
Abstract
Histone three lysine 27 (H3K27) methyltransferase enhancer of zeste homolog 2 (EZH2) is a critical epigenetic modifier, which regulates gene transcription through the trimethylation of the H3K27 residue leading to chromatin compaction and gene repression. EZH2 has previously been identified to regulate human bone marrow-derived mesenchymal stem cells (MSC) lineage specification. MSC lineage specification is regulated by the presence of EZH2 and its H3K27me3 modification or the removal of the H3K27 modification by lysine demethylases 6A and 6B (KDM6A and KDM6B). This study used a bioinformatics approach to identify novel genes regulated by EZH2 during MSC osteogenic differentiation. In this study, we identified the EZH2 targets, ZBTB16, MX1, and FHL1, which were expressed at low levels in MSC. EZH2 and H3K27me3 were found to be present along the transcription start site of their respective promoters. During osteogenesis, these genes become actively expressed coinciding with the disappearance of EZH2 and H3K27me3 on the transcription start site of these genes and the enrichment of the active H3K4me3 modification. Overexpression of EZH2 downregulated the transcript levels of ZBTB16, MX1, and FHL1 during osteogenesis. Small interfering RNA targeting of MX1 and FHL1 was associated with a downregulation of the key osteogenic transcription factor, RUNX2, and its downstream targets osteopontin and osteocalcin. These findings highlight that EZH2 not only acts through the direct regulation of signaling modules and lineage-specific transcription factors but also targets many novel genes important for mediating MSC osteogenic differentiation.
Introduction
O
Recent investigations have identified the epigenetic regulation of chromatin as a key mechanism in dictating lineage-specific differentiation of MSC. The structure of chromatin can be regulated through methylation, acetylation, phosphorylation, SUMOylation, and ubiquitination of histone tails. Methyltransferases and demethylases are thought to be responsible for the switch between repressive H3K27 domains and active H3K4 domains. Given the diverse array of histone modifications, their various combinations form the “histone code,” which can influence the recruitment of effector proteins and transcription factors to chromatin, determining the functional state of chromatin [14 –16].
The epigenetic modifier enhancer of zeste homolog 2 (EZH2) exists within a multisubunit complex known as the polycomb repressor complex 2 (PRC2), which contains suppressor of zeste-12 (SUZ12), yin yang 1 (YY1), and embryonic ectoderm development [17]. EZH2 is a histone three lysine 27 (H3K27) methyltransferase that contains a carboxy-terminal catalytic SET domain responsible for the mono-, di-, or trimethylation modification of H3K27 residues of histone tails [18]. EZH2 H3K7 tri methylation (me3) modification facilitates the recruitment of the PRC1 during chromatin remodeling, compaction of chromatin and gene repression [19 –21]. During development, EZH2 is required for neural crest-derived cartilage and bone formation and anterior/posterior skeletal patterning in mice [22,23]. In postnatal skeletal tissue, reports have identified EZH2 as having a role in regulating MSC lineage differentiation. EZH2 was found to promote adipogenesis by disrupting Wnt/β-catenin signaling through the direct repression of pro-osteogenic Wnt genes, Wnt1, -6, -10a, and -10b, in mouse peripheral preadipocytes [24].
More recently, enforced expression of EZH2 in human bone marrow-derived MSC promoted adipogenesis in vitro and inhibited osteogenic differentiation potential in vitro and in vivo [25]. Conversely, inhibition of EZH2 enzymatic activity and knockdown of EZH2 gene expression inhibited adipogenesis and promoted osteogenic differentiation by MSC [25]. EZH2 has been reported to be regulated through phosphorylation at Thr 487 by cyclin-dependent kinase 1 (CDK1). CDK1 phosphorylation suppresses EZH2 activity, promoting MSC osteoblast differentiation in vitro [26]. Chromatin immunoprecipitation (ChIP) analysis has revealed that the presence of EZH2 and its H3K27me3-associated modification is reduced at the transcription start site (TSS) of key osteogenic transcription factor RUNX2 during osteogenic differentiation [25,26]. However, it is still unclear whether EZH2 directly targets other critical genes and pathways during MSC osteogenic differentiation. The present study aimed to identify novel EZH2 targets in osteogenesis through a bioinformatics approach using publically available ChIP-on-chip, ChIP-Seq, and microarray data sets.
Materials and Methods
Bioinformatic analysis
Bioinformatic analysis was performed to assess gene expression, EZH2 occupancy, H3K27, and H3K4 methylation in three independent data sets. These data sets compared primary cultured human bone marrow-derived MSC versus in vitro differentiated osteoblasts (OB) differentiated with dexamethasone 100 nM (Dex) and 10 mM β-glycerophosphate and with the addition of 50 μg/mL ascorbic acid for 28 days (GSE9451 [27]; [26]) or 50 nM ascorbic acid for 16 days (GSE27900 [28]). GSE9451 data set contained three independent MSC lines. These MSC lines were differentiated under osteogenic inductive conditions to generate paired OB for microarray analysis.
ChIP data sets
ChIP-on-chip data for genes bound by EZH2 in undifferentiated MSC and not bound by EZH2 in OB were obtained from Wei et al. [26]. H3K27me3 and H3K4me3 ChIP-Seq data were obtained from GEO (Gene Expression Omnibus,
Microarray data sets
To assess the differential expression of the genes identified in the ChIP data sets, two data sets were used, both conducted on Affymetrix GeneChip Human Genome U133 plus 2.0 arrays. Microarray (Affymetrix U133A) GEO data set GSE9451 [27] was normalized using RMA (RMA Express program
Isolation and culture of MSC
Bone marrow aspirates were isolated from the posterior iliac crest of healthy human adult donors, following informed consent (SA Pathology normal bone marrow donor program, Royal Adelaide Hospital Human Ethics Number 940911a). MSC were isolated and cultured using plastic adherence [34]. Culture media: Alpha modified Eagle's medium (α-MEM) (Sigma Aldrich, Inc., St Louis, MO) supplemented with 10% (v/v) fetal calf serum (SAFC Biosciences, Melbourne, VIC), 50 U/mL, 5 μg/mL penicillin, streptomycin (Sigma Aldrich, Inc.), 1 mM sodium pyruvate (Sigma Aldrich, Inc.), 100 μM L-ascorbate-2-phosphate (Wako Pure Chemical industries, Richmond, VA), 2 mM
Retroviral transduction overexpression
The human EZH2 coding region of the gene was ligated into pRUF-IRES-GFP retroviral vector using polymerase chain reaction primers to amplify the coding region with Xho1 restriction sites [25]. The pRUF-IRES-GFP-EZH2 construct was transfected into the HEK293T viral packaging cell line together with PGP and VSVG (viral envelope proteins, SBI System Biosciences, Mountain View, CA), and viral supernatant was used for infection of MSCs and sorted by fluorescence-activated cell sorting (FACS: Beckman Coulter Epics Ultra cell sorter, Lane Cove, NSW, AUS). MSC were passaged, and at passage 5 were seeded for osteogenic differentiation assays and RNA assays.
