
Abstract
Induced pluripotent stem cells (iPSCs) are an attractive cell source for cartilage regeneration, but current in vitro chondrogenic differentiation protocols yield insufficient results. In search for shortcomings of iPSC chondrogenesis, this study investigated whether SOX9 protein was adequately regulated during multiphase chondrogenic differentiation of two human iPSC lines in a comparable manner like during mesenchymal stromal cell (MSC) chondrogenesis. Upon generation of intermediate mesenchymal progenitor cells (iMPCs), SOX9 was induced and reached variable protein levels compared to MSCs. Along with an altered condensation behavior, iMPC cartilage formation was less robust compared to MSCs and better in the iMPC line with higher SOX9 protein levels. Despite efficient Smad-2/3 phosphorylation, TGF-β-driven chondrogenic stimulation downregulated SOX9 protein in iMPCs rather than increasing levels like in MSCs. Chondrogenesis was further improved by cotreatment with TGF-β + BMP-4, which appeared to shorten the duration of the SOX9 protein decline. However, this was insufficient to overcome heterogenic outcome and came at the expense of undesired hypertrophy. In iMPCs, but not MSCs, high levels of the SOX9-antagonizing hsa-miR-145 correlated with low SOX9 protein quantity. Thus, considerable iMPC heterogeneity with variable SOX9 protein levels, an altered condensation pattern, and low early SOX9 inducibility appeared as critical shortcomings of iPSC chondrogenesis. We suggest consistent quality of intermediate cell populations with high SOX9 protein induction as important indicators to obtain robust cartilage formation from iPSCs. The impact of this study is the identification of a SOX9 protein regulation opposite to MSC chondrogenesis that will now enable a selective adaptation of the currently limited protocols to the specific needs of iPSCs.
Introduction
T
Induced pluripotent stem cells (iPSCs) are frequently considered to be a ubiquitous cell source for cell therapies and tissue engineering strategies for any tissue. In contrast to adult stem cells, iPSCs possess an unlimited proliferative capacity that allows generating infinite cell quantities, thus meeting the clinical demand for the vast cell numbers anticipated to be needed for cell-based repair procedures. On the other hand, their developmental immaturity makes efforts to differentiate iPSCs into therapeutically valuable cells, such as chondroprogenitors or chondrocytes, challenging.
For bone marrow stromal cells (BMSCs) that contain multipotent mesenchymal stem cells, TGF-β is the required and sufficient chondro-inducer [1]. IPSCs, however, need more sophisticated strategies. The degree of difficulty in iPSC chondrogenesis is demonstrated by the delay between Yamanaka's report on the derivation of human iPSCs (hiPSCs) in 2007 [2] and the first report on successful chondrogenesis of hiPSCs in 2011 [3]. Moreover, weak proteoglycan staining intensities and lack of robust collagen type II deposition by chondrogenically differentiated hiPSCs, in several recent studies [3 –7], indicate the complexity of generating cartilage from hiPSCs in vitro. Thus, the current challenge is to develop strategies that robustly and reproducibly generate cartilage from hiPSCs, which meets the high quality standards regarding extracellular matrix composition, phenotype stability of the cells, and mechanical stability necessary for clinical application.
So far, varying methods for in vitro chondrogenesis of iPSCs have been reported and there is currently no generally accepted standard protocol. A common basis for many protocols is the attempt to reproduce embryonic cartilage development [8,9]. This strategy requires sequential induction of multiple developmental phases and, thus, production of an intermediate phenotype representing a metastable state of mesenchymal progenitors. These progenitors are subsequently induced into the chondrogenic lineage [4,8 –11]. Such iPS-derived mesenchymal progenitor cells (iMPCs) resemble BMSCs by expressing a similar surface marker profile (CD44+, CD73+, CD90+, CD105+, CD34−, CD45−) and a general capacity for in vitro differentiation into the three mesenchymal lineages (cartilage, bone, fat) [12 –15].
We and others recently directly compared iMPCs and BMSCs from the same donor and showed that despite evident similarities, iMPCs are not completely equivalent to BMSCs [12,15]. The most striking differences were their DNA methylation profile, which was still more closely related to iPSCs than to BMSCs, and their inferior inducibility to differentiate into the three mesenchymal lineages. Stimuli that easily induced BMSC differentiation were less efficient for iMPCs. In the current study, we focused on chondrogenic differentiation and searched for limitations that impede successful, efficient, and reproducible iMPC chondrogenesis.
SOX9 is commonly designated as the chondrogenic master transcription factor, since it is expressed in all chondrocytes [16]. SOX9 is required for the activation of chondrogenic gene expression [17] by binding to regulatory elements of cartilage-specific genes (COL2A1, COL9A1, COL11A, ACAN) [18 –24]. SOX9 target gene activation is often mediated by cooperative binding together with L-SOX5 and SOX6, which in combination with SOX9 are thus referred to as SOX trio [22,25]. Functional SOX9 knockout causes severe chondrodysplasia [26,27], and in a mouse model in which Sox9 was inactivated in limb buds before mesenchymal condensation, cartilage and subsequent bone formation were completely abrogated [28]. When, however, Sox9 was ectopically expressed under the Prx1 limb-specific promoter, Sox9 stimulated the formation of ectopic cartilage elements protruding from the limb buds [29]. This indicates that SOX9 is both necessary and sufficient to induce chondrogenesis in mesenchymal cells.
During in vitro chondrogenesis of BMSCs, SOX9 expression is thought to correlate with successful differentiation [30] and BMSCs with elevated SOX9 levels already before chondrogenic differentiation may perform better in chondrogenic assays [31]. We hypothesized that insufficient induction of the chondrogenic master regulator SOX9 is a relevant shortcoming of chondrogenesis of hiPSCs.
The aim of this study was, therefore, to elucidate whether differentiation of iPSCs into iMPCs induced sufficient basal expression of SOX9 to levels observed for culture-expanded BMSCs. Moreover, we assessed whether iMPCs can as efficiently respond to stimulation with TGF-β and BMPs and whether chondrogenic stimulation robustly amplifies SOX9 levels as known for BMSC chondrogenesis. This is important for future attempts to efficiently and reproducibly generate high-quality cartilage tissue from hiPSCs for drug screening, disease modeling, and tissue regeneration.
