
Abstract
The placenta is an organ that is formed transiently during pregnancy, and appropriate placental development is necessary for fetal survival and growth. Proper differentiation of the labyrinthine layer of the placenta is especially crucial, as it establishes the fetal–maternal interface that is involved in physiological exchange processes. Although previous studies have indicated the importance of inhibitor of differentiation/inhibitor of DNA binding-2 (Id2) helix-loop-helix transcriptional regulator in mediating cell differentiation, the ability of Id2 to regulate differentiation toward the labyrinthine (transport) lineage of the placenta has yet to be determined. In the current study, we have generated labyrinthine trophoblast progenitor cells with increased (SM10-Id2) or decreased (SM10-Id2-shRNA) Id2 expression and determined the effect on TGF-β-induced differentiation. Our Id2 overexpression and knockdown analyses indicate that Id2 mediates TGF-β-induced morphological differentiation of labyrinthine trophoblast cells, as Id2 overexpression prevents differentiation and Id2 knockdown results in differentiation. Thus, our data indicate that Id2 is an important molecular mediator of labyrinthine trophoblast differentiation. An understanding of the regulators of trophoblast progenitor differentiation toward the labyrinthine lineage may offer insights into events governing pregnancy-associated disorders, such as placental insufficiency, fetal growth restriction, and preeclampsia.
Introduction
T
Numerous molecular events regulating placental development are conserved in both humans and mice. Both human and rodent placentas consist of analogous cell types that are involved in placental transport processes [3 –11]. In rodents, the placenta comprises two zones: the junctional zone and the labyrinth. The placenta is further subdivided into three predominant cell lineages: labyrinthine cells, spongiotrophoblasts, and trophoblast giant cells [3,6,12 –14]. Different trophoblast lineages are derived via differentiation of trophoblast progenitor cells that perform specialized functions during gestation [3 –5,12]. The labyrinthine cells mediate the physiological fetal–maternal exchange processes, particularly gas, waste, and nutrients [3 –5,9,10,12,15,16].
Transport across the labyrinthine layer of the placenta is the main means by which the fetus is able to obtain the appropriate nutrients for growth and development [5,12,14,17 –20]. Thus, the differentiation of placental progenitors into labyrinthine cells is of critical importance for assuring fetal survival and well-being. In addition to offering insights into signals regulating differentiation of labyrinthine trophoblast cells, murine knockout and in vitro studies have been invaluable in advancing our understanding of genes that are required for proper placental development [5,6,15,16,21 –25].
Morphological differentiation involves branching and fusion of labyrinthine trophoblast stem (TS) cells, leading to formation of a multinucleate exchange surface that is functionally suited for placental transport of nutrients, particularly glucose [3,6,12,26,27]. The molecular events governing labyrinthine trophoblast differentiation have been reported to involve alterations in the expression of several transcription factors, including Id2, Cdx2, and Gcm1 [21,28 –36].
Several transcription factors belonging to the helix-loop-helix (HLH) family are essential for human and rodent placental development and for guiding trophoblast differentiation. Most HLH proteins possess a highly conserved basic region that allows DNA binding and transcriptional regulation. In the placenta, the basic-HLH (bHLH) transcription factors, Mash2, Hand1, and Stra13 direct trophoblast differentiation along the giant cell lineage [29,32,37 –40]. In contrast, bHLH transcription factor Tfeb has been shown to be essential for the vascularization and functional differentiation of the labyrinthine trophoblast lineage of the placenta [41,42]. Additionally, spatiotemporal regulation of the expression of bHLH transcription factors and the ability of HLH inhibitor of differentiation/inhibitor of DNA binding (Id) proteins to modulate these proteins have been reported to be necessary for proper placental development and cell differentiation [29,20,43 –46].
Id proteins are widely expressed throughout development and function in the determination of cell specification into specialized lineages [29,30,43,44,47 –49]. Id proteins lack the basic DNA-binding region of bHLH transcription factors. Instead of binding DNA, Ids are capable of binding to bHLH proteins, thus inhibiting bHLH-induced transcription that is necessary for differentiation [29,48 –50].
Four different Id isoforms (Id1–Id4) have been identified and Id1, Id2, and Id3 expression has been reported in the human and rodent placentas [28 –30,43,48,49,51,52]. In particular, Id2 is known to be an important regulator of placental differentiation, since Id2 mRNA and protein are the highest in proliferative TS cells and decline during differentiation into lineage-specific trophoblast subtypes in both humans and rodents [16,30,53]. Additionally, previous studies in human and rodent cultures have indicated that sustained Id1 and Id2 expression can prevent differentiation into giant cells and extravillous trophoblasts [29,30,43].
Our previous studies have demonstrated a dramatic downregulation of Id2 expression during TGF-β-induced differentiation of the labyrinthine-specific trophoblast progenitor cell line, SM10 [15]. In the current study, we have generated SM10-Id2 clonal cell lines and determined the importance of Id2 in mediating TGF-β-induced morphological, functional, and molecular differentiation. We have furthermore analyzed the effects of RNAi-mediated Id2 knockdown on the differentiation process. Our results suggest that Id2 downregulation is necessary for labyrinthine trophoblast differentiation.
Materials and Methods
Materials
Cell culture media [RPMI1640/
Cell culture and differentiation
Mouse SM10 cells were maintained in RPMI 1640/
Design of Id2 short hairpin sequences
All the Id2 siRNA and scrambled siRNA sequences were designed against the coding region of mouse Id2 gene (BC053699) using siRNA Wizard™ that was available online. Of the several recommended siRNA sequences, the sequences with the most favorable thermodynamic properties were chosen [57,58]. The siRNA sequences were converted into shRNA sequences using Block-it™ RNAi Designer with loop sequences 5′ TTCAAGAGA3′ or 5′CCACAC3′ [59,60]. Two previously published Id2 siRNA sequences were additionally converted into shDNA sequences [61].
Cloning of Id, GFP, and shDNA into lentiviral vector
The human Id2 cDNA was excised from pCDNA3-Id2, and the GFP cDNA was excised from pLv-CMV-GFP-V5 using BamHI and ApaI [45]. Restriction products were resolved by electrophoresis on 1% agarose gels and visualized by ethidium bromide staining. Appropriate restriction fragments were gel extracted and ligated as recommended by the manufacturer. Positive ligations were analyzed by restriction enzyme and western blotting analysis. Correct cloning of pLv-Id2 was further confirmed by sequencing using CMV-F and V5-R primers (Table 1). Id2 shDNA and scrambled shDNA sequences (Table 2) were cloned into the lentiviral expression plasmids using the BLOCK-iT Lentiviral RNAi Expression System. Briefly, shDNA sequences were annealed and cloned into linearized pENTR™/U6 vector as recommended by the manufacturer. Cloning products were analyzed by restriction enzyme analysis (BamH1, EcoRV), and positive clones were used for LR recombination into pLenti6/Block-it-DEST vector. Recombination reactions were performed as recommended by the supplier and confirmed by restriction enzyme analysis (BamHI, KpnI). Correct cloning into pLenti6/Block-it-DEST vector was further confirmed by sequencing using the U6-F and V5-R primers (Table 1).