siRNA transfection
Passage 5 MSC were seeded in either 24-well plates (1.9 cm2) at 3 × 104 or 96-well plates (0. 32 cm2) at 3 × 103 cells per well. Early the next day, MSC were treated with 12 pmol siRNA (Life Technologies, Scoresby VIC). Negative control siRNA (4390843), EZH2 #1 (s4916), EZH2 #2 (s4918), FHL1 #1 (n261445), FHL1 #2 (n261446), MX1 #1 (s9100), and MX1 #2 (s9099) siRNAs with Lipofectamine RNAi MAX were used for this study (Invitrogen/Life Technologies, Scoresby VIC, AUS). siRNA were added to cells as specified by the manufacturer. Seventy-two hours post-transfection, media were removed and culture growth media, adipogenic, or osteogenic inductive media were added [25].
In vitro osteogenic differentiation assay
siRNA-treated MSC in 96-well plates were cultured in either noninductive culture media or osteogenic inductive media (α-MEM supplemented with 5% (v/v) FCS, 2 mM
In vitro adipogenic differentiation assay
siRNA-treated MSC in 96-well plates were cultured in either noninductive culture media or adipogenic inductive media (α-MEM supplemented with 10% [v/v] FCS, 2 mM
Real-time polymerase chain reaction analysis
Culture expanded MSCs infected with pRUF-IRES-GFP and pRUF-IRES-GFP-EZH2 were seeded at 5.64 × 104 per/well in six-well plates in the presence of either noninductive or osteogenic inductive media as previously described [25]. siRNA-treated MSC were cultured in the presence of noninductive, adipogenic, or osteogenic inductive media in 24-well plates. Total RNA was extracted using TRIzol reagent (Invitrogen/Life Technologies) and converted to cDNA by reverse transcription. Gene expression was assessed by real-time polymerase chain reaction (RT-PCR) amplification using specific primer sets (Table 1), SYBR Green/Rox PCR master mix (Qiagen, Doncaster, Chadstone Centre, VIC, AUS), and Rotor-Gene 6000 Real-Time Thermal Cycler (Corbett Research, Mortlake, NSW, AUS). Changes in gene expression were calculated relative to β-actin using the 2-dCT method [25,35].
EZH2, methyltransferase enhancer of zeste homolog 2; OP, Osteopontin; OC, Osteocalcin; β-ACTIN, Beta Actin; PCR, polymerase chain reaction.
Western blot analysis
Passage 5 MSC were seeded in either 24-well plates (1.9 cm2) at 3 × 104 or 96-well plates (0. 32 cm2) at 3 × 103 cells per well in duplicate. MSC were treated with siRNA and cultured under osteogenic differentiation conditions for 14 days. Whole cell lysates (40 mg) were separated on SDS gel as previously described [25]. Membranes were probed with anti-EZH2 mouse IgG (Acc2 Cell Signaling Tecnology, Inc./Genesearch Pty. Ltd., Arundel, QLD, AUS; 1/1,000 dilution), anti-MX1 rabbit IgG (Abcam/Sapphire Bioscience Pty. Ltd., Waterloo, NSW, AUS; ab95926, 1/500 dilution), anti-FHL1 rabbit IgG (Abcam/Sapphire Bioscience, ab133661, 1/1,000 dilution), anti-H3K27me3 rabbit IgG (Merk Millipore, Kilsyth, VIC, AUS; 1/1,000 dilution), and anti-β-actin mouse IgG (Cell Signaling Tecnology, Inc., 8H10D10; 1/1,000 dilution). Secondary detection was performed using anti-Rabbit-Alk Phos (Millipore; 1/10,000) and anti-Mouse-Alkphos (Millipore; 1/10,000) antibodies.
BrdU analysis
MSC at passage 5 were seeded at 2 × 103 cells per well in 24-well plates and cultured overnight at 37°C. Cell proliferation ELISA, BrdU protocol, was followed as described in the instruction manual version August 2007 (Roche applied biosciences, Mannheim, GER). On the sixth day, the BrdU reaction was measured on the Ultra Micro plate reader EL808 (Bio-Tek Instruments, Winooski, VT) at 450 nm.
ChIP analysis
Passage 5 MSC were seeded at 2 × 106 cells per flask (75 cm2) and induced under normal growth medium and/or osteogenic differentiation media for 14 days. ChIP was adapted from the Abcam (Abcam, Melbourne, VIC, AUS) crosslinking chromatin immunoprecipitation (X-ChIP) protocol. Briefly, chromatin was cross-linked with a final of 0.75% formaldehyde. Adherent cells were detached using 1 × trypsin EDTA, and the remaining cells were scraped from the bottom of the flask. Cells were lysed with 400 μL of FA lysis buffer (50 mM HEPES KOH pH7.5, 140 mM NaCl, 1 mM EDTA pH8, 1% Triton X-100, 0.1% Sodium deoxycholate, 0.1% SDS, and protease inhibitors). DNA was sheared with probe sonication on ice. Sonicated samples were used for immunoprecipitation [36].
Immunoprecipitation
Antibodies against anti-rabbit H3K27me3 (1 μg, Merck, Millipore 07-449, Bayswater, VIC, AUS,
IL2, Interleukin 2.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Paired student's t-test was used to assess statistical significance for mRNA expression for microarray and adipogenic RT-PCR experiments. Unpaired Student's t-test was used to assess statistical significance for siRNA knockdown of EZH2/MX1 and EZH2/FHL1 experiments. One-way ANOVA with Dunnett's multiple comparison test was used to assess statistical significance for siRNA and BrdU assays. Two-way ANOVA with Sidak's multiple comparison test was used for ChIP and EZH2 overexpression studies.
Results
Identification of novel EZH2 targets during MSC osteogenic differentiation
Initially, we examined three independent data sets to identify novel genes that may be switched on during osteogenic differentiation by removal of EZH2 binding and its associated H3K27me3 mark. We identified 99 genes that were reported to be bound by EZH2 in MSC, but not following osteogenic differentiated in vitro [26], and were found to have a loss of the repressive modification H3K27me3 during osteogenic differentiation of MSCs in the independent ChIP-Seq data set GSE35576 [28]. Assessment of differential gene expression in the independent microarray data set GSE9451 [27] identified that, of these 99 genes, 6 genes (ZBTB16, HOPX, ROR2, MYADM, FHL1, MX1) were significantly upregulated (>2-fold change; P ≤ 0.05, LIMMA), while no genes were significantly downregulated, under osteogenic differentiation, when compared with undifferentiated MSC (Fig. 1). Of these, two (ZBTB16, FHL1) gained the activating modification H3K4me3 under osteogenic differentiation, one gene lost the H3K4me3 mark in OB (HOPX), one gene (MYADM) had the H3K4me3 mark in both MSC and OB, and two genes (MX1, ROR2) did not have the H3K4me3 mark in either MSC or OB (Table 3). These findings suggest that ZBTB16, HOPX, ROR2, MYADM, FHL1, and MX1 are potential EZH2 targets that may play a role in regulating osteogenic cell fate determination. Two genes, ZBTB16 and FHL1, were selected for subsequent validation studies that met all the selection criteria. MX1, a known mediator of early mesenchymal differentiation, was also assessed as a positive control.