Materials and Methods
Cell culture
The iPS cell line D1 was generated from fibroblasts of a healthy donor (University Medical Center Mannheim, Germany; ethics committee approval No 2009-350N-MA) [32]. The iPS(IMR90)-4 cell line (abbreviated iPS-IMR) from Wicell was originally reprogrammed from IMR90 human fetal fibroblasts by the Thomson group [33]. IPS cells were routinely cultured on Matrigel® hESC-qualified matrix (Corning Life Sciences) with mTeSR™-1 medium (STEMCELL Technologies) and medium for D1-iPCs was supplemented with 100 μg/mL Normocin™ (CAYLA-InvivoGen). Medium was exchanged every day. Upon confluency, iPSCs were detached from the plates using 1 U/mL dispase (STEMCELL Technologies) and reseeded at the appropriate density with mTeSR-1 medium supplemented with 10 μM ROCK inhibitor Y27632 for D1-iPSCs (Miltenyi Biotec GmbH).
To generate iMPCs, medium of 50%–70% confluent iPS cell colonies on Matrigel® was switched from iPS expansion medium to mesenchymal stromal cell (MSC) expansion medium (DMEM high glucose w/o
BMSCs were isolated from fresh bone marrow aspirates obtained from four patients undergoing total hip replacement or osteotomy, as previously described [34]. Written consent was obtained from all tissue donors and the study received approval from the local ethics committee. Cells were seeded into gelatin-coated flasks with the MSC expansion medium described above.
For 4-day chondrogenic induction experiments, high-density cultures were initiated at passage 3. 5 × 105 iMPCs or BMSCs were seeded into 48-well plates with chondrogenic basal medium consisting of DMEM high glucose supplemented with 0.1 μM dexamethasone, 0.17 mM ascorbic acid 2-phosphate, 5 μg/mL transferrin, 5 ng/mL sodium selenite, 1 mM sodium pyruvate, 0.35 mM proline, 1.25 mg/mL bovine serum albumin (all from Sigma-Aldrich), 5 μg/mL insulin (Lantus®; Sanofi-Aventis), and 100 U/mL penicillin/100 μg/mL streptomycin supplemented with 10 ng/mL transforming growth factor-beta (TGF-β; PeproTech) and with or without the addition of 100 ng/mL bone morphogenetic protein (BMP)-4 (R&D Systems) as indicated. Medium was changed on day 2 and samples were harvested for further analyses on day 4 or day 7.
For 6-week chondrogenic micromass pellet culture, 5 × 105 iMPCs or BMSCs were centrifuged to form a micromass pellet and cultured in chondrogenic basal medium supplemented with 10 ng/mL TGF-β with or without the addition of 100 ng/mL BMP-4. Medium was exchanged thrice a week.
Quantitative polymerase chain reaction
RNA was extracted with TriFast (peqGOLD; Peqlab) following standard guanidinium thiocyanate/phenol extraction protocols. Polyadenylated mRNA was isolated from total RNA using oligo d(T)-coupled magnetic beads (Dynabeads; Dynal, Life Technologies) according to the manufacturer's instruction and reverse-transcribed using Omniscript® (Qiagen). Transcript levels were analyzed by quantitative PCR using LightCycler™ technology (Roche Diagnostics) and the following primers: SOX9 forward GTACCCGCACTTGCACAAC, SOX9 reverse TCGCTCTCGTTCAGAAGTCTC, SOX5 forward ATGCGGCAGTACTTCAATGT, SOX5 reverse CCTCTTCCTCGTCGTACTCA, RPL13 forward CATTTCTGGCAATTTCTACAG, RPL13 reverse CAGGCAACGCATGAGGAAT. Relative gene expression was calculated by the ΔCt method using RPL13 as a reference gene.
MiR analysis by quantitative polymerase chain reaction
A 10 ng of total RNA was used for cDNA synthesis with the TaqMan®MicroRNA Reverse Transcription Kit. Relative expression levels were determined through quantitative polymerase chain reaction using TaqMan MicroRNA Assays (both Applied Biosystems, Life Technologies) and the LightCycler technology according to the manufacturer's instructions. MiR detection signals were normalized to snRNU6 (U6) and relative expression levels were calculated using the ΔCt method.
Western blotting
Whole cell lysates were prepared with PhosphoSafe™ Extraction Reagent (Novagen, Merck Millipore) supplemented with 1 mM Pefabloc® (Sigma-Aldrich). Protein concentration was determined with Bradford reagent (Sigma-Aldrich) and bovine serum albumin standards. Thirty microgram protein of each sample was separated by denaturing sodium dodecyl sulfate–polyacrylamide gel electrophoresis and proteins blotted onto a nitrocellulose membrane.
The lower part of the membrane was immunostained for 1 h with mouse monoclonal anti-human beta-Actin antibody (GeneTex; 1:10,000 in 5% skim milk) and bands were detected with a peroxidase-conjugated goat anti-mouse IgG secondary antibody (1:5,000; Jackson ImmunoResearch) using ECL detection (Roche). The upper part was stained for 2 h with rabbit polyclonal anti-Sox9 antibody (1:2,000; Merck Millipore ab5535) followed by a peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:10,000; Jackson ImmunoResearch).
Phosphorylated Smad2 protein was detected with a rabbit monoclonal anti-phospho-Smad2 (Ser465/467) antibody (1:250, overnight at 4°C, #3108 Cell Signaling Technology Europe) and total Smad2/3 protein with a rabbit monoclonal anti-Smad2/3antibody (1:250, overnight at 4°C; #8685 Cell Signaling Technology Europe), respectively. Phosphorylated Smad1/5/9 was detected with a rabbit monoclonal anti-phospho-Smad1 (Ser463/465)/Smad5 (Ser463/465)/Smad9 (Ser465/467) antibody (1:250, overnight at 4°C, #9511 Cell Signaling Technology Europe). Total Smad1/5 was detected with rabbit monoclonal antibody to Smad1 (1:500, overnight at 4°C, [EP565Y] Abcam) and a rabbit monoclonal antibody to Smad5 (1:1,000, overnight at 4°C, [EP619Y] Abcam).
Immunohistochemistry
Constructs were washed with PBS, fixed in 4% formaldehyde, dehydrated, and embedded into paraffin. Five-micrometer-sections were stained with monoclonal mouse anti-human collagen type II antibody (MP Biomedicals/Quartett), or mouse anti-human collagen type X antibody (MP Biomedicals/Quartett) according to standard immunohistochemistry protocols [35]. Alkaline phosphatase (ALP) activity on paraffin sections was stained through the conversion of the ALP substrates NBT and BCIP.
