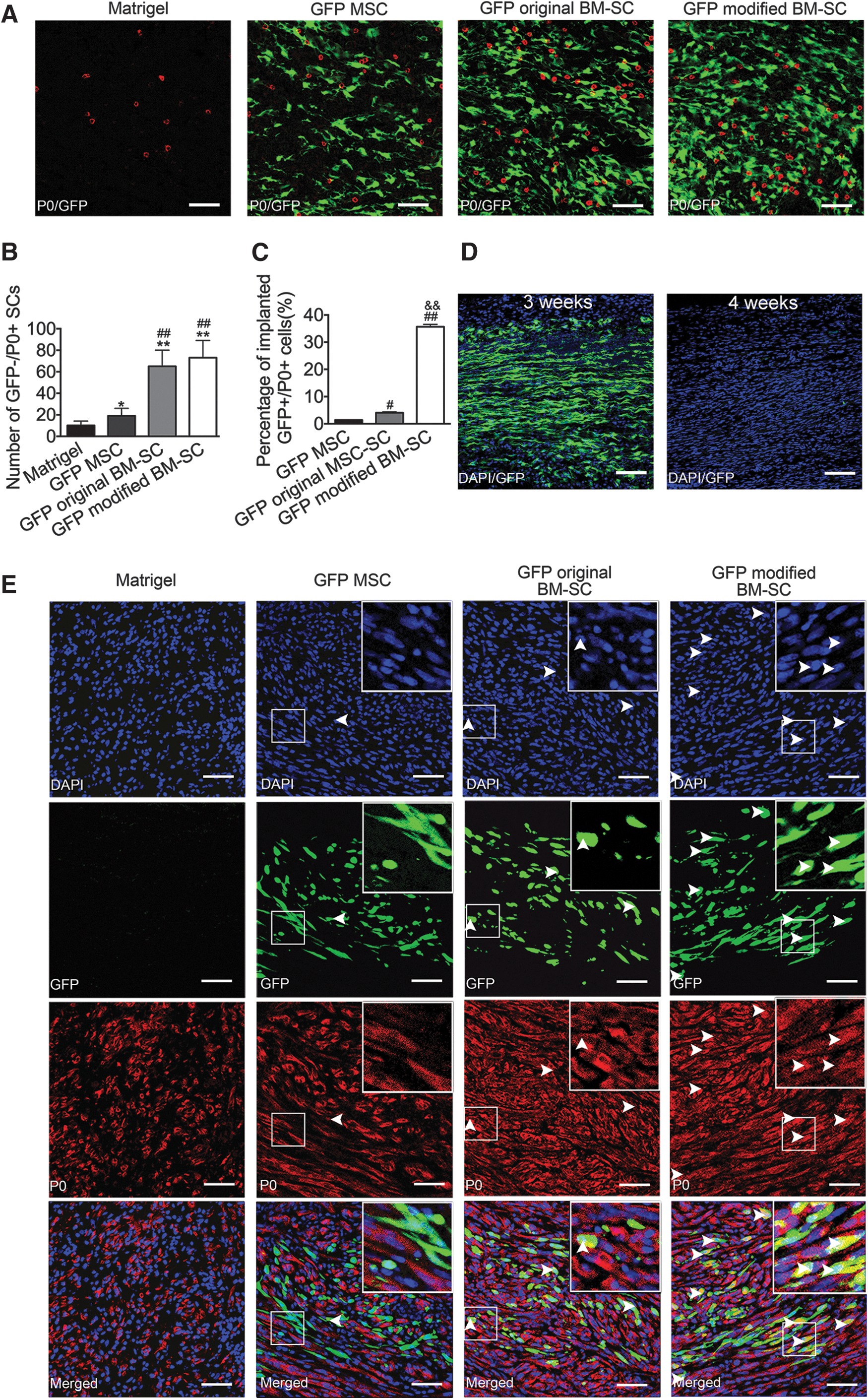
Laser scanning confocal microscopy of transplanted GFP-labeled cells.