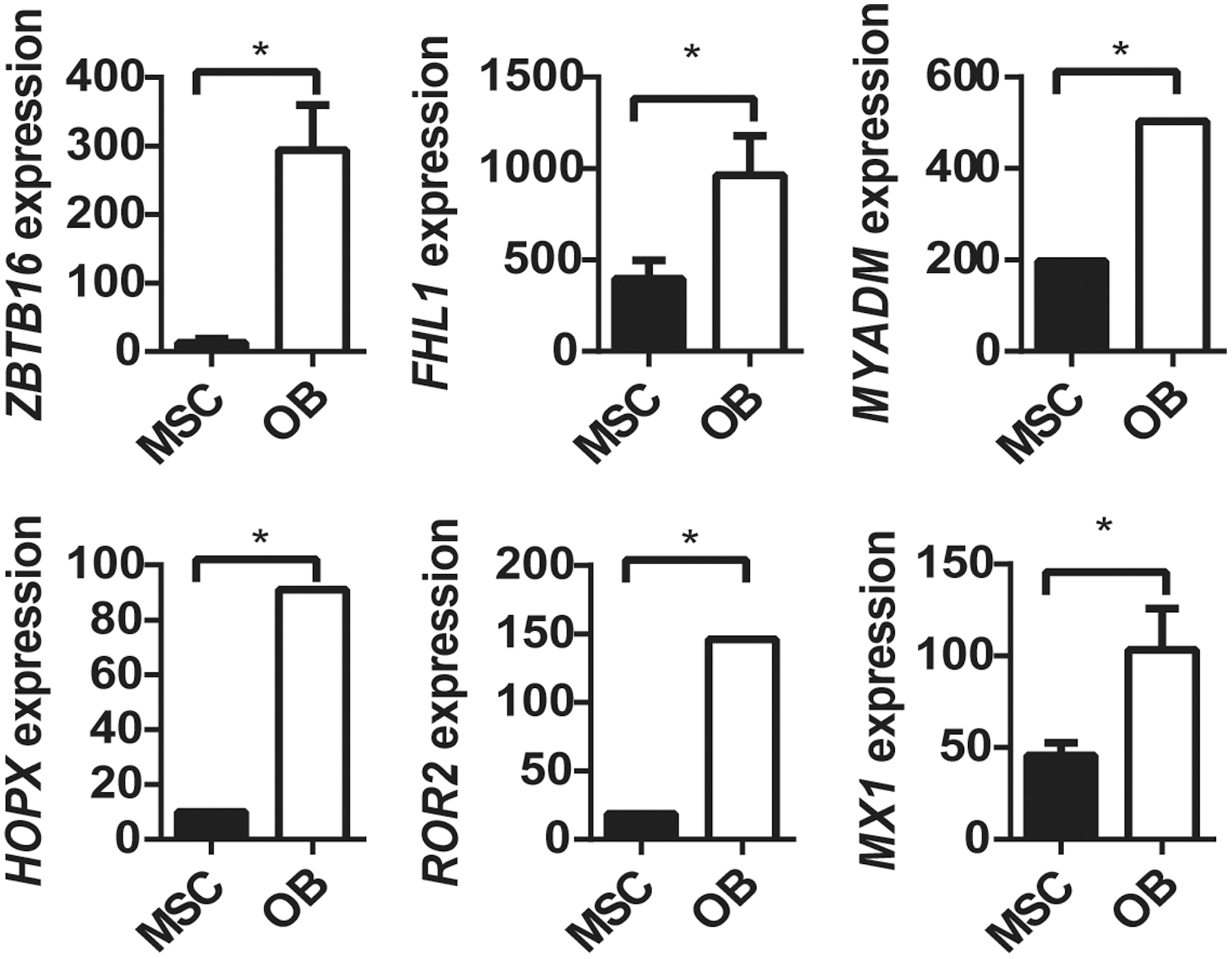
Genes with upregulated expression and loss of EZH2 during osteoblast differentiation associated with loss of H3K27 methylation and/or gain of H3K4 methylation. Genes upregulated expression with loss of EZH2 and H3K27me3 in OB.
Bioinformatics analysis of data-based GEO data set GSE9451 identifying genes bound by EZH2 in MSC, but not in osteoblasts (OB), associated with loss of EZH2 binding and H3K27me3 and/or gain of H3K4me3, during osteogenic differentiation into OB. Fold change is represented as OB/MSC (* P < 0.05). GEO, gene expression omnibus; MSC, mesenchymal stem cell.
Gene expression levels of ZBTB16, HOPX, ROR2, MYADM, FHL1, and MX1 were assessed in MSC cultured under adipogenic inductive conditions (Supplementary Fig. S1; Supplementary Data are available online at
EZH2 directly represses ZBTB16, MX1, and FHL1 gene expression in undifferentiated MSC
To confirm the bioinformatics analysis, manual ChIP analysis was performed for ZBTB16, FHL1, and MX1, using anti-EZH2, -H3K27me3, -H3K4me3, or IgG control antibodies on genomic DNA isolated from human MSC cultured under normal growth conditions or in osteogenic inductive media. Primers targeting the TSS of ZBTB16, MX1, and FHL1 were used to assess the presence of epigenetic modifying enzymes and modifications. P16 was used as the positive control, and GAPDH was used as the negative control for the EZH2 and H3K27me3 ChIP analysis (Supplementary Fig. S2A, B). GAPDH was used as the positive control and the IL2 was used as the negative control for the H3K4me3 ChIP (Supplementary Fig. S2C).
The TSS of ZBTB16, MX1, and FHL1 were significantly enriched for EZH2 binding in undifferentiated MSC compared with MSC cultured under osteogenic conditions (Fig. 2A). This correlated to a significant enrichment of the gene silencing mark H3K27me3 at the TSS of ZBTB16, MX1, and FHL1 in undifferentiated MSC compared with OB (Fig. 2B). In contrast, the active mark H3K4me3 was present at the TSS of all genes in OB correlating with an increase in gene expression during osteogenesis (Fig. 1). Importantly, EZH2 and the H3K27me3 modification were no longer present at the TSS of ZBTB16, MX1, and FHL1 during osteogenesis.