Statistical analyses
For each group, medians, first and third quartiles, maximal and minimal values, as well as extreme values (>three-fold interquartile range) were calculated. To evaluate the statistical significance of the observed differences, a Kruskal–Wallis test with post hoc Mann–Whitney-U Signed-Rank tests were conducted. Only significant Kruskal–Wallis tests were analyzed post hoc. A two-tailed significance value of P ≤ 0.05 was considered statistically significant. Correlation between SOX9 mRNA and protein as well as SOX9 protein and microRNA levels was assessed according to Spearman correlation analysis. Data analysis was performed with SPSS software.
Results
Variable SOX9 protein levels in iMPCs compared to BMSCs
In iPS-D1 cells, SOX9 mRNA, barely detectable by quantitative RT-PCR (Fig. 1A), started to be upregulated around day 10 of iMPC generation, and reached significantly higher mRNA levels at day 14 (5.6-fold, P ≤ 0.05). Similar results were obtained with the independent iPS-IMR cell line (Supplementary Fig. S1A; Supplementary Data are available online at

SOX9 levels in D1-iMPCs.
Western blot analysis detected no SOX9 protein in iPS-D1 cells, whereas iMPCs expressed variable SOX9 protein levels in four independently derived D1-iMPC populations in passage 3 (Fig. 1D). On average, however, D1-iMPCs revealed significantly less SOX9 protein than four different BMSC donor populations (passage 3; Fig. 1D, E). Relative SOX9 levels in D1-iMPCs ranged from 3.8 to 26.5 (band intensity relative to β-Actin), thus varying 7.0-fold (Fig. 1E).
In BMSCs, SOX9 protein levels were between 35 and 119 and varied only 3.4-fold. In IMR-iMPCs, SOX9 protein levels were as high as in BMSCs and less heterogenic than in D1-iMPCs (Supplementary Fig. S1C, D). Importantly, SOX9 protein levels did not correlate with mRNA levels in iMPCs (D1: r = −0.2, P = 0.8; IMR: r = 0.0, P = 1.0) nor in BMSCs (r = −0.6, P = 0.4) according to Spearman correlation analysis (Fig. 1F) suggesting that SOX9 mRNA levels should not be used to judge SOX9 activity. In conclusion, SOX9 protein levels in iMPCs seem afflicted with cell line-dependent variability and individual D1-iMPC populations started into subsequent chondrogenesis from lower and more heterogeneous SOX9 protein levels than IMR-iMPCs and BMSCs.
SOX9 downregulation by chondrogenic stimulation in iMPCs despite efficient Smad phosphorylation
To test whether iMPCs were capable to efficiently respond to the chondro-inducer TGF-β, we analyzed phosphorylation of Smad 2/3, which is induced by the canonical TGF-β pathway, to regulate target gene expression. D1-IMPCs and BMSCs were treated with TGF-β in chondrogenic medium for 4 days and analyzed for Smad 2/3 protein levels and phosphorylation. Western blot analysis showed phosphorylated Smad 2 on day 4 of chondrogenesis for all BMSC donor populations (Fig. 2A). In line, all four tested D1-iMPC populations showed a considerable induction versus day 0 and responded with high levels of phosphorylated Smad 2 on day 4 of chondrogenesis (Fig. 2B). Interestingly, the condensation behavior of iMPCs that formed microscopic nodules deviated from BMSCs that formed large aggregates (Supplementary Fig. S2).
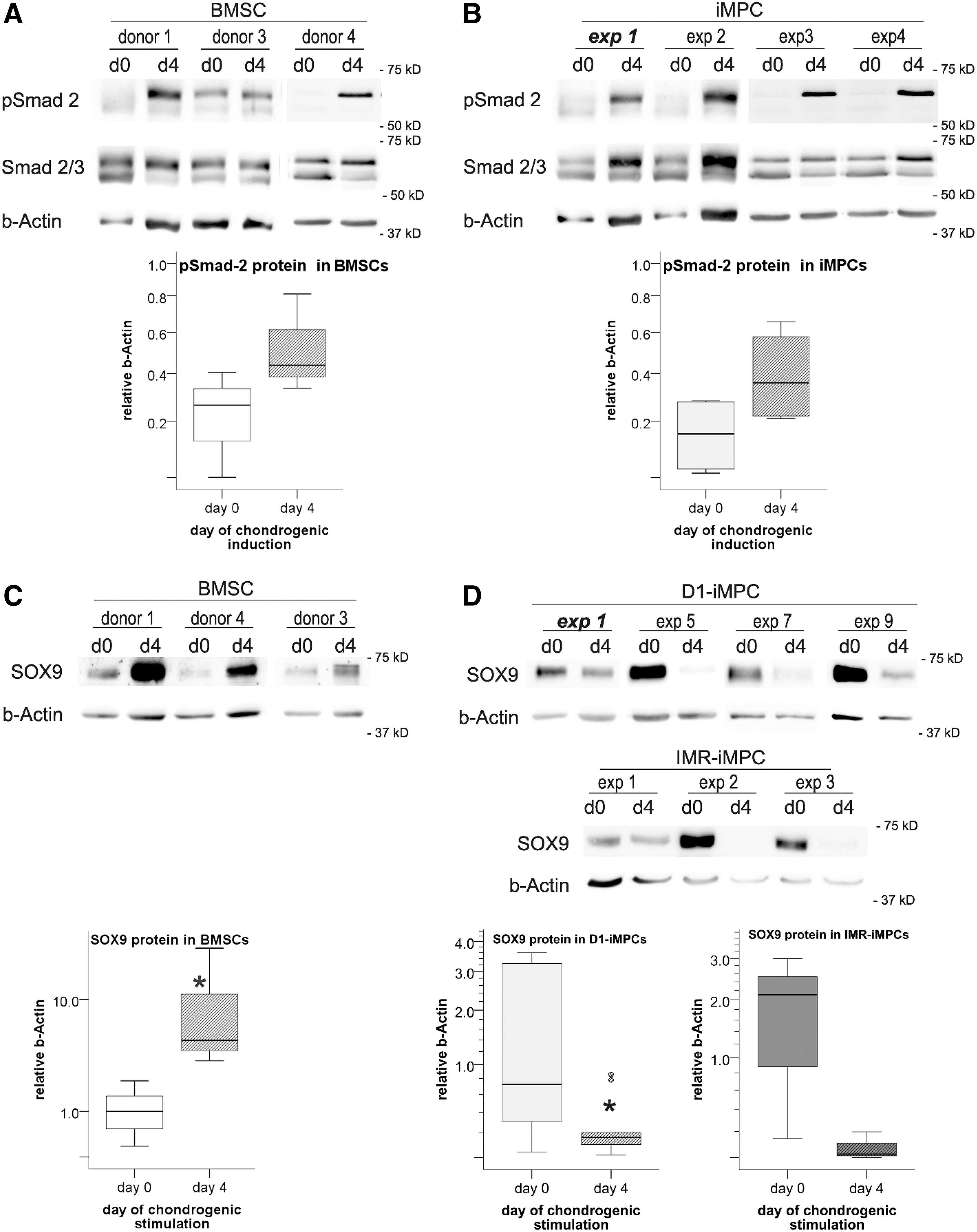
Smad 2 phosphorylation in BMSCs
Having established that iMPCs can adequately react to TGF-β, we next asked whether TGF-β in chondrogenic medium upregulates SOX9 protein in iMPCs equally well as in BMSCs. Western blot analysis demonstrated that BMSCs had robustly upregulated SOX9 protein levels 4 days after chondrogenic induction (9.3-fold, P ≤ 0.05; Fig. 2C). In iMPCs, in contrast, SOX9 levels were lower after 4 days of chondrogenic induction compared to day 0 for both D1-iMPCs (5.4-fold, P ≤ 0.5, n = 10; Fig. 2D) and IMR-iMPCs (23.8-fold, P = 0.13, n = 3). Thus, within the first 4 days of chondrogenesis, iMPCs downregulated SOX9 protein rather than inducing it like BMSCs, thus unraveling an opposite early SOX9 reaction to standard chondrogenic conditions of the two cell types.