Inspection of the transverse axial sections and immunostaining with anti-NF-200 revealed notable axon regenerative improvements in the GFP+ modified BM-SC group (Fig. 7A), with axon numbers of 3,324 ± 342 versus 1,299 ± 218 in the GFP+ original BM-SC group (P < 0.05), 902 ± 87 in the GFP+ MSC group (P < 0.05), and 469 ± 53 in the Matrigel group (P < 0.01) (Fig. 7C). Immunostaining with anti-P0 antibody showed that the total myelin area (in mm2) was higher in the GFP+ modified BM-SC group (1.62 × 10−1 ± 0.16 × 10−1) than in the GFP+ original BM-SC group (1.41 × 10−1 ± 0.12 × 10−1) and the GFP+ MSC group (0.93 × 10−1 ± 0.01 × 10−1) (P < 0.05 for each) and was eightfold higher than the Matrigel group (0.23 × 10−1 ± 0.04 × 10−1) (P < 0.01) (Fig. 7B, D). These results demonstrated that the transplantation of modified BM-SCs into the transected sciatic nerves at 4 weeks significantly improved axon regeneration and myelination compared with original BM-SCs.
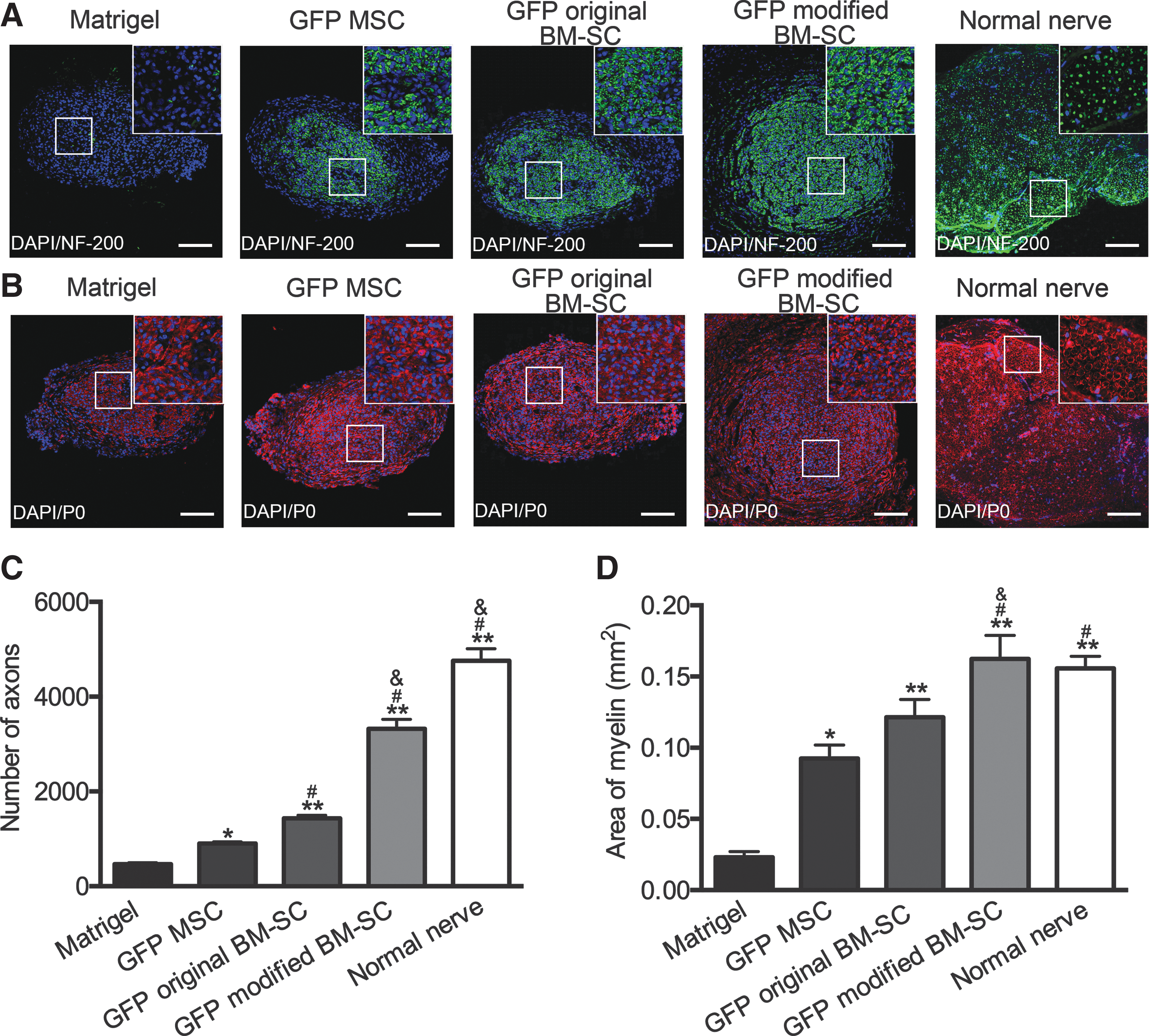
Effects of transplanting modified BM-SCs on axon regeneration and myelination.
