
Abstract
Traumatic spinal cord injury (SCI) leads to a cascade of secondary chemical insults, including oxidative stress and glutamate excitotoxicity, which damage host neurons and glia. Transplantation of exogenous neural stem/progenitor cells (NSPCs) has shown promise in enhancing regeneration after SCI, although survival of transplanted cells remains poor. Understanding the response of NSPCs to the chemical mediators of secondary injury is essential in finding therapies to enhance survival. We examined the in vitro effects of glutamate and glutamate receptor agonists on adult rat spinal cord-derived NSPCs. NSPCs isolated from the periventricular region of the adult rat spinal cord were exposed to various concentrations of glutamate for 96 h. We found that glutamate treatment (500 μM) for 96 h significantly increased live cell numbers, reduced cell death, and increased proliferation, but did not significantly alter cell phenotype. Concurrent glutamate treatment (500 μM) in the setting of H2O2 exposure (500 μM) for 10 h increased NSPC survival compared to H2O2 exposure alone. The effects of glutamate on NSPCs were blocked by the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptor antagonist GYKI-52466, but not by the N-methyl-D-aspartic acid receptor antagonist MK-801 or DL-AP5, or the mGluR3 antagonist LY-341495. Furthermore, treatment of NSPCs with AMPA, kainic acid, or the kainate receptor-specific agonist (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propanoic acid mimicked the responses seen with glutamate both alone and in the setting of oxidative stress. These findings offer important insights into potential mechanisms to enhance NSPC survival and implicate a potential role for glutamate in promoting NSPC survival and proliferation after traumatic SCI.
Introduction
T
A compound of major interest implicated in the pathogenesis of SCI is the excitatory amino acid glutamate. Recently, glutamate has emerged as an important regulator of neural stem cell proliferation, survival, and migration in the developing central nervous system [9]. The effect of glutamate on neural stem cells has varied between studies and likely depends on a number of factors, including the age and source of the stem cells, in addition to the expression and functionality of the various glutamate receptor subtypes. For example, glutamate has been shown to increase proliferation of neocortical ventricular zone cells, while decreasing proliferation of NSPCs in the subventricular zone of embryonic mouse organotypic slices [10]. In contrast, exposure to glutamate or glutamate receptor agonists has been shown to induce proliferation and enhance survival of subventricular zone (SVZ) stem cells derived from perinatal rat brains [11] and promote migration of embryonic mouse cortical-derived neural progenitor cells [12].
Much of the current literature surrounding the effects of glutamate on neural stem cells has focused on cells derived from the developing embryonic or fetal brain. However, glutamate may also play an important role in the regulation of adult neural stem cells after traumatic injuries of the spinal cord where high levels of the excitatory neurotransmitter are released shortly after the initial physical trauma. Despite this, little is known about the effect of glutamate and its various receptors on neural stem cells derived from the adult spinal cord [13,14]. A thorough investigation is necessary given the potential age and region-specific differences in stem cell responses to glutamate. Also, the potential for glutamate to enhance survival of neural stem cells makes it of great interest to examine the effect of glutamate in the setting of other components of the secondary injury of SCI, which are known to impair NSPC survival such as oxidative stress [13].
This study examined the effects of glutamate and various glutamate receptor agonists on adult rat spinal cord-derived NSPCs both alone and in the setting of oxidative stress in vitro. We show for the first time that (i) glutamate increases survival and proliferation of adult spinal cord-derived NSPCs through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptors, (ii) concurrent glutamate exposure attenuates oxidative stress-induced cell death in adult NSPCs, and (iii) glutamate-mediated protection against oxidative stress is dependent on AMPA/kainate receptors.
Methods
Isolation and culture of NSPCs
Cryogenically preserved NSPCs previously isolated from the region of the central canal of the spinal cord of transgenic adult female Wistar rats expressing green fluorescent protein were used in this study. Cryogenically preserved NSPCs previously isolated from the forebrain of the same animals from which spinal cord-derived cells were obtained were used for comparison studies. The methods of isolation were previously described by our laboratory [7]. Cells were grown as free-floating neurospheres in growth media containing neurobasal media (Gibco-Invitrogen), B27 neural supplement (Gibco-Invitrogen), 2 mM
Experimental treatments
For the hydrogen peroxide (H2O2) studies, 30% H2O2 (Fisher Scientific) was added to the well to reach a final concentration ranging between 100 and 500 μM H2O2. For the glutamate studies,
To determine the receptor subtypes involved in glutamate-mediated effects on NSPCs, the following glutamate receptor antagonists were used: MK-801 (10 μM; a noncompetitive N-methyl-D-aspartic acid (NMDA) receptor blocker; Sigma), DL-AP5 (100 μM; a competitive NMDA receptor blocker; Tocris), GYKI-52466 (100 μM; a noncompetitive AMPA receptor blocker, which has also shown effects at antagonizing kainate receptors; Sigma), and LY-341495 (1 μM; a metabotropic mGluR3 antagonist; Tocris). The following glutamate receptor agonists were also used: NMDA (NMDA receptor agonist; Sigma), AMPA (AMPA receptor agonist; Tocris), kainic acid (kainate receptor agonist; Tocris), (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propanoic acid (ATPA) (a GluR5 kainate receptor agonist with limited AMPA receptor affinity; Tocris), and N-acetylaspartylglutamate (NAAG) (a metabotropic mGluR3 agonist; Tocris).