Forward (F) and reverse (R) RT-PCR primer sequences, amplification temperature (Tm°C), expected size of the amplification product, and the reference for the primer sequence sources are shown. Genes for RT-PCR amplification primers are bolded. Genes for PCR amplification from genomic DNA are in italics. The size of the amplification product may vary, depending on the size of the insert, and these primers were additionally used to confirm correct cloning of Id2 and short hairpin constructs into the lentiviral vectors (*). The primer sequence was obtained from Invitrogen (C).
RT-PCR, reverse transcription-polymerase chain reaction.
The number after Id2 indicates the first coding region nucleotide recognized by the siRNA. Sense and antisense DNA sequences for the top (T) strand and the bottom (B) strand oligos, loop regions, and complete Id2 coding region targeted by the siRNA are shown. The Id2 shDNA sequences against the coding region of the mouse Id2 gene used in this study were generated using the siRNA Wizard™ and Block-iT™ RNAi designers and confirmed by NCBI nucleotide Blast.
Viral production and transductions
The viral supernatant was produced as recommended by the supplier. Briefly, 6 × 106 293FT cells were plated 24 h before transfection. The cells were transfected with the recommended amount of ViraPower mix and 10 μg of lentiviral expression vector using Metafectene transfection reagent. Sixteen hours later, the transfection media were removed and cells were provided fresh media. The virus containing media was collected at 60 h and applied to cells with 12 μg/mL of polybrene. Infections were allowed to proceed for 12 h, after which the cells were supplied with fresh media. All infections were performed using fresh virus on SM10 cells seeded the previous day.
Establishing Id2 and GFP expressing SM10 stable cell lines
Cells were plated at 3.3 × 103 cells/mL in 60 mm dishes. Twenty-four hours post-seeding, SM10 cells were infected with freshly isolated pLv-Id2 or pLv-GFP virus in media containing 12 μg/mL polybrene. After selection in blasticidin (8 μg/mL), Id2 and GFP stable clones were established by limiting dilution of 0.5 cell/well. Positive clones were analyzed by PCR amplification of genomic DNA using CMV-F and V5-R primers flanking the Id2 and GFP genes and by western blotting analysis of Id2 (Table 1 and Fig. 3).
Reverse-transcription-PCR
Cells were harvested by trypsinization and centrifuged at 2,000 rpm for 5 min. The media were aspirated, and total RNA was extracted using the RNeasy Kit (Qiagen) according to the manufacturer's instructions. To remove any contaminating DNA, the samples were treated with RNase-free DNaseI for 30 min at 37°C. For RT-PCR analysis, 5 μg of total RNA was reverse transcribed using random primers and PCR was amplified for 40 cycles at appropriate temperatures for each primer set (Table 1) [62 –64]. Briefly, PCR was performed using 2.5 U of Taq DNA polymerase, 1× PCR buffer without MgCl2, 1.875 mM MgCl2, 200 μM dNTPs, and 200 nM reverse and forward primers. Co-amplification of β-actin in each sample was used for semiquantitative normalization. After the initial denaturation at 94°C for 5 min, all PCRs were performed under the following conditions: 95°C denaturation for 1 min, specific primer annealing temperature (Table 1) for 1 min, and primer extension at 72°C for 1 min for a total of 40 cycles. The PCR products were resolved by electrophoresis on 1% agarose gels and visualized by ethidium bromide staining.
Real-time qPCR
Total RNA was collected and processed as described earlier. RNA (2 μg) was reverse transcribed using random primers, and samples were diluted 1:2. Real-time qPCR was performed using an ABI Prism 7700 thermal cycler (Applied Biosystems). Id2 and β-actin amplification was detected using a sequence-specific TaqMan MGB probe and Id2 or β-actin-specific primers. Briefly, each reaction contained 900 nM antisense primers, 900 nM sense primers, a 250 nM probe, 1 × Taqman Universal Master Mix (10 μL), and cDNA sample (1 μL) in a final reaction volume of 20 μL. Samples from three independent experiments were analyzed in triplicate. All samples were run simultaneously on 96-well reaction plates as follows: PCR amplification included an initial phase of 10 min at 95°C, then 40 cycles of 15 s at 95°C, and 1 min at 60°C. Expression levels of Id2 were determined by normalization to β-actin (endogenous control) in each sample using the comparative CT method 2−ΔΔCt [53,65]. All real-time qRT-PCR experiments included a no-template (H2O) control. To quantify decreased Id2 expression, we analyzed changes in Id2 expression by qPCR. Undifferentiated TS cells were used as a positive control for semiquantitative RT-PCR and qPCR analyses.
Western blotting and immunostaining
Western blotting was performed as previously described using rabbit polyclonal anti-Id2 antibody at 1/500 overnight at 4°C overnight in blocking buffer [5% (w/v) non-fat milk, pH 7.4 in 1× PBS containing 0.05% Tween-20 (PBST)] [54,66]. The membrane was washed with PBST, probed with HRP-conjugated goat anti-rabbit IgG secondary antibody at 1/10,000 for 1 h at room temperature, and visualized using Enhanced Chemi-Luminiscence reagent according to the manufacturer's instructions. For immunostaining, the SM10 cells were seeded at 3 × 104 cells/mL in 35 mm culture dishes. Twenty-four hours post-seeding, the cells were infected and subsequently treated with TGF-β or vehicle for 72 h. Cells were rinsed and fixed with 3.7% paraformaldehyde in 1× PBS. Fixation was quenched in 10 mM glycine in 1× PBS for 10 min, and cells were rinsed with 1× PBS. Cells were permeabilized and blocked in buffer containing 3% goat serum, 250 mM KCl, 20 mM HEPES pH7.8, 0.1% glycine, and 0.5% Triton X-100 in 1× PBS for 30 min. The cells were incubated with anti-Id2 antibody at 1/200 overnight at 4°C in blocking buffer; rinsed and probed with Alexfluor 594 F(ab′)2 fragmented goat anti-rabbit IgG antibody at 1/1,000 for 1.5 h; and incubated with 1 μg/mL Hoechst dye for 1 min to stain the cell nuclei. The coverslips were mounted using Vectashield mounting media, and images were visualized by epifluorescence microscopy and MetaMorph software.
Luciferase reporter transactivation
SM10 cells were plated at 5 × 104 cells/mL in a six-well plate. Twenty-four hours post-seeding, the cells were transfected for 24 h with 1 μg E7-TK-luc and 0.1 μg pRLSV40 using Metafectene transfection reagent, as previously described [53,67,68]. The cells were placed in fresh media and 5 ng/mL TGF-β was added for 72 h, after which the cells were rinsed in 1× PBS and analyzed using the Dual Luciferase Reporter system according to the manufacturer's recommendation. All E7-TK-Luc values were normalized to pRLSV40.
3[H]-2-deoxyglucose uptake
Glucose uptake was assayed as previously described [15,53,69]. Briefly, the cells were plated at 2 × 104 cells/mL in 24-well plates. Twenty-four hours post-seeding, the cells were differentiated for 72 h. The media were then aspirated, and the cells were washed three times with pre-warmed 1× PBS. The cells were pulsed for 10 min with 1 μCi of 3[H]-2-deoxyglucose in transport buffer (25 mM HEPES, 0.8 mM MgSO4, 140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2) at 37°C. Glucose uptake was quenched by washing cells with ice-cold transport buffer. The cells were detached by adding 600 μL of 0.03% sodium dodecyl sulfate in H2O, and a 400 μL aliquot was analyzed for radioactivity by liquid scintillation counting. Cells were counted by trypan blue exclusion using the Beckman Coulter cell analyzer and Vi-Cell 1.01 software. Uptake values were normalized to cell counts. For the RNAi analyses, the cells were infected with 0.5 mL virus containing media and briefly selected (3 days) in media containing blasticidin (8 μg/mL).