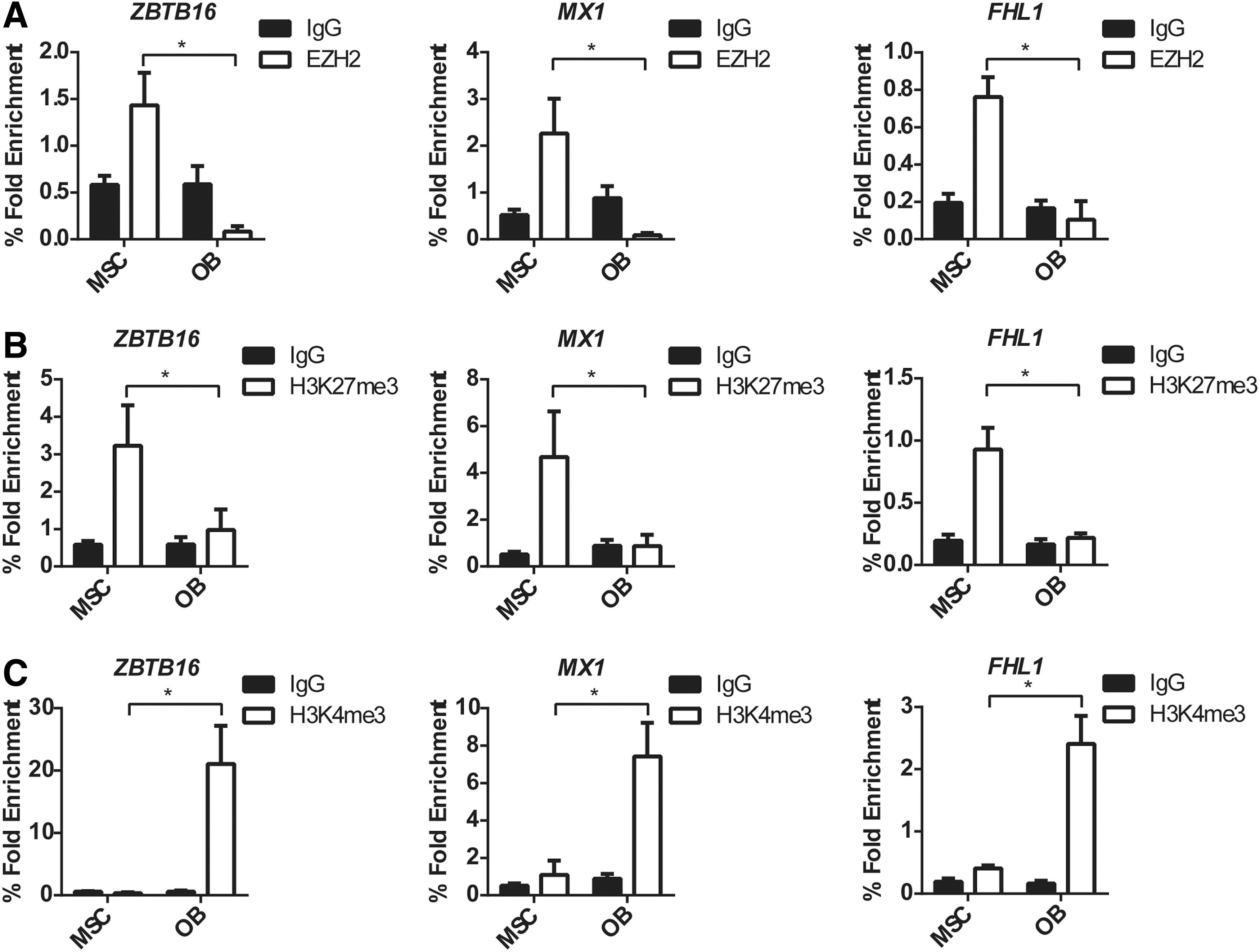
EZH2, H3K27me3, and H3K4me3 ChIP analysis of MSC and osteoblast confirmed modification patterns identified by bioinformatics analysis. Manual ChIP analysis confirms EZH2 regulates ZBTB16, MX1, and FHL1 in osteogenic differentiation. MSC (nondifferentiated) and osteogenic differentiated OB were immunoprecipitated with anti-H3K27me3, anti-EZH2, anti-H3K4me3, and anti-IgG control antibody, and RT-PCR was used to measure enrichment.
We have previously reported that EZH2 inhibits human MSC osteogenic differentiation in vitro and in vivo [25]. In the present study, the role of EZH2 in regulating ZBTB16, MX1, and FHL1 gene expression was confirmed using stably transduced EZH2 overexpressing MSC lines and vector control MSC (Fig. 3A), cultured under normal growth conditions or osteogenic inductive conditions. EZH2 overexpressing MSC was found to exhibit significantly lower expression levels of the osteogenic master regulatory factor, RUNX2, during osteogenic differentiation when compared with vector control MSC (Fig. 3A, B). Furthermore, EZH2 overexpressing MSC demonstrated repressed gene expression levels of ZBTB16, MX1, and FHL1 following osteogenic induction, when compared with vector control MSC (Fig. 3C). In parallel studies, two independent siRNA were used to knockdown gene expression of EZH2 in MSC cultured under osteogenic inductive conditions (Fig. 4A). Knockdown of EZH2 in MSC increased expression of RUNX2 (Fig. 4B) and significantly increased the expression of ZBTB16, MX1, and FHL1 compared with MSC treated with scramble control siRNA (Fig. 4C).