Inefficient iMPC chondrogenesis can be enhanced by BMP-4 treatment
To assess whether iMPCs would still be able to form cartilage, despite initial SOX9 downregulation, iMPCs were subjected to standard micromass pellet culture in chondrogenic medium supplemented with TGF-β for 6 weeks. Immunohistochemical analysis revealed collagen type II deposition in some parts of the micromass pellets of three out of eight independently derived D1-iMPC populations (Fig. 3) and in all six tested IMR-iMPCs (Supplementary Fig. S3A). This demonstrated that a fraction of the iMPCs in the pellets was able to differentiate into chondrocytes depositing cartilaginous matrix. However, pellets from the remaining five D1 populations showed no evident collagen type II deposition, whereas such complete failure was not observed with IMR-iMPCs.
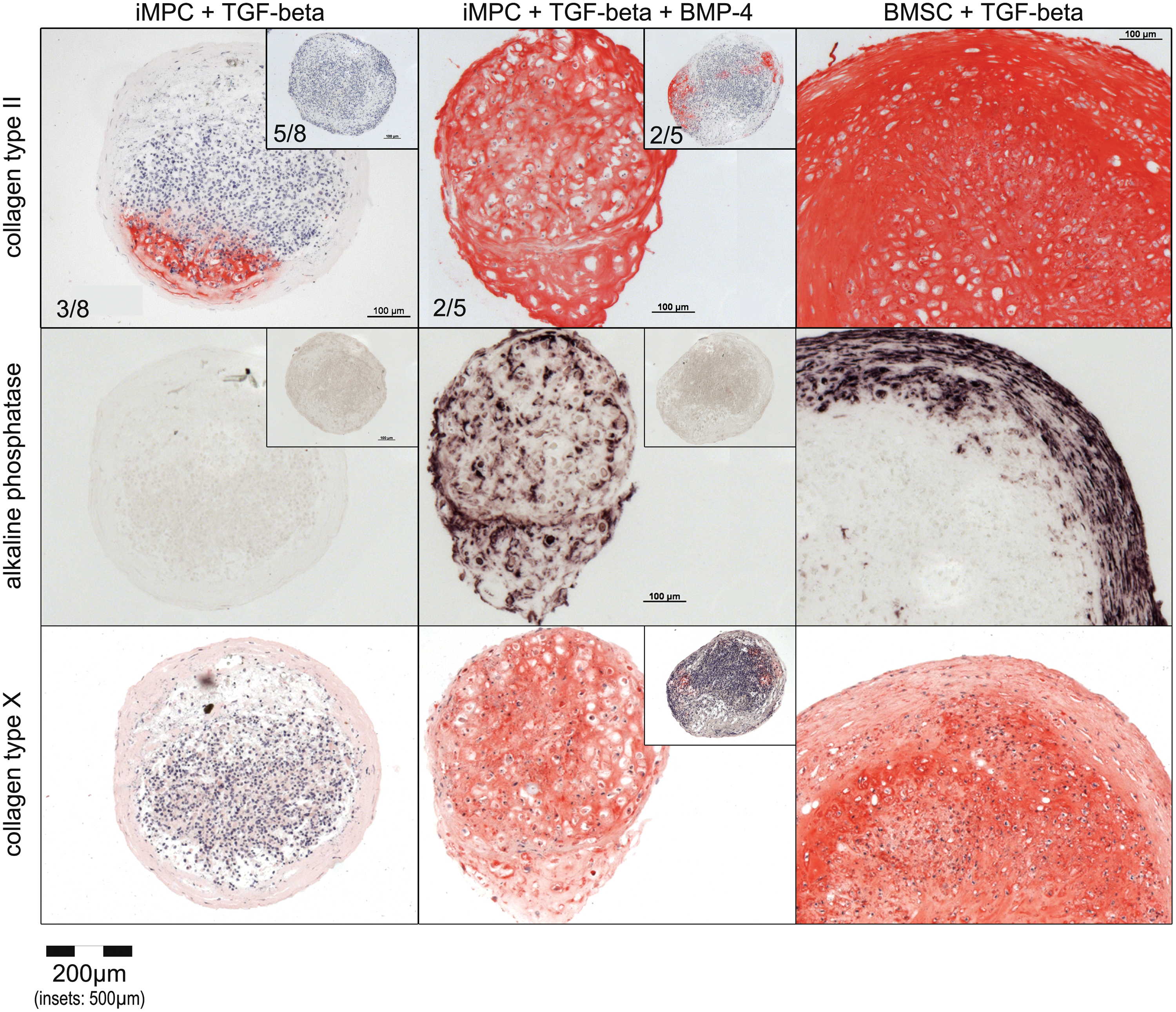
Chondrogenic differentiation of D1-iMPCs and BMSCs after 6 weeks of chondrogenic micromass culture. Pellets consisting of 500,000 cells were treated for 6 weeks with TGF-β (+ BMP-4) in chondrogenic medium as indicated. Representative immunohistological staining of serial sections for collagen type II and X as well as alkaline phosphatase activity staining of pellets from two independently derived D1-iMPC populations (exp 1, insets: exp 2) and 1 BMSC donor population are depicted. Numbers indicate the quantity of independent experiments per total experiments performed with a similar outcome. Color images available online at
All BMSCs subjected to 6 weeks of chondrogenic micromass culture with TGF-β deposited collagen type II homogeneously throughout the entire pellet (Fig. 3). This clearly demonstrated that standard micromass pellet culture and stimulation with TGF-β robustly induced cartilage formation from BMSCs, while iMPCs need different chondrogenic stimuli.