Cell viability
Cell viability was assessed using live/dead staining (Invitrogen). Briefly, NSPCs were incubated with 10 μM ethidium homodimer and 5 μM calcein-AM (diluted in Hank's balanced salt solution containing Ca and Mg) for 10 min at room temperature and then imaged using a Nikon Eclipse TE 300 microscope. Five random images were taken for each well at 10× magnification. The number of calcein+ cells was counted in each image to assess cell survival. The percentage of ethidium homodimer+ cells relative to total cells was calculated to determine percent cell death. Membrane integrity was analyzed using the lactate dehydrogenase (LDH) release assay (Roche).
Immunostaining
Cell differentiation and proliferation were assessed using immunocytochemical staining, as we have previously described. After a 96-h treatment with 500 μM glutamate, cells were fixed with 4% paraformaldehyde for 20 min at room temperature and washed with 0.1 M phosphate-buffered saline (PBS). Cells were then blocked with 10% normal goat serum with 0.3% Triton-X 100 and 1.5% bovine serum albumin (depending on the primary antibody) for 1 h at room temperature. Afterward, cells were incubated with the primary antibody overnight at 4°C. The following primary antibodies were used: Ki67 (1:1,000; Abcam) for proliferating cells, nestin (1:100; BD Science) for NSPCs, glial fibrillary acidic protein (GFAP) (1:200; Millipore) for astrocytes, RIP (1:20; Developmental Studies Hybridoma Bank) for oligodendrocytes, and βIII-tubulin (1:500; Biolegend) for neuronal progenitor cells. Cells were then washed with 0.1 M PBS, incubated with fluorescent Alexa 568 secondary antibody (1:500; Invitrogen) for 1 h, and washed with PBS. To counterstain for cell nuclei, cells were incubated in Hoechst and washed with PBS.
Immunocytochemical staining was imaged using a Nikon Eclipse TE 300 microscope. For Ki67 staining, five random images were taken for each well at 10× magnification and the percentage of Ki67+ cells was quantified per image. For all other antibodies, 10 random images were taken for each well at 20× magnification and the percentage of positive cells was quantified per image.
Statistical analysis
All data are presented as mean ± standard error of the mean and were analyzed using SigmaStat 3.1 software. Statistical differences between multiple groups were assessed using one-way or two-way analyses of variance and Bonferroni's or Dunnett's post-hoc corrections. Differences between two groups were assessed using a two-tailed Student's t-test. A P-value of <0.05 was set as the significance level for all tests.