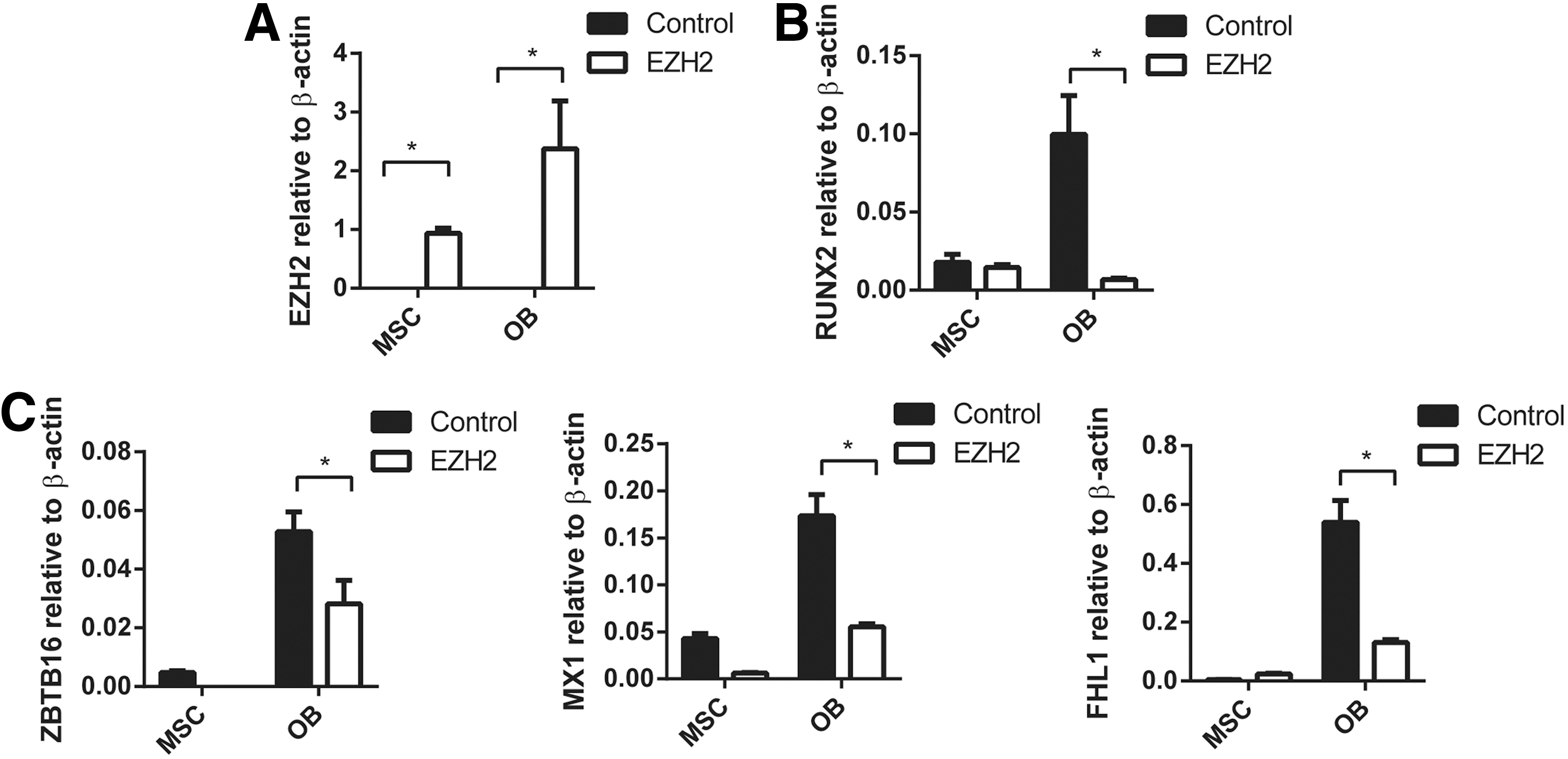
Enforces expression of EZH2 inhibits ZBTB16, MX1, and FHL1 expression in MSC and OB. pRUF-IRES-GFP-EZH2 overexpressing or pRUF-IRES-GFP control MSC were treated with no inductive media (MSC) or osteogenic differentiation media (OB) for 14 days. RT-PCR analysis:
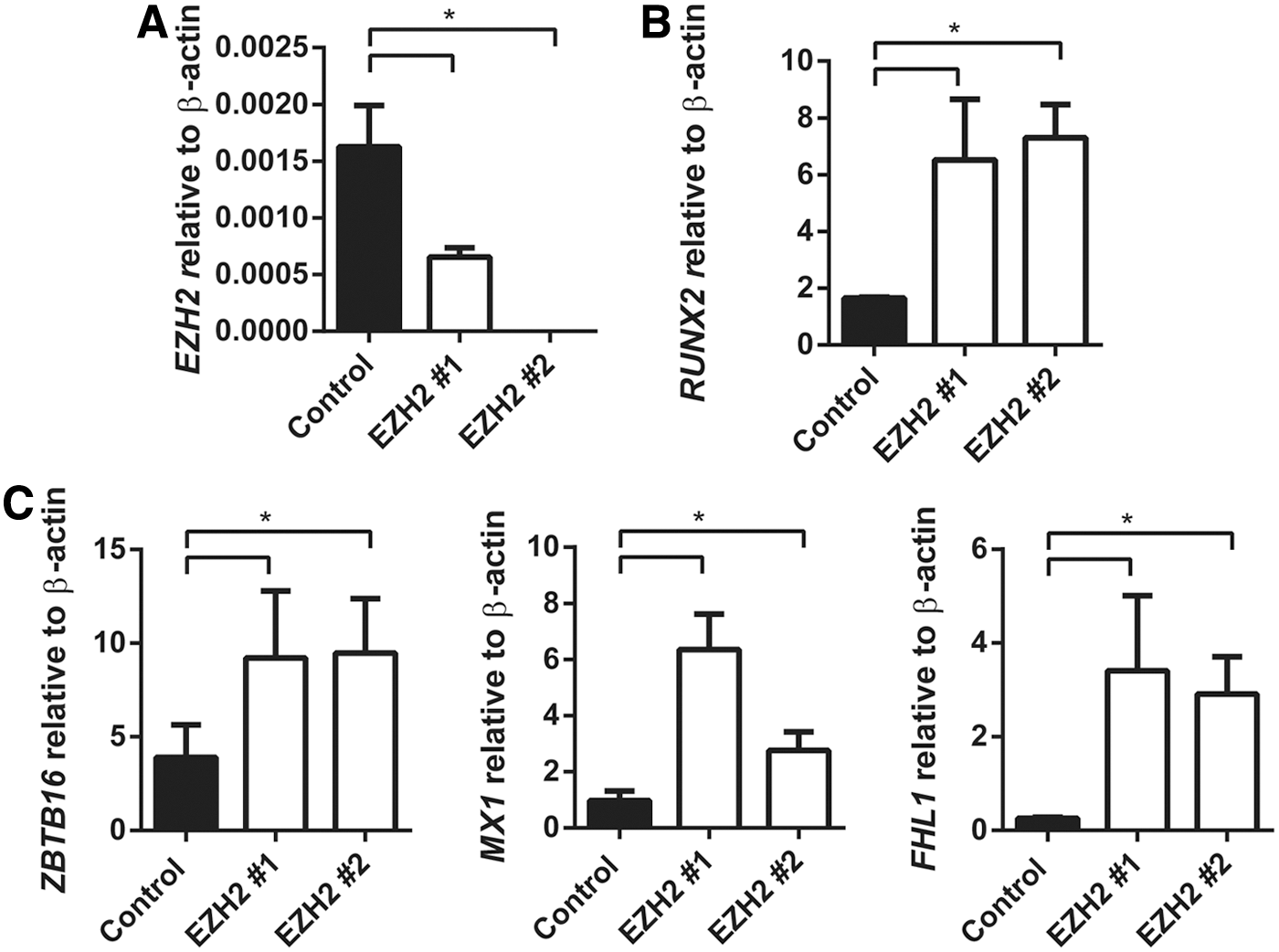
siRNA knockdown of EZH2 promotes expression of ZBTB16, MX1, and FHL1 in OB. MSC were treated with negative control siRNA, EZH2 siRNA #1, and EZH2 siRNA #2. MSC were cultured for 14 days with osteogenic differentiation media.