BMPs are frequently used to enhance chondrogenic differentiation of pluripotent stem cells [11,36]. When we performed chondrogenic micromass culture of iMPCs in the presence of TGF-β + BMP-4, four out of five tested D1-iMPC populations were capable to at least partially differentiate and deposit collagen type II containing cartilaginous matrix after 6 weeks (Fig. 3). Two of the five D1-iMPC populations formed cartilaginous matrix throughout the entire pellet. Increased chondrogenesis in the presence of TGF-β + BMP-4 was also observed with IMR-iMPCs (Supplementary Fig. S3A).
To assess whether BMP-4 promoted undesired hypertrophic differentiation, D1-iMPC pellets were stained for ALP activity and for collagen type X deposition. Indeed, when cotreated with BMP-4, cells in the pellets tended to comprise a hypertrophic morphology, strongly activated ALP, and deposited collagen type X in line with enhanced hypertrophic cartilage formation (Fig. 3). In total, the success rate for at least partial chondrogenesis of D1-iMPCs under cotreatment with TGF-β + BMP-4 was 80% (4/5) and, thus, considerably higher than that observed under treatment with TGF-β without BMP-4 in chondrogenic medium (38%, 3/8). Thus, BMP-4 was capable to enhance iMPC chondrogenesis, but was not an ideal chondrogenic stimulus, due to still partial success and stimulation of undesired hypertrophy.
Combined use of TGF-β + BMP-4-stimulated Smad 1/5/9 phosphorylation beyond Smad 2/3 phosphorylation in both D1-iMPCs and BMSCs (Fig. 4A). However, SOX9 levels were still downregulated or remained low on day 4 of D1-iMPC chondrogenesis (Fig. 4B). After 7 days of stimulation, SOX9 protein levels tended to be higher under cotreatment with TGF-β + BMP-4 than without BMP in both D1- and IMR-iMPCs (Fig. 4C and Supplementary Fig. S3B), indicating that downregulation is transient and culture conditions produced a considerable delay of SOX9 induction compared to BMSCs.
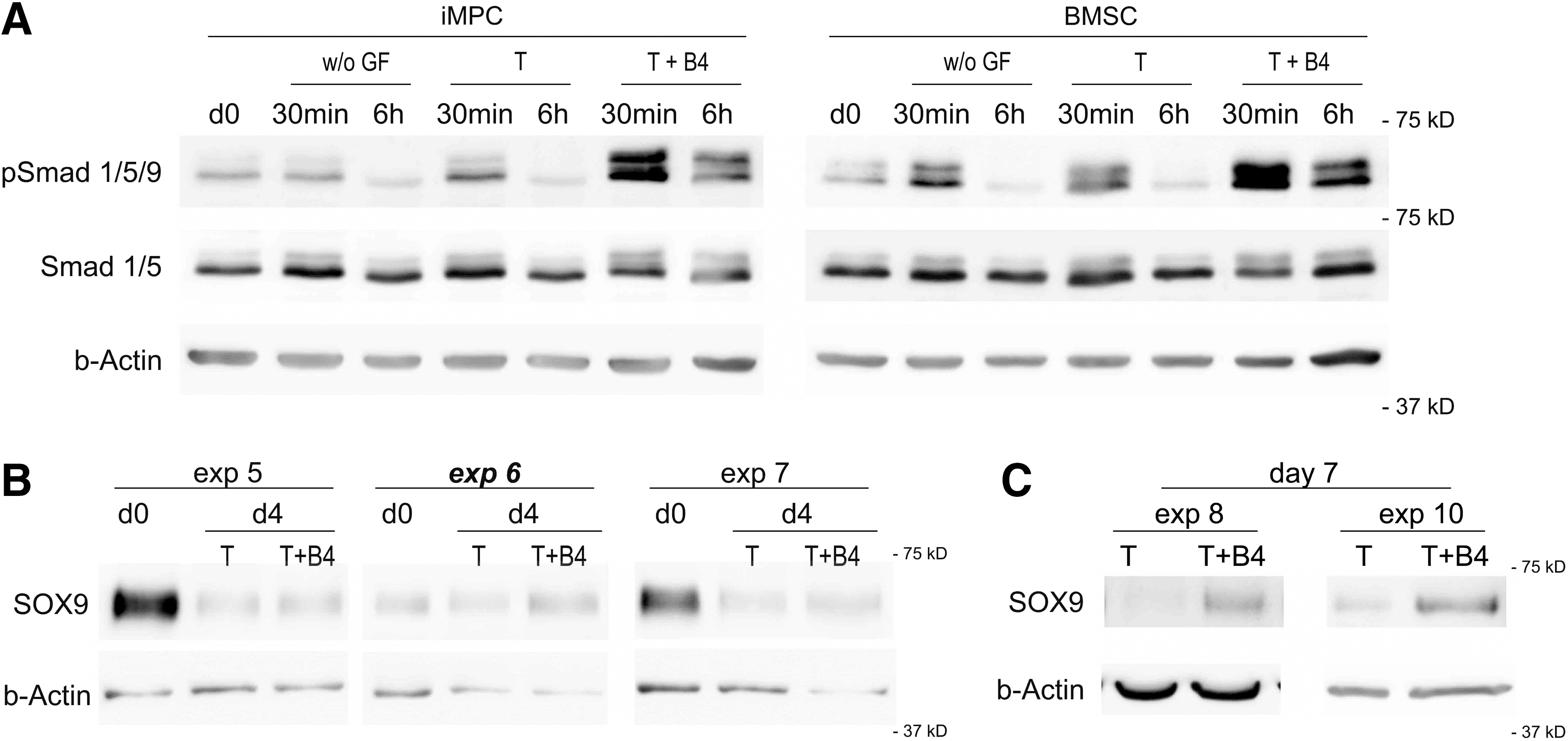
Smad 1/5/9 phosphorylation in D1-iMPCs as well as in BMSCs
High miR-145 levels in iMPCs correlate with low SOX9 levels
To elucidate why high SOX9 mRNA levels in some iMPCs translate into comparably low SOX9 protein levels, we investigated expression of the SOX9 targeting microRNA hsa-miR-145, which was shown to suppress SOX9 translation [37 –44]. Quantitative PCR revealed that hsa-miR-145 and the coexpressed hsa-miR-143 levels were strikingly variable in D1-iMPCs (Fig. 5A, B).