Results
Comparison of adult rat spinal cord and brain-derived NSPC responses to oxidative stress and glutamate exposure
Adult NSPCs are present in both the subventricular zone of the forebrain and the periventricular region of the central canal in the spinal cord. We determined if there were any differences in the response to oxidative stress and glutamate exposure between cells derived from either source. After a 10-h exposure to H2O2, both spinal cord and brain-derived NSPCs showed a similar dose-dependent decrease in live cells (calcein+; Fig. 1A) and an increase in LDH release (Fig. 1B).
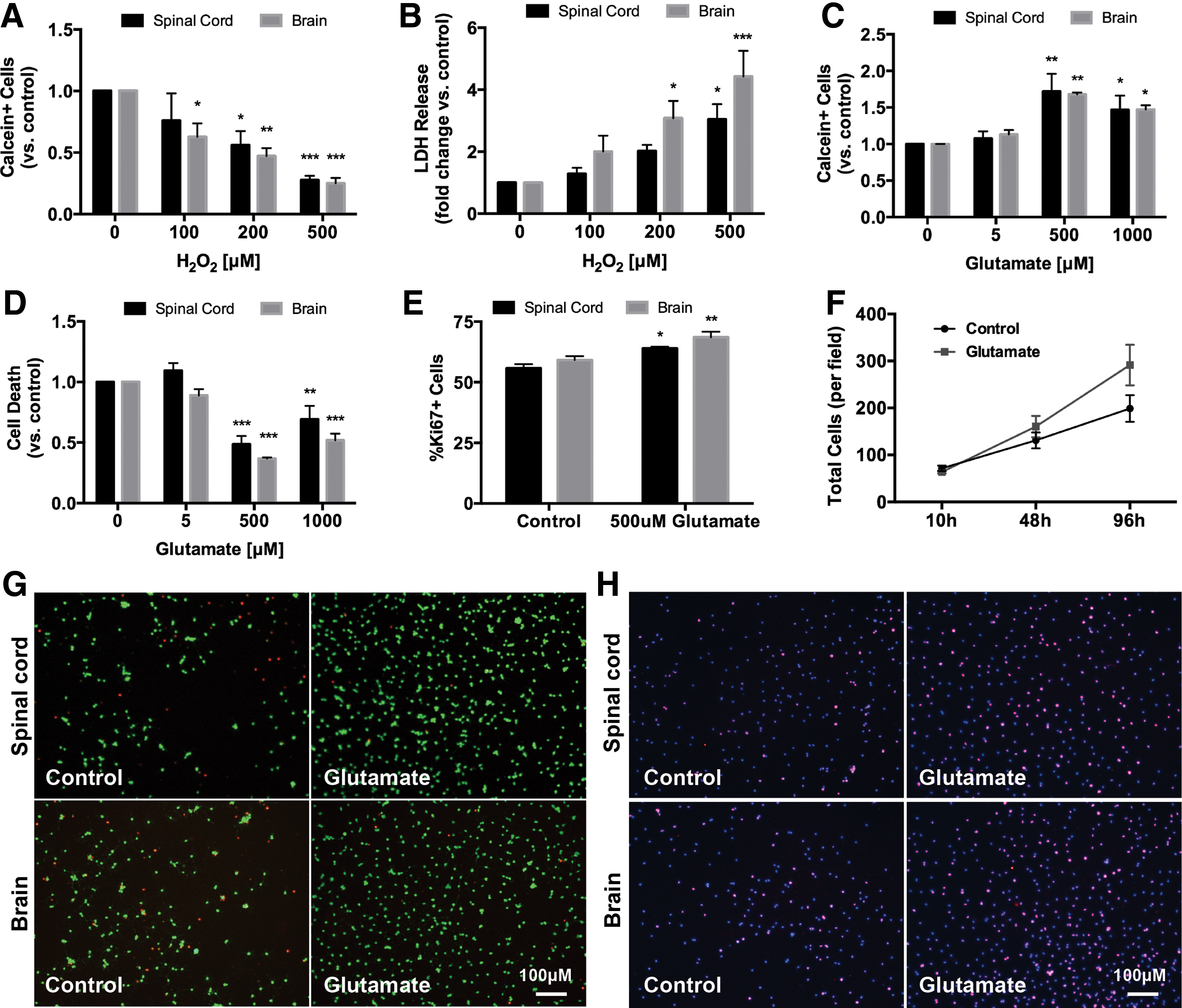
Comparison of adult brain and spinal cord-derived NSPC responses to oxidative stress and glutamate exposure. Both brain and spinal cord-derived NSPCs had a dose-dependent decrease in the number of live cells (calcein+)
To examine the effect of glutamate on NSPCs, cultures were exposed to glutamate (5–1,000 μM) for 96 h after which point there was a significant increase in the number of live cells labeled with calcein (Fig. 1C). We wanted to determine whether this increase in live cells was due to an effect on survival, proliferation, or a combination of both. We found a significant decrease in the percentage of dead cells labeled with ethidium homodimer under glutamate treatment (Fig. 1D). In addition, there was an increase in the expression of the proliferative marker Ki67 in both brain and spinal cord-derived cells treated with 500 μM glutamate for 96 h (Fig. 1E). To further assess the effect of glutamate on NSPC proliferation and expansion, we quantified total cell density at multiple time points under 500 μM glutamate treatment. We found that short exposures to glutamate (≤48 h) did not significantly increase total cell number compared to controls, however, cell density was increased after 96 h of treatment (Fig. 1F). Overall, these results suggest that chronic high-dose glutamate exposure enhances NSPC proliferation but also maintains and promotes survival of these cells.
Given that adult rat brain and spinal cord-derived NSPCs had a very similar response to oxidative stress and glutamate exposure, we focused on spinal cord-derived NSPCs for all subsequent experiments.
Effect of glutamate on adult spinal cord-derived NSPC phenotype