Phalloidin staining
SM10 cells were seeded at 3 × 104 cells/mL in 35 mm culture dishes. Twenty-four hours post-seeding, the cells were infected and subsequently treated with 5 ng/mL TGF-β or vehicle. The cells were processed as described earlier for immunostaining. Cells were incubated with 6.6 μM rhodamine-conjugated phalloidin in blocking buffer for 1 h, rinsed in 1× PBS, and incubated with 1 μg/mL Hoechst dye for 1 min to stain the cell nuclei. Coverslips were mounted using Vectashield mounting media, and images were visualized by epifluorescence microscopy and MetaMorph software.
Statistical analysis
Experiments were independently replicated at least three times with similar results. Quantitative data from experiments are represented as the average ± SEM. Statistical analysis was performed by the Statistical Consulting Center at Wright State University by using one sample t-test for glucose uptake experiments and two-sample t-test for qPCR analyses. P values for 95% confidence interval were calculated. P < 0.025 was considered significant and is denoted by *.
Results
Id2 is the only Id family member that exhibits decreased RNA expression on differentiation of the labyrinthine trophoblast progenitor cell line, SM10
Previous reports have shown expression of several Id isoforms in rodent and human trophoblast cells [28 –30,43,52]. We have previously demonstrated that the expression of Id2 is dramatically reduced during differentiation of labyrinthine-specific, trophoblast progenitor SM10 cells [15]. To determine the presence of other Id isoforms in SM10 cells and their response to TGF-β or vehicle treatment, the expression of Id1–Id4 in SM10 cells was analyzed by RT-PCR and qPCR. Expression of Id1 was minimally detectable in undifferentiated and differentiated SM10 cells (Fig. 1A). Rcho-1 and TS cells served as controls and were analyzed for Id1 expression, and only the Rcho-1 cells expressed Id1. In contrast to Id1, Id2 expression was readily detectable in vehicle-treated SM10 cells and dramatically downregulated in response to TGF-β, as previously described (Fig. 1C). Quantification of Id2 expression by real-time qPCR analysis indicated a significant 13-fold reduction in Id2 levels in the SM10 cell line in response to TGF-β (Fig. 1D). Low levels of Id3 expression were detected in the SM10 cell line; however, addition of TGF-β did not change the level of expression (Fig. 1B). Similar to Id1, Id3 could also be detected in Rcho-1 cells that were used as a control, but expression was absent in TS cells (Fig. 1B). Expression of Id4 could not be detected in TGF-β or vehicle-treated SM10 cells (data not shown).
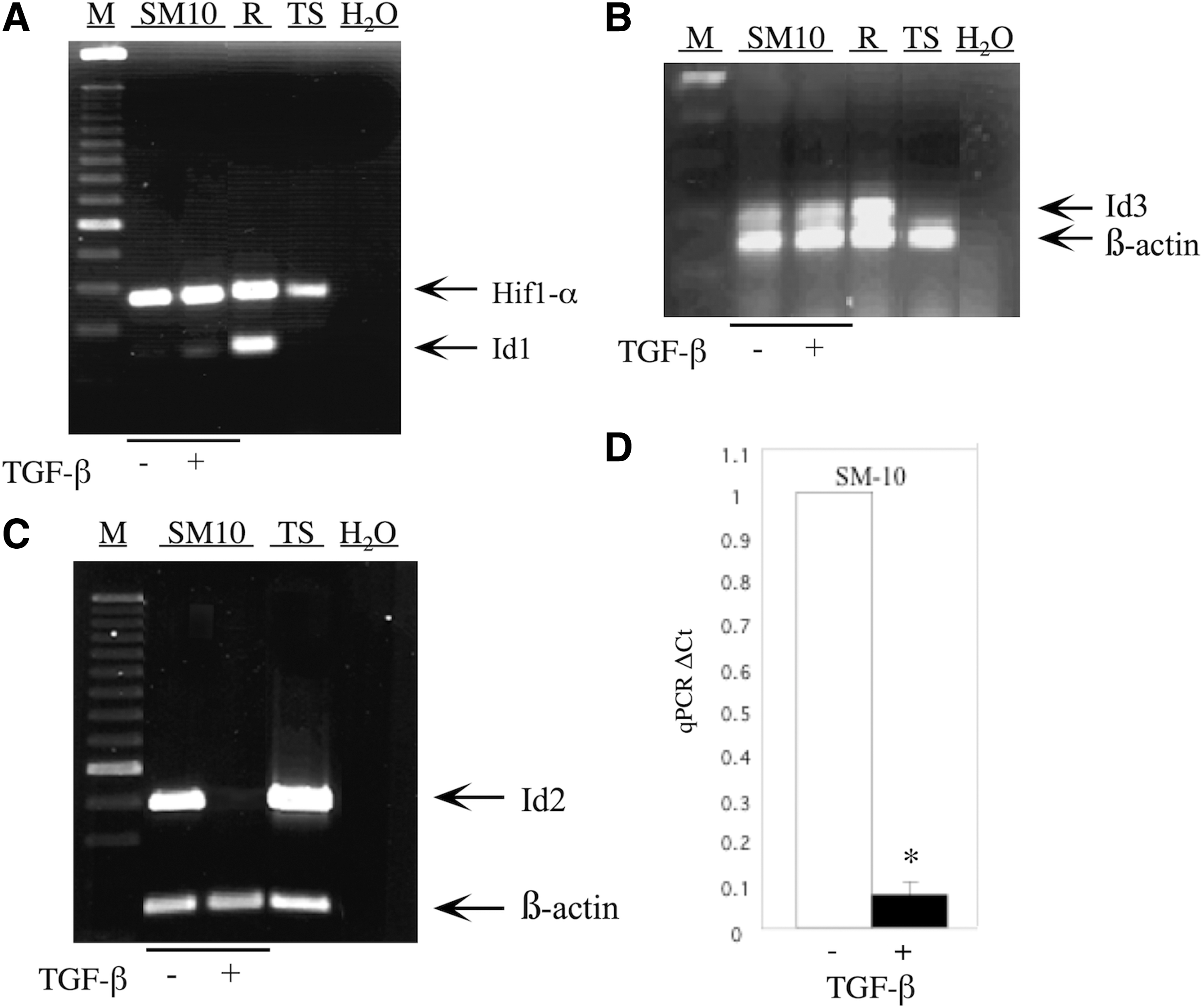
Id2 expression is inhibited on differentiation of the labyrinthine trophoblast progenitor cell line, SM10. Total RNA from TGF-β or vehicle-treated, SM10 cells was RT-PCR amplified with appropriate primers (Table 1) and resolved on a 1% agarose gel. Expression of Id1
Id2 protein is decreased in differentiated SM10 cells
In addition to mRNA downregulation during trophoblast progenitor cell differentiation, Id2 protein levels during differentiation were also investigated. To analyze changes in Id2 protein levels during differentiation of progenitors into labyrinthine cells, Id2 protein levels were investigated via immunofluorescent staining of TGF-β and vehicle-treated SM10 cells. Id2 protein was readily detected in vehicle-treated SM10 cells (Fig. 2C); however, its presence was nearly abolished in response to TGF-β treatment (Fig. 2D). The color combination of Id2 immunofluorescent staining and nuclear images (Fig. 2E, F) additionally indicated increased nuclear concentration of Id2 in the undifferentiated SM10 cells (Fig. 2E).