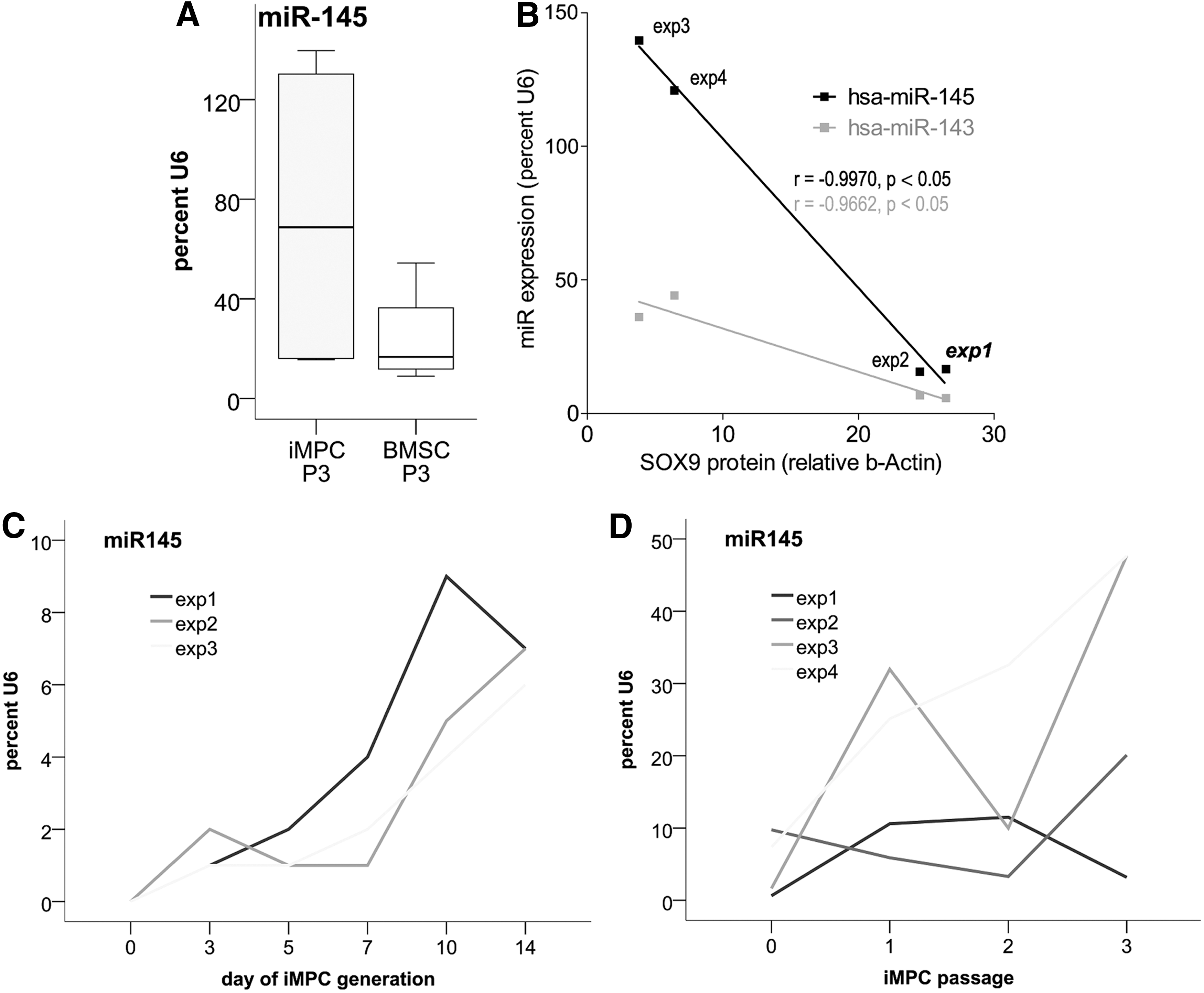
MiR-145/143 expression in iMPCs.
In IMR-iMPCs and in BMSCs in contrast, SOX9 protein levels were less heterogeneous along with low variability of hsa-miR-145 expression (compare Fig. 5A with Fig. 1D, E for BMSCs, Supplementary Fig. S1C, D, and data not shown for IMR-iMPCs). In IMR-iMPCs, the mean miR-145 expression was significantly lower than in D1-iMPCs (8.6-fold, P ≤ 0.05) and reached only 5.9% U6 compared to 50.3% U6 in D1-iMPCs (data not shown). Interestingly, low miR-145 expression in IMR-iMPCs was accompanied by high SOX9 protein levels that were as high as in BMSCs (Supplementary Fig. S1). D1-iMPC populations with high levels of hsa-miRs-145 and 143 expressed low SOX9 protein levels, whereas D1-iMPC populations with low levels of hsa-miRs-145 and 143 expressed higher SOX9 protein levels (Fig. 5B). Note that of all D1-iMPC populations, exp 1 showed the best chondrogenic differentiation (Fig. 3), had high SOX9 protein levels (Fig. 1D), and low miR-145 levels (Fig. 5B). Pearson correlation analysis verified a highly significant negative correlation between hsa-miR-145/143 expression and SOX9 protein levels (hsa-miR-145: r = −0.997, P ≤ 0.05; hsa-miR-143: r = −0.966, P ≤ 0.05) in D1-iMPCs, but not in BMSCs.