We examined the phenotype of adult spinal cord NSPCs immediately after plating (0 h) and after the 96-h treatment with either control media or 500 μM glutamate to determine if glutamate induced differentiation into a specific neural lineage (Fig. 2). Initial NSPC cultures had a high expression of the neural precursor cell marker nestin (85.4 ± 0.5%) and low expression of the oligodendrocyte marker RIP (6.3 ± 3.9%), neuronal precursor marker beta III tubulin (2.2 ± 1.4%), and astrocyte marker GFAP (0.4 ± 0.6%). This phenotypic profile did not significantly change after the 96-h treatment with either control media or glutamate (500 μM). The majority of NSPCs remained nestin+ (87.4 ± 5.5% control vs. 91.2 ± 1.8% glutamate) and the percentage of RIP+ cells was similar between both conditions (6.2 ± 3.1% control vs. 6.9 ± 0.7% glutamate). Both control and glutamate-treated cells had a low expression of beta III tubulin (2.3 ± 0.7% vs. 1.4 ± 0.1%) and GFAP (0.2 ± 0.2% vs. 0.4 ± 0.1%). Therefore, treatment with 500 μM glutamate does not alter the phenotypic profile of adult spinal cord NSPCs.
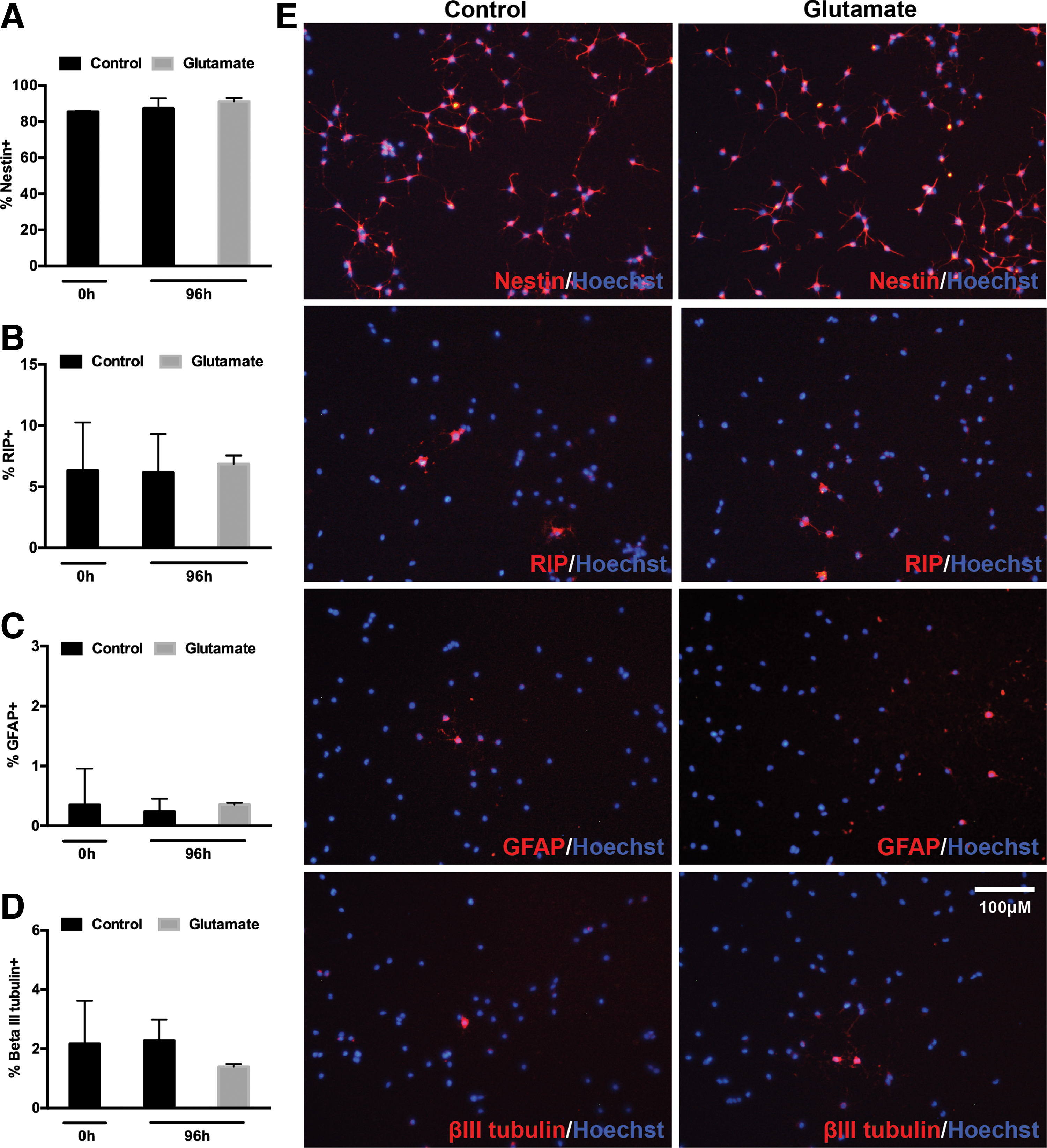
Phenotype of spinal cord NSPCs after 96 h of glutamate treatment. Initial NSPC cultures (0 h) had a high expression of nestin
The survival and proliferative effects of glutamate on adult spinal cord-derived NSPCs are mediated by AMPA/kainate receptors
To determine which glutamate receptors were involved in the survival and proliferative effects seen after 96 h of glutamate treatment, we concurrently exposed cells to the following receptor antagonists in the presence of 500 μM glutamate for 96 h: MK-801 (10 μM; a noncompetitive NMDA receptor antagonist), DL-AP5 (100 μM; a competitive NMDA receptor antagonist), GYKI-52466 (100 μM; a noncompetitive AMPA receptor antagonist, which has also been shown to antagonize kainate receptors), and LY-341495 (1 μM; an mGluR3 receptor antagonist). Antagonism of NMDA receptors (with either MK-801 or DL-AP5) or mGluR3 (with LY-341495) did not block the effects of glutamate on NSPCs. In contrast, AMPA/kainate receptor antagonism with GYKI-52466 blocked the increase in live cells (Fig. 3A), decrease in cell death (Fig. 3B), and increase in total cell number (Fig. 3C) induced by the 96-h glutamate treatment.
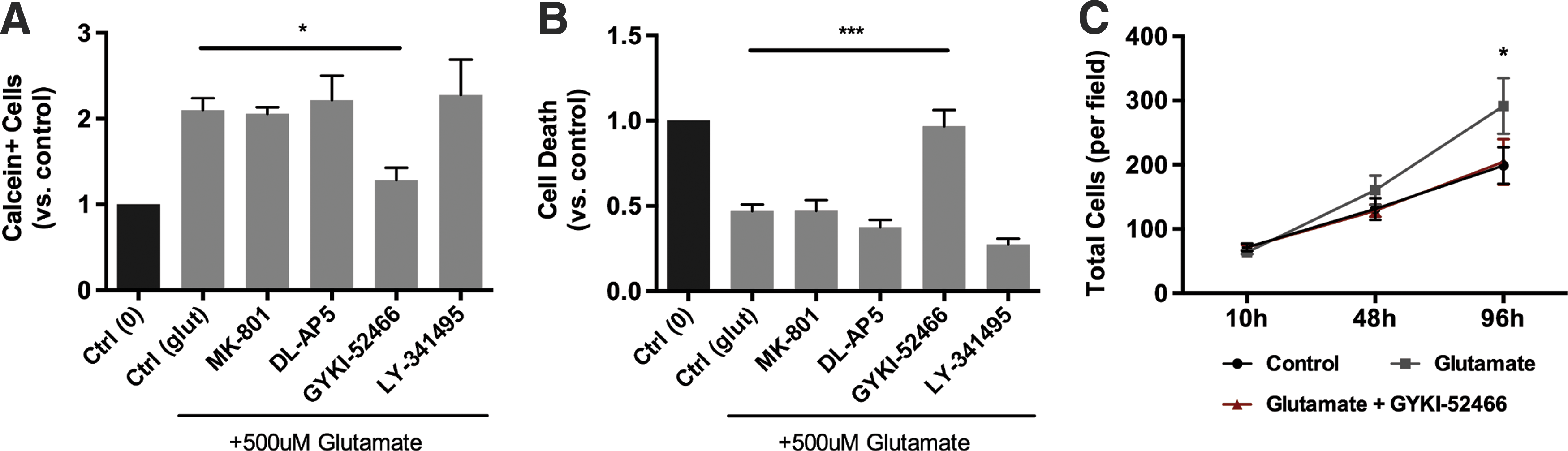
Effect of various glutamate receptor antagonists on glutamate-induced survival and proliferation of spinal cord NSPCs. Spinal cord-derived NSPCs were treated with the following glutamate receptor antagonists in the presence of 500 μM glutamate for 96 h [