Discussion
In this study, we reported for the first time that the combined application of GLUC, PROG, and insulin induced MSCs to adopt characteristics of mature SCs in vitro. The modified BM-SCs had increased expression levels of mature SC markers and significantly enhanced the stability of the phenotypic and functional characteristics of SCs even after the inductive factors were withdrawn. Most importantly, the transplantation of modified BM-SCs improved the stimulatory effects of early axon regeneration and myelination in vivo.
Consistent with previous studies [11,13,16], our results showed that the original BM-SCs resemble immature SCs and maintain the morphological and phenotypic characteristics of SCs for only a short time after inductive factors are removed, indicating that classical current-inducing reagents may not affect myelination [31]. Therefore, it is necessary to add new inducing reagents to the classical differentiation conditions to produce more mature, stable SC-like cells. For SC culture, PROG enhances neurotrophic synthesis and myelination, specifically by stimulating the expression of P0 and PMP22 [19,21,22]. However, the substitution of PROG for HRG in the classical induction of MSCs does not increase the S100 and P0 expression levels [16]. In addition, PROG does not stimulate promoter 2 of PMP22 [22] and does not influence SC growth [24,32]. GLUC activates PMP22 promoters 1 and 2 [20], and the combined application of PROG and GLUC induces a twofold increase in the activity of myelin gene promoters [24]. Furthermore, combined treatment with PROG and insulin results in the strong synergistic stimulation of cell proliferation, neurotrophin synthesis, and myelin formation [23]. Therefore, we used a combined induction therapy of GLUC, PROG, and insulin to optimize the differentiation culture conditions. The modified BM-SCs treated with the addition of GLUC, PROG, and insulin showed substantially upregulated P0 and PMP22 expression and downregulated GFAP expression, suggesting that they differentiated into mature SC-like cells [33]. Intriguingly, the modified BM-SCs showed slightly decreased expression of GDNF, NGF, and BDNF compared with original BM-SCs, further implying that the modified BM-SCs were more mature than the original BM-SCs [34].
In this study, after removal of the inducing reagents, modified BM-SCs developed a spindle-like elongated morphology. Moreover, the modified BM-SCs showed increased GFAP expression and decreased PMP22 and P0 expression, indicating that they had dedifferentiated into immature SC-like cells [33,35]. We propose that modified BM-SCs dedifferentiate into immature SCs and show increased secretion of neurotrophic factors after the induction medium is withdrawn, as do the host-derived SCs in the process of Wallerian degeneration [36,37]. Therefore, we evaluated the mRNA and protein expression of neurotrophic factors in modified BM-SCs. After the induction reagents were withdrawn for 5 days, the expression of BDNF, NGF, and GDNF in modified BM-SCs at the mRNA level was elevated. In addition, the NGF protein secretion level significantly increased after 2 days and remained elevated throughout the study. Meanwhile, BDNF production was increased after 16 days in the modified BM-SCs. Nevertheless, at 5 days after the induction reagents were withdrawn, neurotrophin expression in the original BM-SCs was remarkably reduced to a level nearly equivalent to that in MSCs, indicating that the original BM-SCs had turned into MSCs. These observations further verified that modified BM-SCs dedifferentiated into immature SC-like cells [36,37] and enhanced the expression and/or secretion of neurotrophic factors for longer durations.