MiR-145 expression rose homogeneously during the initial 14 days of D1-iMPC generation (Fig. 5C) and heterogeneity developed during the subsequent passages 0–3 of maturation culture (Fig. 5D). In this phase, iMPCs also adopted a typical MSC surface marker profile and turned positive for CD73, CD90, CD105 remaining negative for CD34, CD45 at passage 3. Passage 0 iMPCs, however, comprised low fractions of CD105-positive cells (data not shown). Moreover, P0 iMPCs were incapable of reproducible CFU-F formation and did not, in this respect, fulfill the criteria for MSCs. Thus, iMPCs were still in a transient phase between iPSCs and MSCs at P0.
These data suggest that inhibition of SOX9 mRNA translation by hsa-miR-145 may be one potential parameter for the divergence between SOX9 mRNA and protein levels in different D1-iMPC populations that could limit iMPC chondrogenesis.
Discussion
In search for shortcomings of robust chondrogenesis during multiphase chondrogenic differentiation of human iPS cells, this study investigated whether SOX9 protein was sufficiently induced in a comparable manner like in BMSCs. We show that, although SOX9 mRNA was induced during iMPC generation and reached levels seen in BMSCs, SOX9 protein levels were heterogeneous between independently derived iMPC populations. Chondrogenesis had to start out with lower SOX9 protein levels in D1-iMPCs than in IMR-iMPCs and BMSCs. Although efficient Smad 2/3 phosphorylation was observed by chondrogenic stimulation with TGF-β, SOX9 protein levels, surprisingly, were considerably downregulated in iMPCs, while SOX9 protein rose in BMSCs. Thus, iMPCs appeared to respond by an opposite early SOX9 reaction under standard chondrogenic conditions compared to BMSCs. Along with ineffective SOX9 regulation by chondrogenic stimulation, iMPCs formed cartilage less efficiently in vitro compared to BMSCs.
Treatment with BMP-4 improved chondrogenic differentiation of iMPCs, but at the expense of undesired signs of hypertrophy, and still only 40% of D1-iMPC populations deposited cartilaginous matrix throughout the entire pellet. Thus, treatment with TGF-β + BMP-4 was not sufficient to overcome heterogenic outcomes of iMPC chondrogenesis, but may have shortened the period of depressed SOX9 protein levels relative to TGF-β alone. Hsa-miR-145 expression, which negatively correlated with SOX9 protein levels in D1-iMPCs, may explain part of the discrepancy between SOX9 mRNA and protein levels. Altogether, to overcome iMPC heterogeneity, iMPC generation should be improved to produce iMPCs with a consistent quality regarding SOX9 protein induction. Preferably, stimuli should be adapted to upregulate SOX9 without delay already in the early condensation phase as known for embryonic chondrogenesis.
In this study, we used two iPSC lines that are both derived from fibroblasts. In view of several reports on the epigenetic memory of iPSCs for the tissue of origin [45,46], the use of iPSCs derived from BMSCs or chondrocytes might appear more intuitive. In a previous comparative study, however, the chondrogenic activity of iPSCs derived from BMSCs versus amniotic epithelium was low and did not vary considerably [15], suggesting that epigenetic memory alone cannot rescue limited cartilage formation.
Sequential induction of several developmental phases with production of an iMPC type, here called iMPCs, is a common strategy for in vitro cartilage generation from hiPSCs [4,8 –11]. To generate iMPCs, in this study we used the frequently applied method to culture adherent cells in serum-containing medium until they acquire a mesenchymal phenotype [12,47]. Alternatively, iMPCs are generated by growing the adherent cell population out of embryoid bodies [48]. Direct comparison of both methods yielded equivalent cell populations [12,15] and stimulation with BMSC-secreted factors on a BMSC-produced extracellular matrix did not add apparent quality [15]. Importantly, considerable heterogeneity between independently derived iMPC populations was linked with all these iMPC generation methods demonstrating that reproducible induction of iMPCs with a consistent quality is a common problem. In our study, heterogeneity evolved primarily during iMPC maturation from P0 to P3, as judged from notably variable SOX9 protein and miR-145 quantity at P3.
It is surprising that the current study appears to be the first investigating protein levels of SOX9 during iMPC generation and chondrogenesis. Most importantly, we show that SOX9 mRNA was not predictive of SOX9 protein levels demonstrating that a more detailed analysis beyond mRNA levels of this chondrogenic master inducer is mandatory to eliminate shortcomings of current methods. Previous work detected SOX9 mRNA in iMPCs [36,49,50], which remained constant or increased only marginally during 21 days of iMPC chondrogenesis [4,50,51]. Other studies reported SOX9 mRNA levels to rise at day 14 or day 21 of iMPC chondrogenesis [8,11,12,52] and, thus, much later than during embryo development, where SOX9 upregulation precedes cell condensation [28]. Our study emphasizes that detection of SOX9 mRNA cannot be taken as evidence for successful chondrocyte formation since lack of SOX9 protein appears to be a crucial limit that needs to be overcome for a robust in vitro cartilage formation by iMPCs. Although SOX9 protein levels, as high as in BMSCs before chondrogenic induction, may favor cartilage formation by iMPCs, this seemed not sufficient, because some D1-iMPC populations had a considerable SOX9 quantity, but still a low chondrogenic capacity (compare exp 2 in Fig. 1 with insets in Fig. 3). This demonstrated that SOX9 protein levels of iMPCs at passage 3 are no predictor of cartilage formation.
The major question raised by our results is what hinders iMPCs to efficiently respond to chondrogenic stimuli, upregulate SOX9, and form cartilage in vitro. We excluded the responsiveness toward TGF-β and BMPs as a primary cause for deficient chondrogenesis, because iMPCs phosphorylated Smad 2/3 and Smad 1/5/9 equally well as BMSCs.
The mechanisms underlying SOX9 transcription indeed remain fairly unresolved. Data for human BMSCs and iPSCs are rare, but in other cells (mouse in vivo models, murine chondrocytes, C3H10T1/2 cells, primary mouse and chick limb bud cultures, and SW1353 cells), multiple molecules apart from canonical Smad-mediated TGF-β and BMP signaling have been suggested to be involved in SOX9 transcription, such as ERK-1/2 activated by noncanonical TGF-β-signaling [53] and ATF2 activated by TAK1 (also noncanonical TGF-β signaling) [54] have been indicated to induce SOX9 expression.