To further confirm these findings, we treated NSPCs with various concentrations of the following glutamate receptor agonists for 96 h and assessed whether a similar effect to that seen with glutamate could be elicited: NMDA (10-1,000 μM; NMDA receptor agonist), AMPA (1–500 μM; AMPA receptor agonist), kainic acid (1–100 μM; kainate receptor agonist), and NAAG (1–500 μM; mGluR3 receptor agonist) (Figs. 4 and 5). Treatment with either NMDA (Fig. 4J, K) or NAAG (Fig. 4M, N) for 96 h did not alter the number of live or dead cells compared to controls. Moreover, neither of these agents had an effect on total cell growth (Fig. 4L, O). Treatment with AMPA or kainic acid mimicked the response seen with glutamate and was associated with a similar increase in live cell numbers and decrease in cell death compared to controls, and this approached significance (Fig. 4A, B, D, and E). Both agents also led to an increase in total cell density (Fig. 4C, F). Concentrations of kainic acid >100 μM were toxic to NSPCs after 96 h of treatment. Given that kainic acid is an agonist at both AMPA and kainate receptors, we determined if the effects of kainic acid are mediated through action on AMPA receptors rather than kainate receptors. We thus exposed cells to the GluR5 kainate receptor agonist, ATPA (1–500 μM), which has limited affinity for AMPA receptors. Following 96 h of ATPA exposure (100 μM), there was a similar increase in the number of live cells, decrease in cell death, and increase in total cell density as seen with glutamate (Fig. 4G–I).

Effect of various glutamate receptor agonists on spinal cord NSPCs. Treatment with AMPA (1–500 μM), kainic acid (1–100 μM), or ATPA (1-500 μM) for 96 h led to a dose-dependent increase in the number of live (calcein+) NSPCs
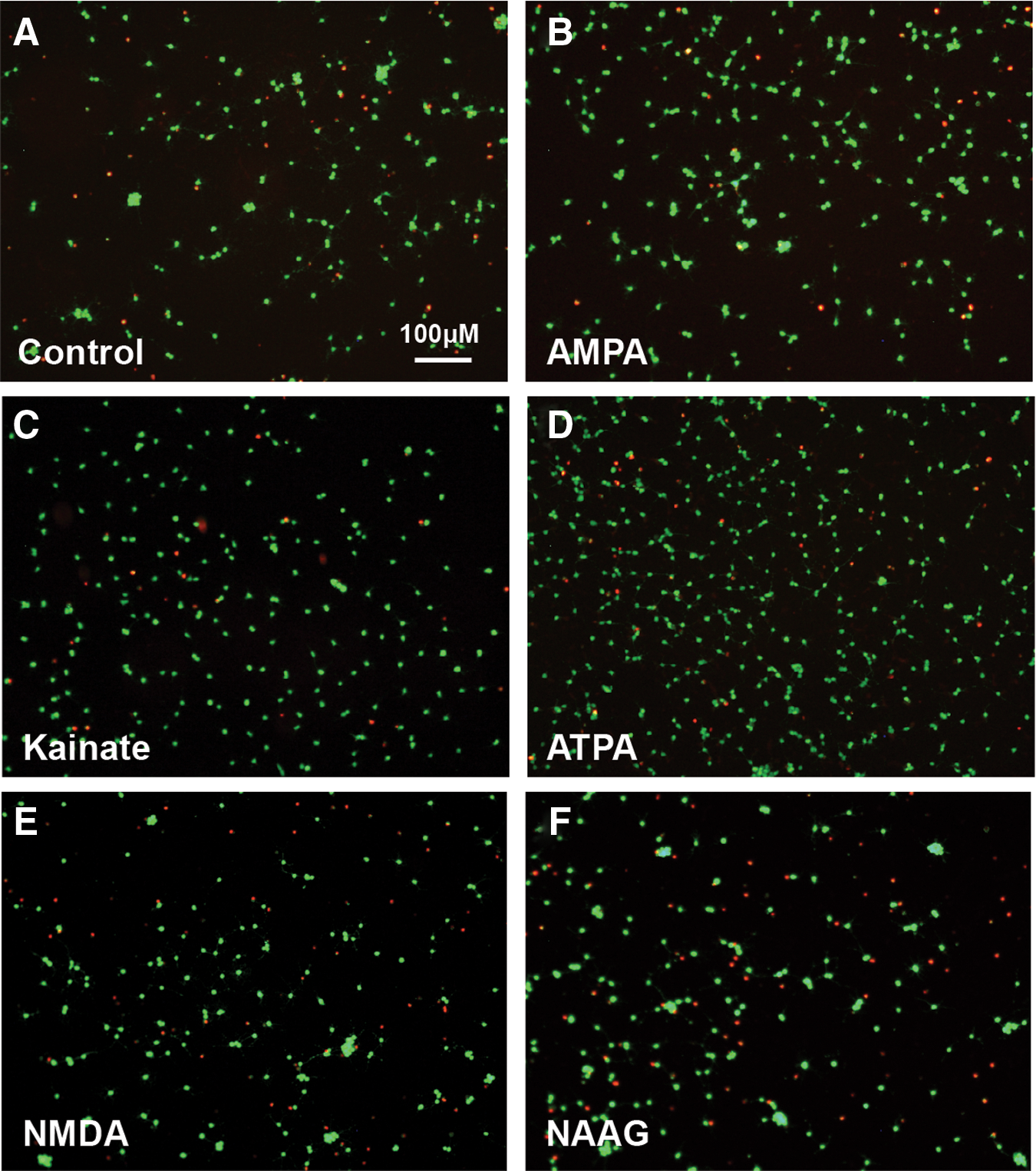
Live and dead cell staining after 96 h of treatment with glutamate receptor agonists. Representative images of calcein (green)/ethidium homodimer (red) staining after a 96-h treatment with the following agents: control media
Glutamate protects NSPCs from in vitro oxidative stress
To better examine the potential survival effects of glutamate on NSPCs, we determined whether glutamate could protect NSPCs from oxidative stress in vitro. NSPCs were exposed to H2O2 (500 μM) for 10 h in the presence or absence of 500 μM glutamate. In the absence of H2O2, a 10-h treatment with glutamate (500 μM) did not affect cell numbers compared to media-treated controls (Fig. 6A; 0 μM H2O2). However, NSPCs treated with 500 μM H2O2 in the presence of glutamate had a significant increase in the number of surviving cells compared to control cells exposed to H2O2 alone (Fig. 6A; 500 μM H2O2). Membrane integrity was also significantly higher in the glutamate-treated group compared to controls when exposed to H2O2 as measured by LDH release (Fig. 6C; 500 μM H2O2). Similar findings were also seen with brain-derived NSPCs (Fig. 6B, D).