Collectively, these studies showed that ZBTB16, MX1, and FHL1 are direct targets of EZH2 binding and activity in undifferentiated MSC leading to suppression of gene expression. However, during osteogenic lineage commitment, ZBTB16, MX1, and FHL1 expression is induced due to the loss of both EZH2 binding and H3K27me3, correlating with the appearance of the H3K4me3 modification.
Targeted knockdown of MX1 and FHL1 inhibits MSC osteogenic differentiation
ZBTB16 has previously been implicated in regulating osteogenesis in human MSC by acting upstream of RUNX2 to promote osteogenic differentiation and the expression of key osteogenic differentiation genes [38,39]. However, the direct role of MX1 and FHL1 in regulating MSC osteogenic differentiation is not as well defined. Two independent siRNA targeting different regions of MX1 or FHL1 were used to assess the role of these factors in MSC osteogenic differentiation. Transient knockdown of MX1 and FHL1 was confirmed with real-time PCR and western blot analysis (Fig. 5A, B). Functionally, siRNA knockdown of MX1 or FHL1 reduced Alizarin red positive mineralized deposits (Fig. 5C) and reduced extracellular calcium (Fig. 5D) compared with scramble control siRNA-treated MSC. RT-PCR analysis of MX1 and FHL1 siRNA-treated MSC revealed a significant reduction in expression of RUNX2, and the mature bone-associated markers, OP and osteocalcin (OC) compared with control MSC, following osteogenic induction (Fig. 5E).

siRNA knockdown of MX1 and FHL1 inhibits osteogenesis. MSC were treated with negative control siRNA, MX1 siRNA #1, MX1 siRNA #2, FHL1 siRNA #1, and FHL1 siRNA #2 and were cultured for 14 days under osteogenic differentiation media.
We next assessed the proliferation rates of MX1 and FHL1 siRNA-treated MSC by BrdU incorporation. After 6 days of culture, there were no significant differences in the proliferation rates between siRNA scramble control-treated MSC compared with MX1 or FHL1 siRNA-treated MSC (Supplementary Fig. S3A). The data demonstrate that MX1 and FHL1 play an important role in promoting MSC osteogenic differentiation, similar to that described for ZBTB16.
Targeted knockdown of EZH2 and MX1 or FHL1 rescues aberrant osteogenic differentiation
MX1 and FHL1 appear to play a functional role in promoting osteogenic differentiation in MSC, where siRNA knockdown of MX1 or FHL1 reduced Alizarin red positive mineralized deposit formation and expression of key osteogenic genes. We next assessed whether siRNA transfection of EZH2 and either FHL1 or MX1 could rescue the increased osteogenic differentiation potential observed in MSC treated with siRNA targeting EZH2 as we have previously described [25]. In the present study, MSC were treated with a combination of siRNA targeting EZH2 and MX1 (EZH2/MX1) or EZH2 and FHL1 (EZH2/FHL1) during osteogenic differentiation. Alizarin red staining of mineralized deposits and extracellular calcium quantitation revealed EZH2/MX1, and EZH2/FHL1 siRNA-treated MSC formed significantly less mineral deposits and extracellular calcium when compared to MSC treated with EZH2 siRNA or scramble control siRNA (Fig. 6A, B).
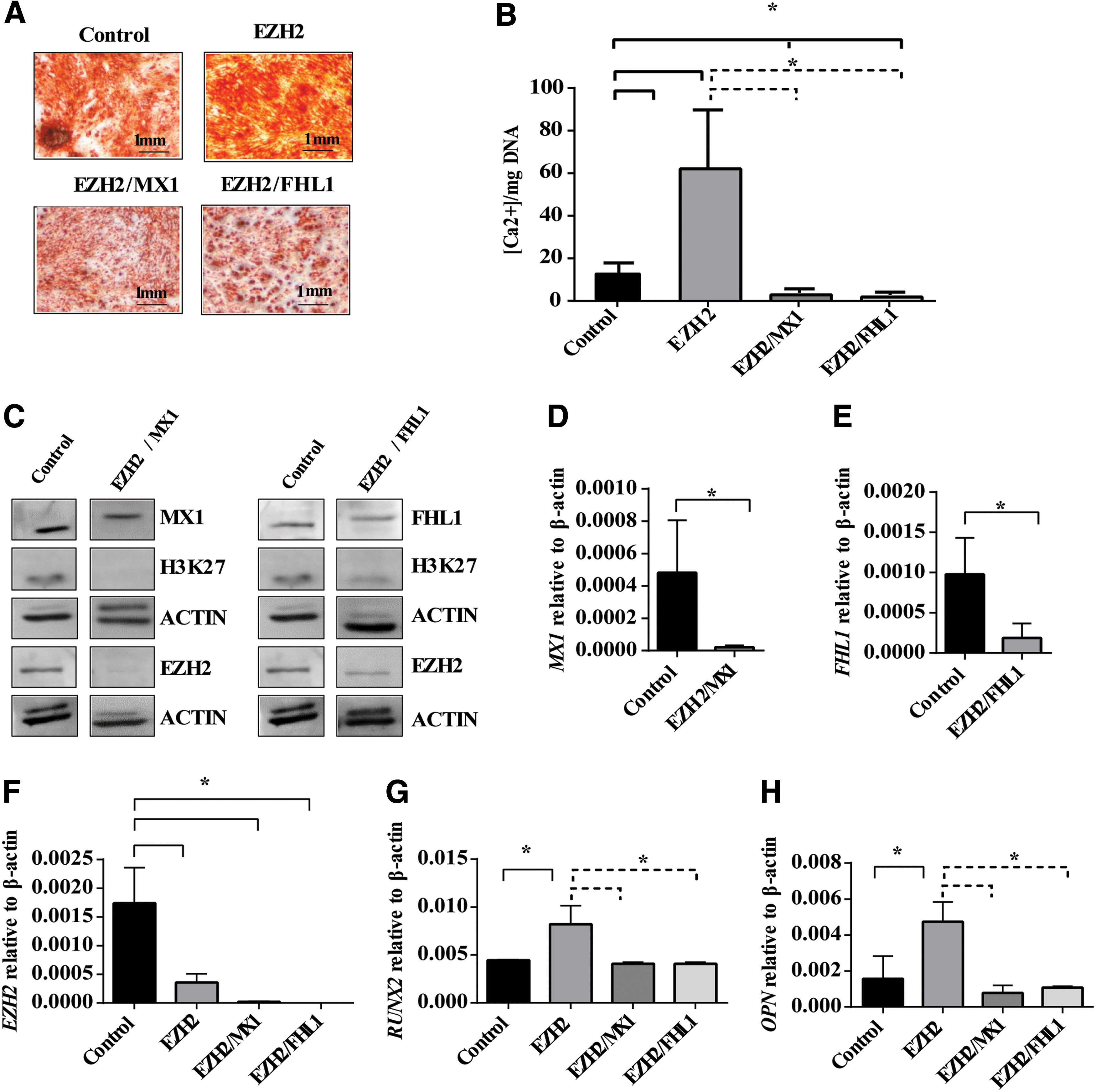
siRNA knockdown of EZH2 and MX1 or FHL1 rescued aberrant osteogenic differentiation. MSC were treated with 12 pmol of negative control siRNA (Control), EZH2 siRNA #1 (EZH2), EZH2 siRNA # 1/MX1 siRNA #1 (EZH2/MX1), and EZH2 siRNA # 1/FHL1 siRNA #2 (EZH2/FHL1) culture under osteogenic differentiation conditions for 14 days.