The transcriptional repressors TGIF and SnoN have been suggested to suppress the TGF-β-induced and Smad-mediated induction of SOX9 [55]. Moreover, Notch and its target genes Hes/Hey have been described to regulate SOX9 transcription [56,57]. β-catenin has been shown to mediate SOX9 protein degradation [58,59] and the SOXC proteins (SOX-4, SOX-11, and SOX-12) have been suggested to both inhibit degradation of β-catenin and SOX9 gene expression [60]. Also, actin cytoskeleton organization and the related signaling pathways (RhoA/ROCK, RALA) have been suggested to be involved in regulating SOX9 gene expression [61 –63], mRNA stability [64], protein levels [65], and activity [66]. Thus, multiple signaling pathways could potentially cause the apparent opposite reaction of iMPCs toward TGF-β-containing chondrogenic medium compared to BMSCs, and it will be an important task to unravel this in future studies.
Frobel et al. reported that the DNA methylation profile of iMPCs seemed to be more closely related to iPSCs than to BMSCs and the SOX9 gene was about 20% hypermethylated compared to BMSCs in this data set [12]. This welcomes to speculate that epigenetic silencing might further impair the induction of SOX9 gene expression during iMPC chondrogenesis.
Hsa-miR-145 is a SOX9-inhibiting microRNA with low expression in iPS cells and exceptionally high expression in those D1-iMPCs with low SOX9 protein levels. Moreover, in IMR-iMPCs that expressed SOX9 protein levels like BMSCs, hsa-miR-145 levels were 8.6-fold lower than in D1-iMPCs. Thus, hsa-miR-145 might be one factor limiting SOX9 translation in some iMPC populations. Furthermore, hsa-miR-145/143 have been reported to be enriched during differentiation of mouse ES cells into multipotent cardiac progenitors and to regulate smooth muscle fate [67,68]. This invites the speculation that iMPC populations with increased hsa-miR-145 levels might misdifferentiate into a nonchondrogenic, for example, cardiomyogenic lineage with a decreased inducibility of SOX9.
Interestingly, the D1-iMPC populations capable of partial chondrogenesis were three fast-growing populations and IMR-iMPCs in general proliferated faster than D1-iMPCs. This suggested that, similar as seen for BMSCs before [69], active cell proliferation reflecting high cell activity is an important requirement for successful iMPC chondrogenesis. Overall, in view of the essential role of SOX9 in chondrogenesis and the complex mechanism of chondrocyte differentiation from progenitor cells, SOX9 protein levels should be put into focus in future studies to optimize differentiation outcome.
In chick embryo development, nondetermined (stage 22) mesenchymal progenitors appear to require to transiently drop out of the cell cycle to enter chondrogenesis [70], a phenomenon which we confirmed to occur in the early phase of BMSC chondrogenesis [69]. In determined chick limb bud explants (stage 24), chondrogenesis can only proceed if explant size is not too small, possibly since smaller explants show increased cell proliferation which may disturb chondrogenesis [71].
A dissociated chick limb bud cell population of apparently uniformly chondrogenic cells was already disturbed in chondrogenesis when as little as 10% of nonchondrogenic mesenchymal limb bud cells were added. Interestingly, the formation of one large aggregate (which we here observe with BMSCs) transformed into nodular condensations in micromass culture in the presence of 10%–30% of nonchondrogenic cells [72], a phenomenon resembling the condensation behavior of iMPCs in this and in previous studies [11,49]. Authors suggested that, rather than the point of cell origin in the limb bud, aggregation versus nonaggregation of cells decided about successful chondrogenesis [72]. Thus, the condensation behavior which strongly deviates between iMPCs and BMSCs may be the key to successful chondroinduction and requires further detailed investigation. Furthermore, the size of micromass culture, transient dropout of cells from the cell cycle, and contamination with nonchondrogenic cells appear to be promising parameters to be optimized to support chondrogenesis. The main regulating signaling pathways of the equivalent processes during embryonic cartilage development include Activin/Nodal/TGF-β, BMPs, FGFs, and the Wnt signaling network [73,74]. Our data suggest that, to find a combination and timing of the multitude of possible modulators for these, SOX9 protein inducibility should be used as a correlate for successful cartilage formation.
A recent study from the Tsumaki group generated chondrocytes from iPS cells by combining a brief mesodermal induction period of 3 days with a subsequent induction of cell condensation [11]. The resulting tiny nodules were then manually harvested and this isolation step was essential for successful chondrogenesis. Deposition of cartilaginous matrix within the nodules showed that indeed only condensing cells had a high chondrogenic activity as seen during embryo development. We also observed iMPCs to form numerous small nodules within the first 4 days of chondrogenic stimulation, but did not isolate the cartilage nodules from remaining monolayer cells. Most likely, this procedure will yield a more homogeneous chondrogenic cell population, which could be crucial for success, but the method is highly time-consuming and laborious, starts from low and unknown cell numbers, and is hardly compatible with high-throughput methods, combination with tissue engineering scaffolds, or generation of sufficient material for therapeutic applications. GMP procedures strongly recommend to start from exactly defined cell quantities and consistent cell quality, where both cannot be provided by uncontrollable cell condensates. Thus, it is mandatory to find adequate stimuli that are capable to robustly and reproducibly induce large numbers of well-defined chondroprogenitors from iPSCs.
In conclusion, we consider the low SOX9 protein inducibility and the heterogeneity of iMPC generation a major limit of current iPSC chondrogenesis protocols. We observed an early decline of SOX9 protein upon chondrogenic stimulation of iMPCs along with an altered condensation behavior and an insufficiently robust cartilage formation by iMPCs in vitro. Cotreatment with TGF-β + BMP-4 improved iMPC chondrogenesis and could yield homogeneous cartilage, but this method still lacked robustness and was achieved at the expense of undesired hypertrophy. In view of an apparently opposite early SOX9 protein modulation in iMPCs and BMSCs, we suggest to use the inducibility of SOX9 protein as an indicator to find improved chondrogenic methods for iPSCs. The impact of this study is the identification of critical shortcomings of hiPSC chondrogenesis that will now enable a selective adaptation of the currently limited protocols to the specific needs of iPSCs.
Footnotes
Acknowledgments
The authors thank Birgit Frey for technical support and Katharina Bomanns for helping with Smad western blotting. Parts of this work were funded by the European Union's Seventh Framework Program (FP7/2007-2013) HydroZONES under grant agreement no. 309962. J.U. was supported by grants from the German Cancer Aid (Max-Eder Research Group), Baden-Württemberg Foundation, and German Research Council (DFG).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.