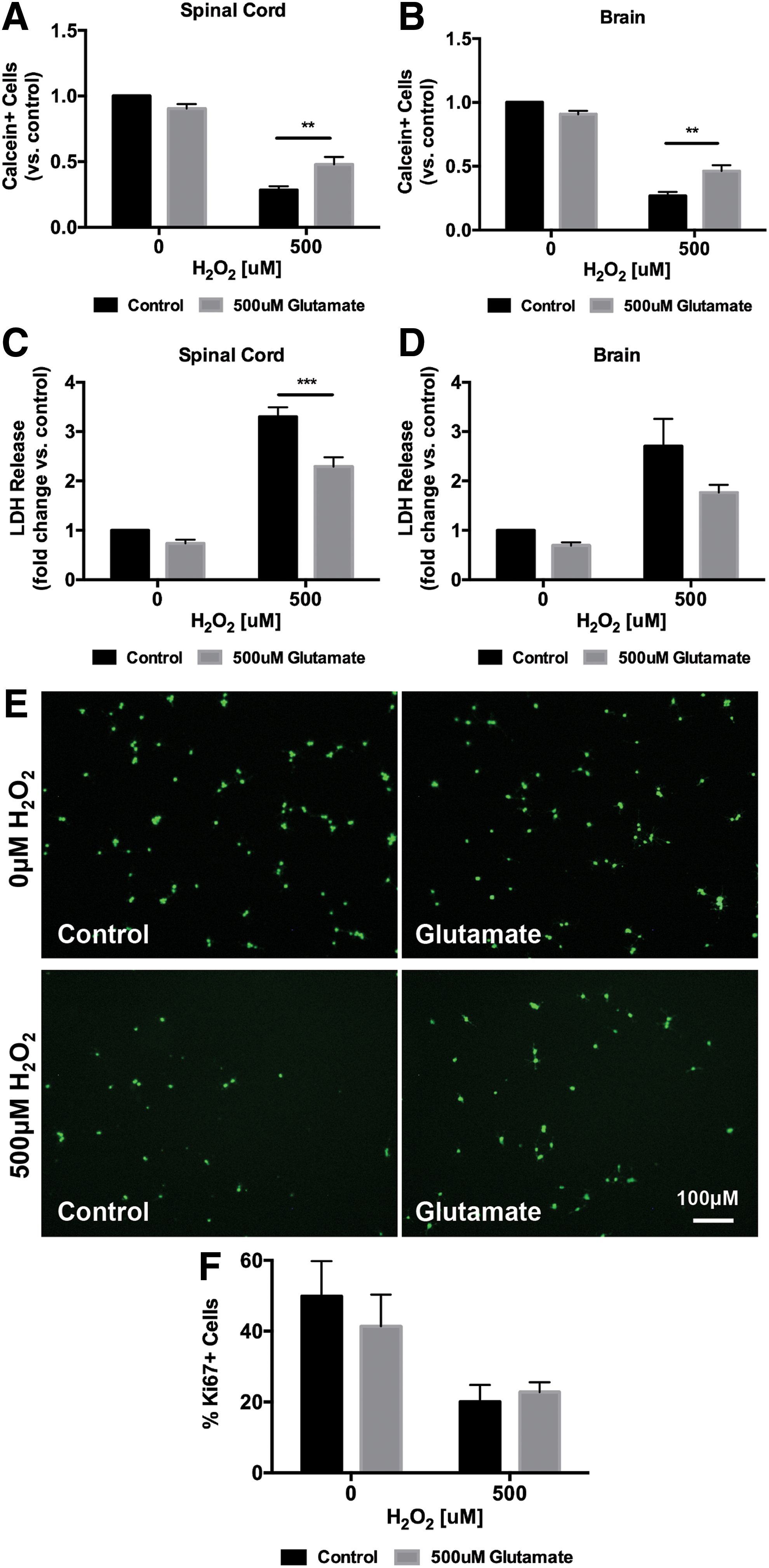
Glutamate attenuates in vitro oxidative stress-induced cell death in spinal cord NSPCs. Exposure to 500 μM H2O2 for 10 h significantly reduced live cell numbers and increased LDH release in both spinal cord and brain NSPCs [
Although we showed that treatment with glutamate (500 μM) for 10 h in the absence of H2O2 does not increase NSPC cell number, we wanted to exclude the possibility that glutamate in the presence of H2O2 could stimulate proliferation and contribute to the increase in live cells we observed. Therefore, to confirm that the increase in live cell numbers seen under glutamate treatment in the presence of H2O2 was due to survival and not proliferation, we stained for the proliferative marker Ki67. In the absence of H2O2, treatment with glutamate (500 μM) for 10 h did not affect the percentage of Ki67+ cells compared to controls. Similarly, in the presence of 500 μM H2O2, there was no difference in the percentage of Ki67+ cells between glutamate-treated cells and controls (Fig. 6F; 20.07 ± 4.73 vs. 22.78 ± 2.79), thus eliminating the possibility that concurrent glutamate treatment enhanced the proliferation of NSPCs in the setting of oxidative stress.