Western blot analysis confirmed a decrease in MX1 or FHL1, EZH2, and H3K27 methylation in EZH2/MX1 or EZH2/FHL1 siRNA-treated MSC compared to scramble control siRNA, following osteogenic induction (Fig. 6C). Real-time PCR analysis confirmed knockdown of EZH2 and MX1 or FHL1 in the EZH2/MX1 or EZH2/FHL1 siRNA-treated MSC, respectively (Fig. 6D–F). Knockdown of EZH2 in MSC increased RUNX2 and OP gene expression during osteogenic differentiation (Fig. 6G, H), as we have previously described [25]. However, knockdown of both EZH2 and MX1 or FHL1 in MSC resulted in repression of RUNX2 and OP gene expression at similar levels to scramble control-treated MSC (Fig. 6G, H). These findings demonstrate that MX1 and FHL1 are important downstream regulators of osteogenic differentiation of MSC. However, osteogenic differentiation is not solely reliant on these factors. Therefore, we suggest EZH2 functions to repress osteogenic differentiation through direct methylation of RUNX2, ZBTB16, MX1, FHL1, and many other genes, which are critical to driving MSC osteogenic differentiation.
Discussion
Our previous work has shown that the histone H3K27 methyltransferase EZH2 is an essential epigenetic modifying enzyme involved in inhibiting osteogenesis [25] and is recruited to the p16/Ink4A locus in response to the bHLH transcription factor Twist-1 to repress senescence [40]. Although we have illustrated that EZH2 can bind to some key osteogenic gene promoters to repress their expression, we have a limited knowledge in regard to what the targets of EZH2 are and how they are involved in inhibiting osteogenesis. To identify novel EZH2 targets that are involved in human MSC osteogenic lineage specification, we conducted a bioinformatics survey of publically available databases to identify putative EZH2 target genes suppressed in undifferentiated human bone marrow-derived MSC and subsequently activated following osteogenic differentiation. The present study revealed that EZH2 directly regulated the pro-osteogenic genes ZBTB16, MX1, and FHL-1 in human bone marrow-derived MSC through H3K27me3.
Upon MSC osteogenic differentiation, EZH2 and H3K27me3 were removed from the promoters of these genes, correlating with the appearance of the epigenetic activation mark H3K4me3. This is in agreement with our recent studies that identified EZH2 as a suppressor of MSC osteogenic differentiation, through the regulation of key osteogenic genes, RUNX2, OP, and OC [25]. Supportive studies demonstrated that inhibition of EZH2 methyltransferase activity following treatment with DZNep, or deactivation of EZH2 through phosphorylation of Thr 487 by CDK1, resulted in the promotion of osteogenesis [25,26]. Furthermore, EZH2 has been reported to regulate the β-catenin signaling pathway by targeting Wnt genes Wnt1, -6, -10a, and -10b in mouse peripheral preadipocytes and MSC to stimulate adipogenesis over osteogenesis [24]. As these Wnt genes are involved in the induction of RUNX2 and downstream osteogenic genes, their inhibition by recruitment of EZH2 to the promoter inhibits osteogenesis. Therefore, EZH2 acts to repress osteogenesis at multiple levels by directly inhibiting WNT genes, key osteogenic transcription factor RUNX2, and its downstream targets such as OP and OC while allowing adipogenic differentiation to occur. Collectively, these findings support the notion that EZH2 is a key regulator of MSC fate determination by selective repression of osteogenic inductive pathways.
The validity of the bioinformatics approach used here was shown by the identification of the putative EZH2 target ZBTB16, a promyelotic leukemia zinc finger transcription factor. ZBTB16 is a negative regulator of cell division in embryogenesis and controls skeletal patterning through repression of HOX, BMP, and Sonic Hedge Hog (Shh) expression. Deletion of ZBTB16 in mice leads to severe skeletal deformities due to impaired limb and axial skeleton patterning leading to transformation of anterior skeletal elements into posterior structures [41 –43]. ZBTB16 is required for stylopod and zeugopod formation during early stages of limb development, before the initiation of cartilage condensation [44].
ZBTB16 has been found to either be deleted or mutated in patients exhibiting bone deformities. A 12-year-old male patient with skeletal defects, including hypoplasia of radius and ulna, short stature, mental retardation, delayed bone age, and mild facial dysmorphism, was identified with a ∼8 Mbp interstitial deletion in the 11q23 chromosomal region, which includes the ZBTB16 gene. This patient also revealed a recessive missense mutation (c. 1849ARG) within the remaining ZBTB16 allele [45,46]. This study indicated that ZBTB16 may play a critical role in the development of the skeleton and bone aging. Furthermore, Ikeda et al. (2005) found ZBTB16 was one of 24 genes upregulated during osteoblastic differentiation of OPLL cells [39].
ZBTB16 has been reported to regulate mesenchymal osteogenic and chondrogenic differentiation, acting upstream of the master transcription factors RUNX2 and SOX9, respectively, in mesenchymal progenitor cell line C3H10T1/2 [47]. Overexpression of ZBTB16 in C3H10T1/2 MSC line promoted osteogenic and chondrogenic differentiation while inhibiting adipogenic differentiation, Furthermore, these ZBTB16 overexpressing C3H10T1/2 MSC readily formed bone and cartilage when implanted into an intratibial osteochondral defect model [47]. Small interfering RNA knockdown of ZBTB16 in human and mouse MSC reduced osteogenic differentiation and expression of collagen 1A1 (COL1A1), alkaline phosphatase, RUNX2, and OC. Furthermore, overexpression of Zbtb16 in a murine MSC line increased Runx2 and Col1A expression during osteogenic differentiation [39]. Collectively, these studies and this current study have identified ZBTB16 as a regulator of osteogenic differentiation.