Id2 protein is decreased in differentiated SM10 cells. SM10 cells were treated for 72 h with TGF-β or vehicle. The cells were paraformaldehyde fixed and probed with anti-Id2 primary and Alexa Fluor 594–conjugated secondary antibodies. The cell nuclei were stained with Hoeschst dye. Epifluorescence microscopy analysis of Id2 immunoreactivity
Id2 overexpression prevents TGF-β-induced downregulation of Id2 RNA, protein, and activity in clonal SM10-Id2
The dramatic downregulation of Id2 mRNA and protein levels in response to TGF-β suggested its involvement in directing labyrinthine trophoblast differentiation. To determine whether Id2 is required for TGF-β-induced differentiation, Id2 and GFP expressing SM10 cell lines were generated. Stable clones (5, 10, and 12) were confirmed by PCR analysis of the genomically integrated Id2 gene using CMV-F and V5-R primers flanking the cDNA sequence (Fig. 3A and Table 1) and by western blot analysis (Fig. 3B). No endogenous Id2 protein could be detected by western blot analysis, as indicated in the GFP control (Fig. 3B). Id2-transfected Cos7 cells served as a positive control (Cos7/Id2). The inability to detect endogenous Id2 in GFP clones is most likely due to antibody sensitivity, as transfected Id2 was readily detectable (Fig. 3B), which is consistent with the manufacturer's specifications.
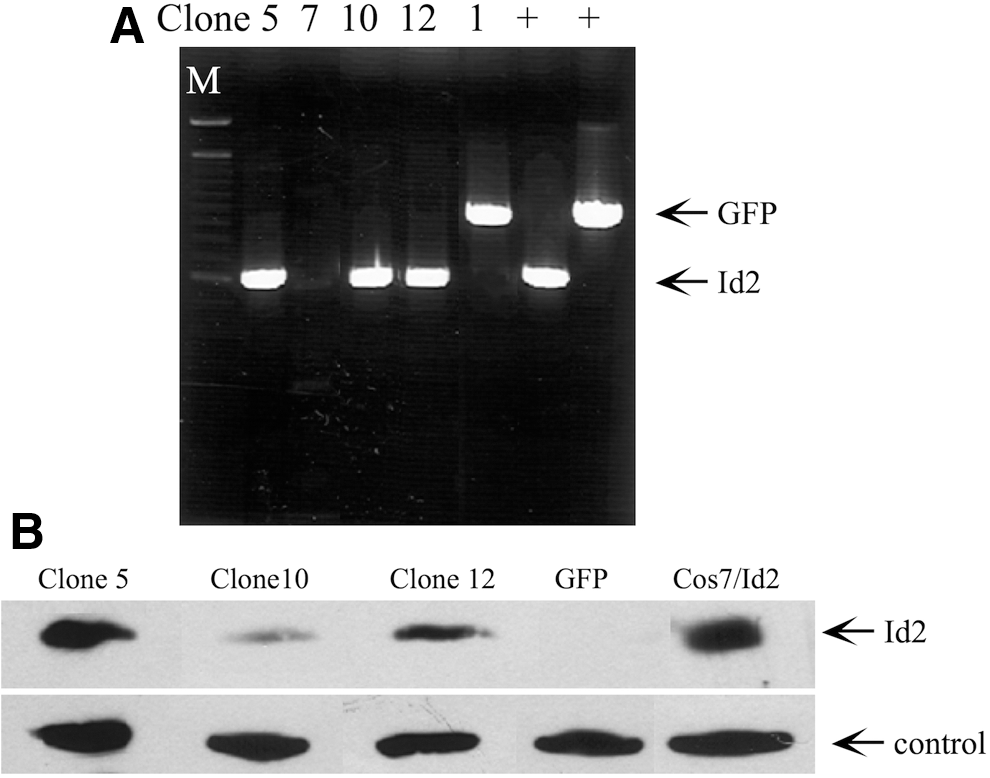
Generation of SM10-Id2 and SM10-GFP stable cell lines.
Id2 mRNA and protein levels in TGF-β differentiated or undifferentiated, vehicle-treated SM10-Id2 and SM10-GFP clones were additionally analyzed for Id2 expression (Figs. 4 and 5). In contrast to SM10-GFP clones, SM10-Id2 clones maintained Id2 mRNA expression after addition of TGF-β, as shown by RT-PCR (Fig. 4A). Western blotting analysis further confirmed that Id2 protein levels were maintained in SM10-Id2 expressing cells (Fig. 4B). Cos7 cells transiently expressing lentiviral Id2 served as a positive control (Fig. 4B). To further evaluate Id2 function, we analyzed E-box transactivation luciferase assays in Id2-SM10 clones using the E7-TK-luciferase reporter, which contains seven E-box sequences in its promoter (Fig. 4C) [70]. Id2 is known to bind to bHLH transcription factors and to inhibit bHLH transcription factor-induced expression of genes containing E-box elements [49,50]. Therefore, a reduced level of Id2 would lead to an increase in E-box luciferase activation. In SM10-GFP clones, in which TGF-β inhibits Id2, we see a corresponding 7.5-fold increase in E-box luciferase activity (Fig. 4A, C). In SM10 cells stably expressing Id2, however, Id2 overexpression abolishes TGF-β-induced transactivation of E7-Tk-luciferase reporter activity (Fig. 4A, C). This result is consistent with the fact that maintenance of Id2 expression, even under differentiating conditions, prevents E-box activation.