Glutamate-mediated protection of adult spinal cord-derived NSPCs against oxidative stress is dependent on AMPA/kainate receptors
To better understand the mechanisms behind glutamate-mediated protection of NSPCs in the setting of oxidative stress, we tested the effect of glutamate in the presence of different glutamate receptor antagonists (Fig. 7A). Cotreatment with MK-801 (10 μM) or DL-AP5 (100 μM) did not abolish the protective effects seen with glutamate in the setting of oxidative stress. However, the survival effects were lost when AMPA/kainate receptors or mGluR3 receptors were antagonized with GYKI-52466 (100 μM) or LY-341495 (1 μM), respectively.

Effect of various glutamate receptor antagonists and agonists on NSPC survival in the setting of in vitro oxidative stress. Spinal cord-derived NSPCs were treated with the following glutamate receptor antagonists in the presence of 500 μM glutamate and 500 μM H2O2 for 10 h [
In line with these findings, we found that concurrent treatment with NMDA (10–1,000 μM) did not protect NSPCs from H2O2-induced oxidative stress, but in fact reduced cell numbers below that of H2O2 control-treated cells (Fig. 7E), although this did not reach significance. Treatment with NAAG (1-500 μM) (Fig. 7F) had no effect on NSPC numbers. In contrast, concurrent treatment with AMPA, kainic acid, or ATPA produced a similar protective effect as that seen with concurrent administration of glutamate (Fig. 7B–D). Kainic acid receptor activation resulted in a more robust survival effect compared with AMPA receptors. Together, these results suggest that glutamate attenuates oxidative stress-induced cell death in NSPCs primarily through AMPA/kainate receptor signaling.
Discussion
Following traumatic SCI, high levels of glutamate are released into the spinal cord milieu leading to extensive neuronal and glial cell death. Multipotent, proliferative NSPCs reside in the region of the central canal in the spinal cord, but provide only limited regenerative capacity following injury [4,15,16]. Transplantation of exogenous adult NSPCs has emerged as a promising therapy to enhance regeneration after SCI in experimental models, however, graft survival is poor [17,18]. The effect of glutamate on the survival and proliferative capacity of this population of stem cells was largely unknown. In this study, we demonstrate that glutamate enhances survival and proliferation and attenuates oxidative stress-induced cell death in cultured adult spinal cord-derived NSPCs. Furthermore, we show that these effects are mediated by non-NMDA ionotropic glutamate receptors. These findings may have important implications for discovering mechanisms to enhance NSPC survival.
Glutamate and glutamate receptor-specific agonists have previously been shown to enhance the proliferation and survival of a variety of populations of brain-derived neural stem cells, including adult hippocampal, embryonic cortical, and perinatal SVZ cells [9]. Little is known about whether glutamate elicits similar responses in adult spinal cord-derived stem cells. Therefore, we compared the effects of glutamate treatment and oxidative stress on NSPCs derived from the SVZ of the brain and the periventricular region of the spinal cord of adult rodents. Both cell types were negatively affected by H2O2 exposure, but had increased survival and proliferation when exposed to high levels of glutamate (500 μM). Thus, we found no regional differences in responsiveness of adult brain or spinal cord NSPCs to glutamate or oxidative stress. As both brain and spinal cord-derived NSPCs are currently being used for transplantation after SCI, our findings provide important comparative insights into their response to different components of the secondary injury cascade.
Brazel et al. have previously shown that glutamate reduces basal levels of apoptosis in perinatal brain-derived neural progenitor cells through kainate and mGluR3 receptors [11]. In this study, we found that glutamate not only reduced cell death in NSPC cultures but also protected NSPCs from oxidative stress-induced cell death. Specifically, concurrent treatment with 500 μM glutamate increased the number of live cells and reduced LDH release following a 10-h exposure to H2O2 (500 μM), compared to cells exposed to H2O2 in the absence of glutamate. This effect was not due to an increase in cell proliferation as the expression of the proliferative marker Ki67 was the same between controls and glutamate-treated cells after 10 h of oxidative stress. Furthermore, 10-h glutamate treatment did not increase the number of NSPCs compared to controls in the absence of H2O2.