Mechanistically, ZBTB16 can act through its localization within nuclear bodies and plays a role in transcriptional activation or repression by regulating chromatin modifications and remodeling [48]. ZBTB16 can act as a transcriptional repressor through the recruitment of nuclear compressors SMRT, N-COR, SIN-3, and class 1 and class 2 histone deacetylases to the transcriptional complex [48 –54]. ZBTB16 mediates transcriptional repression of HoxD gene expression through chromatin remodeling through long range DNA looping and interactions with polycomb protein Bmi-1 on DNA [48]. This study identified EZH2 actively binds ZBTB16 transcriptional start site and this coincides with the presence of H3K27me3 and low ZBTB16 gene expression. However, during osteogenic differentiation, EZH2 and its H3K27me3 modification is removed from the TSS followed by the addition of H3K4me3 and significant activation of ZBTB16. In the current study, we have, for the first time, demonstrated that overexpression of EZH2 in MSC, and thereby inhibiting osteogenic differentiation [25], was associated with reduced expression of ZBTB16. Conversely, siRNA-mediated knockdown of EZH2 promoted osteogenic differentiation [25] and promoted expression of ZBTB16. However, this study is the first to identify ZBTB16 as an EZH2 target in human mesenchymal stem cells.
In the present study, MX1, another known regulator of osteogenesis, was identified as a putative EZH2 target. The MX1 gene encodes a guanosine triphosphate-metabolizing protein that participates in the cellular antiviral response, where it is induced by type I and type II interferons and antagonizes the replication process of several different RNA and DNA viruses [55,56]. In vivo lineage tracing studies, using Mx1-Cre mice crossed with Rosa-YFP reporter mice, revealed that MX1-positive cells were predominantly osteolineage restricted with a small number of adipocytes found to have originated from MX1-positive cells during normal development and or bone fracture healing [57]. While the MX1-positive cell population exhibited trilineage potential, differentiating into adipocytes, osteoblast, and chondrocytes in vitro, MX1-positive cells did not contribute to chondrogenesis in vivo [57], where MX1 is expressed by less than half of the multipotent MSC and seems to be restricted to the osteogenic lineage in vivo. These findings suggest a distinct role of MX1 in murine osteogenic differentiation; however, the role of MX1 in human osteogenic differentiation is not as well defined.
Global wide gene expression analysis and ChIP analysis conducted by multiple groups indicate that human MSC express low levels of MX1, which is targeted by Ezh2 and is significantly upregulated during osteogenic differentiation. The present study demonstrated that siRNA-mediated suppression of MX1 expression decreased osteogenic differentiation of human MSC in vitro. These findings are supported by microarray studies that have identified MX1 as being differentially upregulated in transformed bone-forming human stromal cell lines versus nonbone-forming stromal cell lines [58]. Furthermore, our data suggest that EZH2-mediated repression of MX1 expression in MSC may result in an inhibition of osteogenic differentiation, where removal of EZH2 and subsequent H3K27me3 at the TSS of MX1 is critical for promotion of osteogenic lineage differentiation.
Another gene that was identified as a potential EZH2 target was four-and-a-half LIM domains 1 (FHL1), which encodes a member of the four-and-a-half-LIM-only protein family. FHL1 contains two conserved zinc finger domains with four highly conserved cytosine binding zinc atoms in each zinc finger. FHL-1 contains a TCF/LEF binding site, which can be stimulated by β-catenin or Wnt pathway agonist lithium chloride inducing muscle hypertrophy and repressing chondrogenesis. [59,60]. In support of our results, a recent study [61] identified that overexpression of FHL1 in murine MC3T3-E1 cells promotes osteogenesis. Transfection of Wnt3a or treatment with estrogen in MC3T3-E1 cells significantly increases FHL1 expression and osteogenic differentiation.
Overexpression of FHL1 increased alizarin red staining and extracellular calcium, which was associated with increased expression of osteogenic genes RUNX2, OP, and OC. Treatment with shRNAs targeting FHLl inhibited osteogenic differentiation of MC3T3-E1 cells compared to scramble control. These studies identify the role of FHL-1 in regulating muscle, chondrogenic, and osteogenic lineage specification. Our study identified that FHL1 is upregulated in human OB (Fig. 1B) and that knockdown of FHL1 by siRNA repressed osteogenic differentiation (Fig. 3B) as similar previously described in murine MC3T3-E1 cells. Furthermore, this is the first study to identify FHL1 as a novel EZH2 target during osteogenesis. EZH2 may act to repress FHL1 in MSC, preventing osteogenic lineage specification, suggesting that removal of EZH2 and hence H3K27me3 is critical for osteogenic differentiation.
Through bioinformatics analysis of publically available ChIP-on-chip, ChIP-Seq, and microarray data sets, we identified three novel EZH2 targets that may play a role in the suppression of osteogenesis by EZH2. With the use of overexpression studies, we verified that EZH2 directly inhibits ZBTB16, MX1, and FHL1 expression in MSC. Furthermore, siRNA knockdown of EZH2 in OB promoted the expression of ZBTB16, MX1, and FHL1. Knockdown studies using siRNA targeting MX1 and FHL-1 identified that MX1 and FHL-1 are critical for human MSC osteogenic differentiation and for the expression of key osteogenic transcription factor RUNX2 and its downstream targets. These findings suggest that EZH2-mediated H3K27me3 of ZBTB16, MX1, and FHL1 in MSC prevents osteogenic differentiation, keeping the MSC in a more undifferentiated state. Furthermore, EZH2 does not only act on these genes identified in this study but regulation of RUNX2 and WNT genes is also critical for the lineage specification [24 –26].
Recent studies from our laboratory, and others, have identified the role of the histone H3 lysine 27 demethylase KDM6A and KDM6B in regulating MSC osteogenic differentiation [25,62]. We predict that these demethylases together or individually may be responsible for the removal of H3K27me3 from the TSS of ZBTB16, MX1, and FHL-1 during MSC to osteogenic lineage commitment. Furthermore, as we know KDM6A and KDM6B often associate with the H3K4 methyltransferases, we suggest that the recruitment of these demethylases to the TSS of these genes is critical for the removal of H3K27me3 and the subsequent addition of H3K4me3 modification. Further studies are needed to fully identify the mechanism H3K27me3 removal and the addition of H3K4me3 on these key osteogenic genes. Furthermore it is still unclear how and why is EZH2 recruited to the promoters ZBTB16, MX1, and MX1 and what factors and or signaling pathways are critical for the specific removal of EZH2 from these genes. Studies have identified EZH2 as an important regulator of the HOX genes during skeletal development and neural crest-derived cartilage and bone in mice [22,23]. Therefore, we predict that EZH2 may directly regulate ZBTB16, MX1, and FHL1 during murine and human skeletal development.
Footnotes
Acknowledgments
Funding National Health and Medical Research project grant APP1046053 and Fellowship APP1042677. S.H. Australian Postgraduate Award (APA) and Dawes Top-up scholarship. K.V. was supported by a Mary Overton Early Career Research Fellowship (Royal Adelaide Hospital).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.