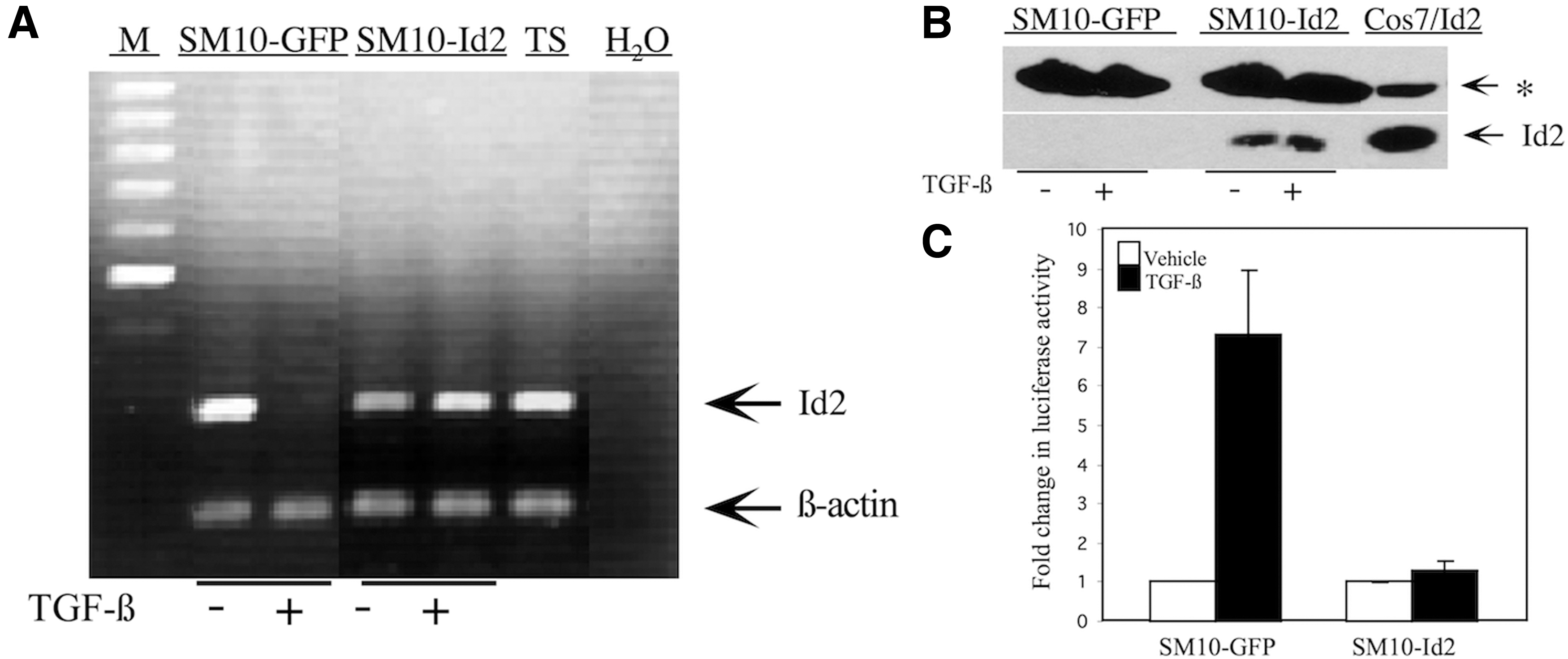
Id2 is expressed and transcriptionally active in clonal SM10-Id2 with TGF-β or vehicle control treatment.

Id2 overexpression prevents TGF-β-induced downregulation of Id2 protein in clonal SM10-Id2. Epifluorescence microscopic analysis of Id2 protein immunoreactivity in SM10-GFP and SM10-Id2 cells treated with TGF-β or vehicle control. Id2 expression was analyzed using anti-Id2 primary antibody and Alexa Fluor 594–conjugated secondary antibody. Cell nuclei were stained with Hoechst dye before imaging, and nuclei are indicated in green
In addition, we examined Id2 protein in our stable clones by immunofluorescent staining. TGF-β and vehicle-treated SM10-GFP and SM10-Id2 cells were stained for nuclei (Fig. 5A–D) or Id2 (Fig. 5E–H). Additionally, color-combined images were examined to assess nuclear localization of Id2 (Fig. 5I–L). As expected for SM10 cells stably expressing GFP, Id2 was downregulated (Fig. 5E, F). Id2 localization, in SM10-GFP cells, was similar to that seen in nontransduced SM10 cells (Fig. 2C, D). Our immunofluorescent staining confirmed that Id2 protein levels were maintained in SM10-Id2 cells, even in the presence of TGF-β (Fig. 5H, L).
Id2 overexpression inhibits TGF-β-induced morphological differentiation
Previous studies have demonstrated that differentiation of labyrinthine trophoblast cells involves morphological changes, such as cell aggregation and colony formation [15,53]. To investigate the effects of Id2 overexpression on TGF-β-induced morphological differentiation, we analyzed cell morphology by using rhodamine-conjugated phalloidin and nuclear staining of TGF-β or vehicle-treated SM10-GFP and SM10-Id2 cells (Fig. 6). No morphological differentiation was detected in cells exposed to the vehicle (Fig. 6A, B). SM10-Id2 cells showed inhibition of morphological differentiation when exposed to TGF-β (Fig. 6D), as compared with SM10-GFP cells exposed to TGF-β (Fig. 6C). Instead of aggregating and forming colonies, the cells retained a more progenitor-like, single-cell morphology (Fig. 6D). Formation of multinucleated colonies in TGF-β-treated SM10-GFP, which is indicative of differentiation, was clearly evident (Fig. 6C); whereas this response was almost completely inhibited by Id2 overexpression (Fig. 6D).

Id2 overexpression inhibits TGF-β-induced morphological differentiation. Epifluorescence microscopy images of rhodamine-conjugated, phalloidin-stained clonal SM10-GFP
Id2 overexpression inhibits TGF-β-induced SM10 functional and molecular differentiation
Functional and molecular differentiation accompanies the morphological differentiation of labyrinthine trophoblast cells. One of the main functions of the labyrinthine layer of the placenta is the physiological exchange of nutrients between the mother and the fetus [4,5,12,14,17 –20]. Thus, the effects of Id2 overexpression on functional differentiation were examined by analyzing glucose uptake in TGF-β or vehicle-treated SM10-GFP and SM10-Id2 cells. TGF-β treatment resulted in a significantly induced twofold increase in glucose uptake into SM10-GFP cells; however, this upregulation was abolished by the overexpression of Id2 in the SM10-Id2 cells, which is suggestive of inhibition of differentiation (Fig. 7).