The implantation of original BM-SCs or undifferentiated MSCs improved both myelination and the number of regenerated fibers [11,12,38]. The paracrine effect of neurotrophin [6,14,15,38] contributes to nerve regeneration. Compared with MSCs, induced SC-like cells secrete more neurotrophic factors [13,15], enhancing endogenous SC proliferation and myelination and improving the regeneration of injured axons [33,39 –42]. In this study, modified BM-SCs showed enhanced secretion of neurotrophic factors for longer durations with low initial amounts compared with original BM-SCs, which may reflect the significant enhancement of early nerve regeneration by the initial release of a low amount of neurotrophin compared with the initial release of a high amount [42]. In a sciatic nerve defect model, the potential myelination capacity of implanted MSCs was evaluated, and myelination was found to be a potentially important factor for nerve regeneration [11,43]. Previous reports have used immunofluorescence to demonstrate that induced mesenchymal stromal cells can myelinate regenerated axons in vitro and in vivo, and these results have been further confirmed by immunoelectron microscopy [11,44]. However, very few (<5%) of implanted stem cells are accompanied by myelination in vivo [43]. In the present study, at 4 weeks after transplantation, a low GFP signal was detected in all GFP-labeled cell groups, while GFP-labeled cells maintained in culture did not show decreased GFP expression (data not shown), indicating that only few GFP-labeled cells survived and that there were some types of paracrine or supportive effects of the GFP-labeled cells on host-derived SC proliferation and myelination at this time point. Although at 3 weeks after transplantation, ∼30% of implanted GFP+ modified BM-SCs expressed detectable levels of P0 (original BM-SCs: <5%); we propose that early nerve regeneration is more strongly influenced by the paracrine effect of neurotrophin than by the differentiation potential of implanted induced MSCs in vivo. Another potential benefit of nerve regeneration with modified BM-SCs is that these cells stimulate the proliferation of endogenous SCs and endogenous myelination in the microenvironment of a damaged nerve.
Previous reports have shown that implanted MSCs survive for more than 4 weeks after transplantation [12,15,43]. However, quantification studies have shown that the percentage of implanted MSCs surviving for up to 4 weeks after transplantation is <1% [45,46]. In the present study, the median survival time of the implanted induced MSCs was ∼3 weeks, which is still too early to evaluate the function of these cells in a long-term experiment for functional recovery of host animals. Several studies have demonstrated that 10 mm nerve defects require ∼3 weeks for connection [47,48]. Herein, we selected the 4-week time point after cell implantation to evaluate nerve regeneration. This enabled us to maximize the sensitivity level for the detection of treatment differences and to identify early axonal events important for the entire nerve regeneration process [49]. However, the long-term safety of modified BM-SCs and their function in nerve regeneration will be further evaluated in future research once the survival of transplanted cells in vivo can be extended. Moreover, the transdifferentiation cocktails remain toxic for the cells [50,51]. Our future studies will address the possibility of reducing the doses of the inducing regents.
The results of the present study have shown that the combined application of PROG, GLUC, and insulin represents a clear improvement over the classical differentiation culture conditions, increasing the stability of induced SCs in terms of morphological, phenotypic, and functional characteristics in vitro and their capabilities of enhancing axonal outgrowth and endogenous myelination in vivo. These results provide insights into the effects of the combined application of PROG, GLUC, and insulin for MSC differentiation into mature SC-like cells, which could be used for creating a microenvironment that promotes axon extension and the clinical treatment of peripheral nerve injuries in the future.
Footnotes
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81471270, 81252899).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.