Using various glutamate receptor antagonists and agonists, we found that the prosurvival and proliferative effects of glutamate on NSPCs were mediated primarily through AMPA/kainate receptors. Kainate receptor activation induced a more robust effect compared to AMPA receptors, thus suggesting that this class of receptors may play a more important role in adult NSPCs. Previous studies have found that there is a high expression of AMPA/kainate receptors in the proliferative ventricular and subventricular zones of the developing embryonic brain [19]. Moreover, cultured neural stem cells have been shown to express functional Ca-permeable AMPA/kainate receptors [11,20]. Despite this, few studies have investigated the role of these receptors on neural stem cell survival and proliferation. Kainate receptor activation has been associated with increased proliferation and survival of neural progenitor cells derived from perinatal rodent brains [11] and fetal spinal cord organotypic slices [20], and activation of GluR5 containing kainate receptors with ATPA has been shown to be neuroprotective in the setting of hypoxic/ischemic stroke [21]. AMPA receptor potentiators or agonists have also been shown to increase proliferation of hippocampal dentate gyrus (DG) cells in the adult rat brain [22] and oligodendrocyte precursor cells in vitro [23].
The exact downstream pathways involved in the survival and proliferative effects seen with AMPA/kainate receptor activation in neural stem cells remain unknown and is an area that warrants further investigation. It has been found that neural stem cells protect mature neurons from glutamate-induced excitotoxicity through secretion of neurotrophic factors [24] or endocannabinoids. [25] While it is possible that secretion of these factors could act in an autocrine manner to enhance survival of NSPCs in the setting of glutamate exposure, it is unlikely that this mechanism alone can explain the robust glutamate-mediated survival and proliferative effects that we and other groups have found in neural stem cells [11]. Future experiments are therefore necessary to elucidate the underlying mechanisms behind this response.
Of the four classes of glutamate receptors, NMDA receptors (NMDARs) have received the most attention when studying secondary injury mechanisms after SCI as activation of these receptors leads to extensive neuronal and glial cell death. Calcium-permeable NMDA receptors are present on neural stem cells [11,26], however, the effect of NMDAR activation varies widely between studies, likely due to differences in stem cell age, source, and experimental conditions. Among stem cell lines derived from the developing embryonic or fetal brain, activation of NMDARs has generally been found to promote proliferation or survival [27,28], although conflicting reports exist [11]. In contrast, NMDAR stimulation in adult brain-derived neural stem cells appears to reduce cell proliferation or survival. Specifically, blocking NMDAR function was found to increase proliferation of cells in the dentate gyrus of the hippocampus [29 –31]. Interestingly, activation or antagonism of NMDARs was found to have no effect on SVZ cells in the adult rodent brain [30]. Consistent with these previous reports, we found that NMDARs were not responsible for the glutamate-mediated survival response of NSPCs. In fact, concurrent exposure to NMDA and H2O2 led to a slight decrease in cell survival compared to treatment with H2O2 alone, suggesting that NMDA may still maintain some toxicity in adult NSPCs in the setting of other lethal factors. Nevertheless, the effect of NMDAR activation on neural stem cell survival in this study was minimal. This may be explained by evidence suggesting that prolonged culture in FGF2-containing media, similar to that used in our study, downregulates the responsiveness of hippocampal neural stem cells to NMDA [32].
In addition to the ionotropic glutamate receptors, neural stem cells also express three different classes of metabotropic receptors (class I–III). In this study, we examined the mGluR3 receptor (a member of the class II family). Previous studies have found that mGluR3 receptor activation promotes proliferation of brain-derived neural stem cells [33,34]. Stimulation of mGluR3 receptors with NAAG has previously been shown to reduce neural precursor cell apoptosis [11]. Furthermore, blockage of mGluR3 receptors in adult mice was associated with decreased proliferation and survival of neural progenitor cells in the subventricular zone and dentate gyrus [35]. We did not see a similar effect as treatment with NAAG did not significantly alter NSPC numbers either alone or in the presence of H2O2. However, our findings do not preclude the possibility that activation of other metabotropic glutamate receptor classes could have an effect on spinal cord NSPC survival or proliferation. Recent evidence suggests that activation of mGluR4 receptors (a member of the class III family) protects embryonic rat cortical neural stem cells from oxidative stress in vitro [36]. The class I mGluR5 receptor has also been implicated in promoting proliferation of human NSPCs [37] and adult hippocampal neural stem cells [38]. An analysis of other metabotropic glutamate receptors in the context of adult spinal cord NSPCs thus warrants further investigation.
Our findings have important implications for understanding the response of adult NSPCs to the secondary injury of SCI. Glutamate may be an important factor that promotes their proliferation and survival after injury and allows them to persist even in the setting of other lethal stresses. Importantly, using microdialysis experiments, Xu et al. found that the concentration of glutamate in vivo after SCI reaches 550 μM, which is similar to the in vitro dose used in this study [39]. Furthermore, transplantation of adult neural stem cells is currently under investigation for use after SCI to improve functional recovery. Given the low rates of survival of these cells after transplantation, determining ways to promote their survival is essential. In this study, we demonstrate the neuroprotective effects of AMPA/kainate receptor activation in NSPCs and thus provide insight into a potential mechanism to explore to enhance survival of transplanted neural stem cells.
Conclusions
In summary, we present the first thorough examination of the effects of glutamate and glutamate-receptor agonists on NSPCs derived from the adult spinal cord. We show that high doses of glutamate enhance survival and proliferation of adult rat neural stem cells and this effect is dependent on non-NMDA ionotropic glutamate receptors. Furthermore, we show that glutamate attenuates in vitro oxidative stress of NSPCs through AMPA/kainate receptors, thus offering important insights into potential mechanisms to enhance NSPC survival.
Footnotes
Acknowledgments
We thank Linda Lee and Rita van Bendegem for their technical assistance and Shannon Weissman for assistance with the cell counts. We also thank the Krembil Foundation, Toronto General and Western Hospital Foundation, Spinal Cord Injury Ontario, and the Ontario-China Research and Innovation Fund for support.
Author Disclosure Statement
No competing financial interests exist.