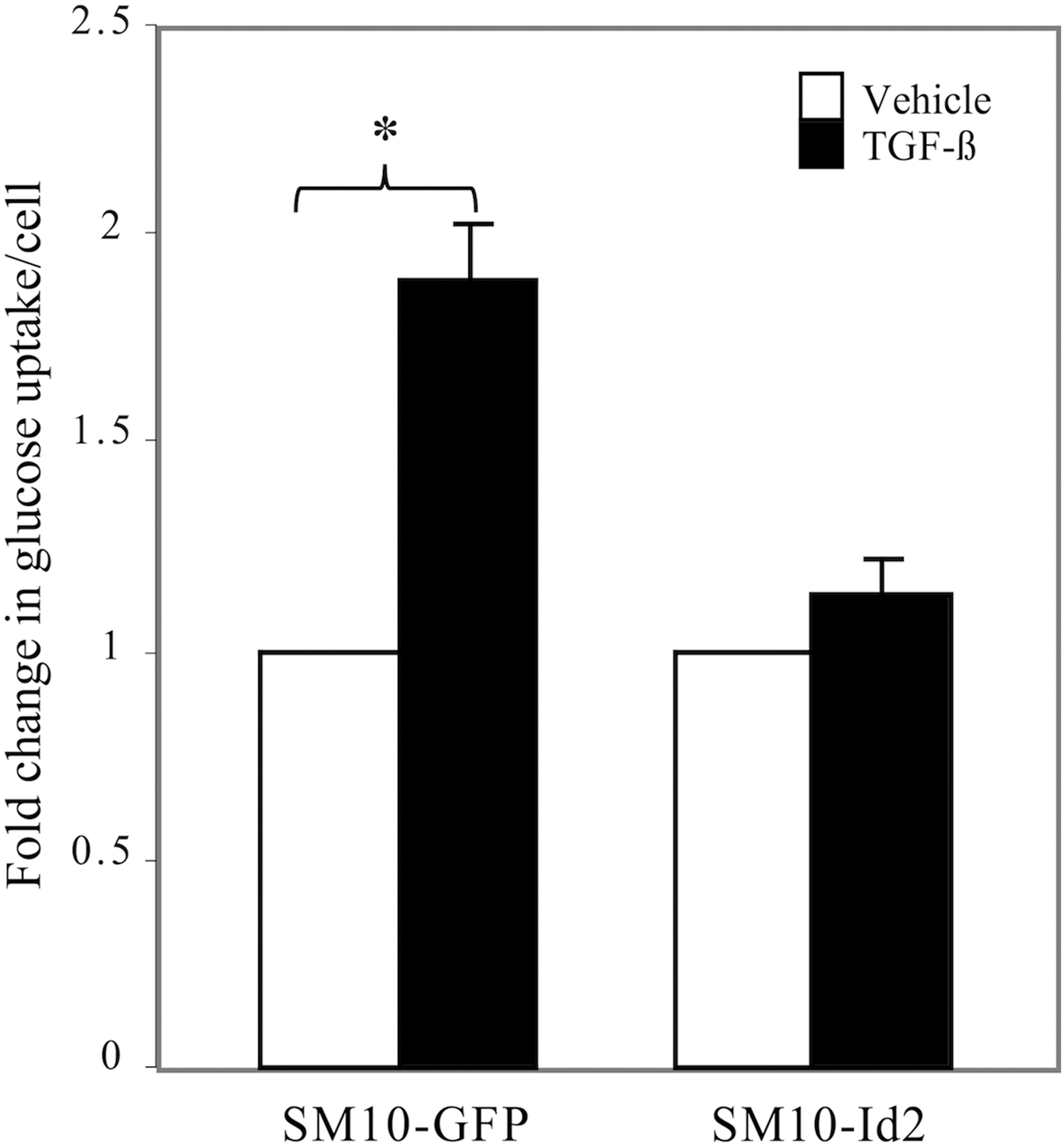
Id2 overexpression prevents the TGF-β-induced increase in glucose uptake. After 72 h of TGF-β or vehicle treatment, clonal SM10-GFP and SM10-Id2 cells were pulsed with 1 μCi/mL of 2-deoxyglucose for 10 min and uptake was measured by liquid scintillation counting. Cells were counted by trypan blue exclusion, and glucose uptake values were normalized to cell number. Fold change in glucose uptake is indicated as a ratio of normalized TGF-β-treated values to normalized vehicle control-treated values. Each experiment was conducted at least three independent times. Error bars represent standard deviations from the mean *P < 0.01.
Likewise, TGF-β-induced molecular differentiation was inhibited in Id2 overexpressing SM10 cells, as the expression of labyrinthine TS cell marker, Cdx2, was maintained in TGF-β-treated SM10-Id2 cells (Fig. 8A) [47,71,72]. In addition, TGF-β treatment induced the expression of a marker of differentiating labyrinthine trophoblasts, Gcm1, in SM10-GFP cells but did not induce Gcm-1 in SM10-Id2 cells, further supporting the maintenance of a progenitor cell-like state of SM10-Id2 cells (Fig. 8B) [35,73,74].

Id2 overexpression results in sustained expression of stem cell marker Cdx2 and repression of labyrinthine differentation marker Gcm1 SM10 cells. Total RNA from subconfluent SM10-GFP and SM10-Id2 cells was RT-PCR amplified with appropriate primers and resolved on a 1% agarose gel. Expression of stem cell-specific marker (Cdx2)
Knockdown of Id2 in SM10 cells results in labyrinthine trophoblast differentiation
Even though overexpression analyses suggested that Id2 is a mediator of TGF-β-induced labyrinthine trophoblast differentiation, Id2 knockdown experiments were additionally designed to confirm that inhibition of endogenous Id2 is necessary for labyrinthine trophoblast differentiation. Several sequences targeting the Id2 coding region were designed. The two most effective Id2-shRNA sequences and their scrambled control sequences were generated and cloned into lentiviral vectors (Table 2). Although both shRNAs displayed similar results, the most effective Id2-shRNA (Id2-369) and its control-scrambled shRNA (Id2-369s) were used in subsequent experiments (Table 2). Genomic integration of the Id2-shRNA (Id2sh) and control scrambled Id2-shRNA (Ssh) sequences into the SM10 cells was confirmed by PCR amplification of genomic DNA of infected SM10 cells using primers flanking the exogenous insert (Fig. 9A and Table 1). Knockdown of Id2 RNA in SM10 cells was confirmed by PCR using primers specific to endogenous Id2 (Fig. 9B and Table 1). Additionally, RNA collected from TS cells and PCR amplified using primers specific to Id2 were used as positive controls (Fig. 9B). Expression of Id2 is abolished by the expression of Id2-shRNA, but not by the expression of scrambled Id2-shRNA (Fig. 9B).
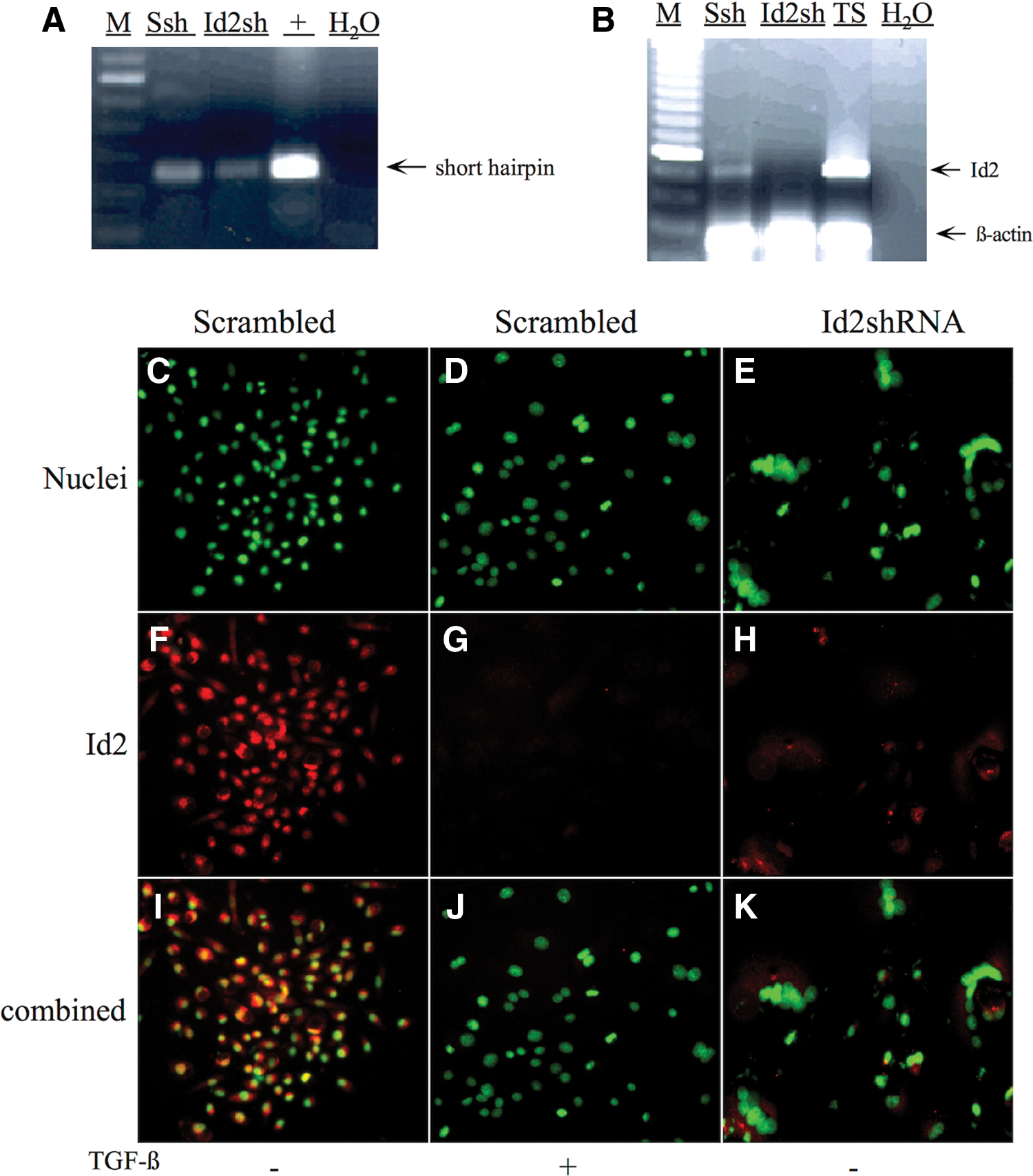
Generation of Id2 knockdown and scrambled control shRNA SM10 cells.
In addition to PCR amplification, immunofluorescent staining for Id2 and nuclear staining was performed to assess abundance and location of Id2 protein in TGF-β or vehicle control-treated SM10 cells expressing scrambled Id2-shRNA or vehicle-treated Id2-shRNA SM10 cells (Fig. 9C–K). It should be noted that repeated attempts to propagate a stable cell line expressing Id2-shRNA were unsuccessful due to the induction of continual terminal differentiation. As expected for SM10 cells with scrambled control Id2-shRNA (Scrambled), Id2 localization was similar to the localization seen in nontransduced SM10 cells (Fig. 9F, G and Fig. 2C, D). The immunofluorescent staining also confirmed that Id2 is reduced in vehicle-treated Id2-shRNA expressing SM10 cells (Id2-shRNA) (Fig. 9H) compared with the vehicle-treated scrambled Id2-shRNA expressing SM10 cells (Scrambled) (Fig. 9F). Interestingly, this reduction in Id2 protein, in the Id2-shRNA expressing SM10 cells (Fig. 9H), is similar to that of the TGF-β-treated scrambled Id2-shRNA expressing SM10 cells (Fig. 9G).
Our Id2 overexpression studies suggested that Id2 may mediate events associated with TGF-β-induced changes in labyrinthine trophoblast cell morphology and that Id2 knockdown promotes labyrinthine trophoblast differentiation. Analysis of cell morphology using rhodamine-conjugated phalloidin and nuclear staining showed that SM10 cells infected with scrambled Id2-shRNA maintained a progenitor-like, single-cell morphology (Fig. 10A) and differentiated as expected when treated with TGF-β (Fig. 10C). In Id2-shRNA expressing SM10 cells, in the absence of TGF-β, the formation of multinucleate cells could be observed 3–4 days post-infection in vehicle-treated controls and this response was maintained up to 10 days, as observed by rhodamine-conjugated phalloidin and nuclear staining (Fig. 10B). Thus, reduced Id2 expression resulted in morphological differentiation of SM10 cells (Fig. 10B). Addition of TGF-β to Id2-shRNA expressing SM10 cells did not have any further differentiating effect on the morphology of Id2-shRNA expressing SM10 cells (Fig. 10D).
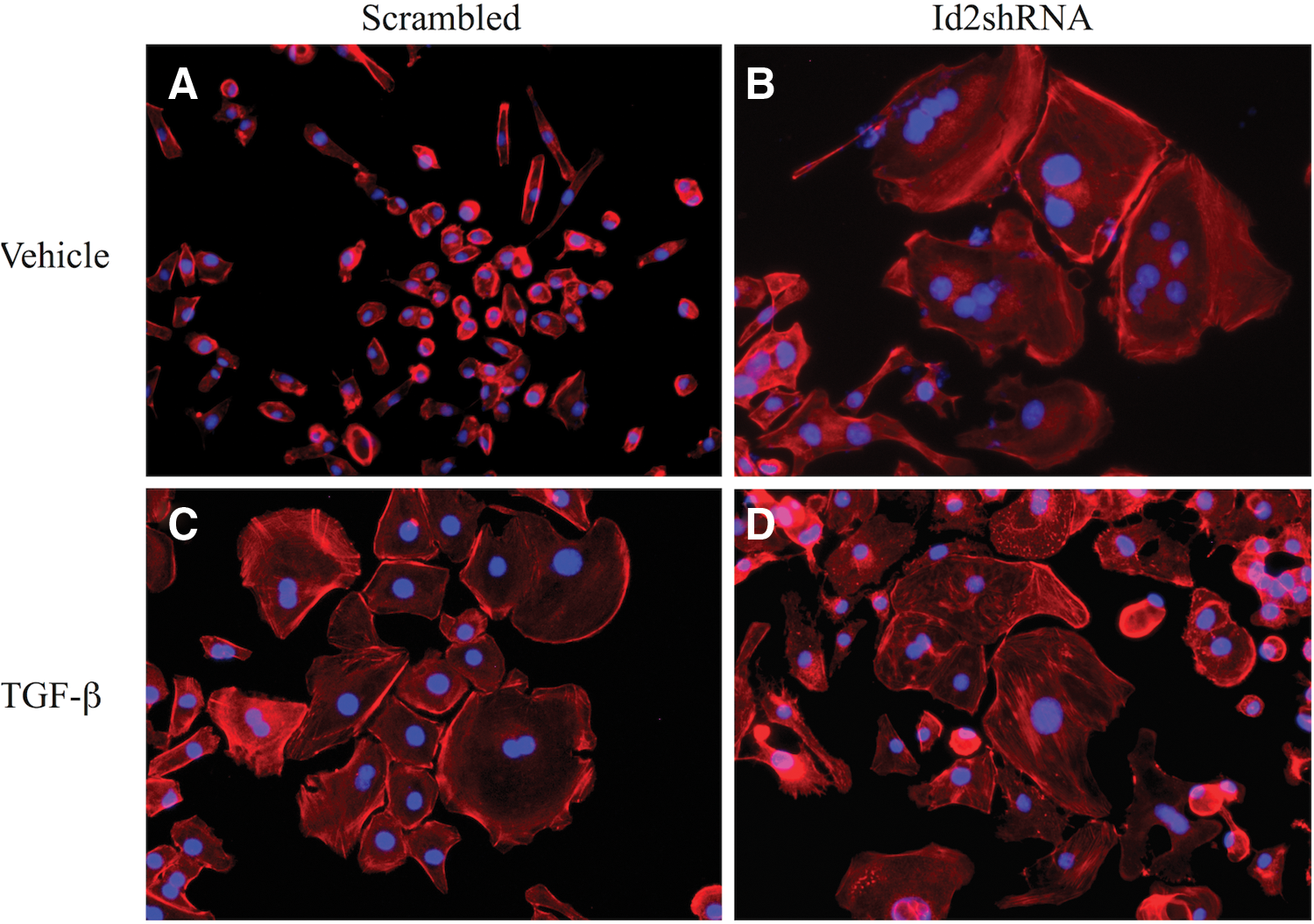
Knockdown of Id2 in SM10 cells results in labyrinthine trophoblast differentiation. SM10 cells were transduced with lentiviral scrambled Id2-shRNA (Scrambled) or Id2-shRNA (Id2-shRNA). Three days post-transduction, infected cells were enriched under (8 μg/mL) blasticidin selection and treated with 5 ng/mL TGF-β or vehicle for 72 h. Epifluorescence microscopy images of rhodamine-conjugated, phalloidin-stained SM10 cells transduced with scrambled Id2-shRNA (Scrambled)
Discussion
Appropriate placental development is essential for fetal survival and a healthy pregnancy. This process is highly regulated and involves differentiation of placental trophoblast cells into lineage-specific subtypes that fulfill several roles as the interface between the mother and the fetus. The proper development of the placental labyrinth is required for establishing the fetal–maternal interface needed for nutrient and waste exchange. Although several studies using knockout mice have been conducted and have been useful for understanding the molecular events of trophoblast differentiation, a detailed examination of the molecular pathways governing placental development has been hindered by the lack of suitable cell culture systems to model labyrinthine trophoblast differentiation [6,25,43].
We have previously shown that the trophoblast progenitor cell line SM10 expresses several labyrinthine-specific lineage markers (Esx1, Tfeb, and Tec) and undergoes TGF-β-induced differentiation [15,16]. Notably, this differentiation was associated with downregulation of Id2 expression [15]. In this investigation, the expression of Id isoforms in the SM10 labyrinthine progenitor cell line and the effect of Id2 overexpression and knockdown on TGF-β-induced differentiation were examined. The expression of Id proteins is essential for normal development and differentiation of several cell types [28,51,75 –80]. Id isoforms are also expressed in the placenta, and this suggests their importance in directing trophoblast self-renewal and differentiation [15,29 –32,43,76]. Even though mice that are deficient in a single Id isoform are viable, double deficiency of Id1, Id2, and/or Id3 isoforms leads to embryonic lethality, implying functional redundancy of expression [48,49,81 –87]. Unfortunately, studies have not yet been conducted on investigating the possible placenta defects in these knockout mice [48,49,81 –87].
Of the four Ids characterized, expression of Id1, Id2, and Id3 has been shown in human and rodent placentas [28 –30,43,48,49,51,52]. The labyrinthine trophoblast progenitor cell line, SM10, was shown to express Id isoforms. However, expression of Id1 and Id3 is constitutive and not responsive to treatment with TGF-β; whereas the expression of Id2 is readily detectable in undifferentiated SM10 cells, but it is drastically reduced on treatment with TGF-β and subsequent differentiation. The lack of Id4 expression in the SM10 cells confirms previously reported observations in placental cells [30,51]. In contrast to previous studies done in placental giant cells that showed a decrease of both Id1 and Id2 on differentiation, only the expression of Id2 mRNA and protein was shown to be downregulated during labyrinthine trophoblast differentiation in the current study and suggests that Id2 is a major mediator of labyrinthine trophoblast differentiation [15,29].
Although Id2 downregulation has been reported during trophoblast differentiation into invasive and transport subtypes, triggers inducing lineage-specific differentiation appear complex and suggest multifaceted control of Id2 expression [15,29,30,32]. Differentiation into the transport lineage by TGF-β has been reported in both humans and rodents [13,15,26,88,89]. Even though the intracellular mediators guiding trophoblast differentiation and Id2 downregulation in response to TGF-β are still being investigated, studies in other epithelial cell models have reported TGF-β-induced downregulation of Id1, Id2, and Id3 [61,90 –92]. Additionally, TGF-β-induced Id2 downregulation has been shown to depend on Smad4, suggesting the possibility of direct regulation of Id2 expression by the TGF-β-activated Smad-signaling pathway [61]. In addition, Sp1 transcription factor binding sites that are capable of interacting with Smad transcriptional complexes have been identified in the promoter of the Id2 gene and may be involved in Smad-dependent regulation of Id2 expression [61,93 –96].
No previous investigations have addressed the effects of Id isoforms, specifically on the labyrinthine trophoblast differentiation. Our studies indicate that overexpression and knockdown of Id2 in the labyrinthine-specific trophoblast cell line SM10 alters TGF-β induced morphological, functional, and molecular differentiation, which is suggestive of its importance in differentiation into the transport lineage. Indeed, Id2 overexpression inhibited TGF-β-induced morphological differentiation and maintained cells in a progenitor-like phenotype. In contrast, morphological differentiation (cell aggregation, colony formation, inability to proliferate) was clearly induced when the Id2 gene expression was knocked down. Considering the changes in phalloidin staining patterns in Id2 overexpressing and Id2 knockdown SM10 cells, Id2 may be involved in regulating the function of cytoskeletal proteins that have been implicated in altering trophoblast cell morphology, including β-catenin and connexins [47,79,97].
In addition to morphological differentiation, functional differentiation was inhibited in Id2 overexpressing SM10 cells, as demonstrated by a lack of increase in glucose uptake on treatment with TGF-β. Three glucose transporters, Glut1, Glut3, and Glut4, have been shown to be present in the placenta [53,98,99]. TGF-β has previously been shown to induce glucose uptake in several models via upregulation of Glut1 [100 –103]. In contrast, Glut3 expression appears to be TGF-β insensitive [103]. Further studies on the effects of TGF-β and Id2 on Glut expression and function are warranted.
In summary, we have demonstrated that Id2 is one of the primary mediators regulating labyrinthine trophoblast progenitor cell differentiation. We have further demonstrated that SM10 cells can be easily transduced to express genes of interest using lentiviral constructs to study development of labyrinthine trophoblasts and can lead to the elucidation of events underlying development. An understanding of the basic molecular pathways of trophoblast differentiation offers valuable insights into the mechanisms that govern placental development. Lineage-specific trophoblast cell lines can also help identify possible subtype-specific placental abnormalities that may lead to pre-eclampsia, fetal growth restriction, and placental insufficiency.
Footnotes
Acknowledgments
Plasmid pcDNA-Id2 was a generous gift of Dr. John D. Norton of the University of Essex, Colchester, the United Kingdom. The lentiviral plasmid, pLv-CMV-GFP-V5, was kindly provided by Dr. Steven Berberich of the Wright State University, Dayton, Ohio. Mouse SM10 cells were kindly provided by Dr. Joan S. Hunt of the Kansas University Medical Center, Kansas City, Kansas. TGF-β2 was a kind gift of Dr. Steven Ledbetter of Genzyme, Inc., Boston, Massachusetts. TS3.5 cells (TS cells) were generously provided by Dr. Janet Rossant, The Hospital for Sick Children, Toronto, Canada. The pE7-TK-luc construct was kindly provided by Dr. Yoshifumi Yokota, the University of Fukui, Fukui, Japan. The authors would like to thank Amy Gultice for her valuable assistance and input with the TS and Rcho cells. This work was supported in part by grants from the Wright State University Research Incentive Program, The Ohio Board of Reagents (T.L.B.), the Biomedical Sciences Ph.D. Program (K.S., R.E.A.), the Wright State University Graduate Council Scholarship (R.E.A.), The Wright State University Endowment for Research on Pregnancy Associated Disorders (
Author Disclosure Statement
No competing financial interests exist.