
Abstract
Mesenchymal stromal/stem cells (MSCs) constitute progenitor cells that can be isolated from different tissues. Based on their immunomodulatory and neuroprotective functions, MSC-based cell-therapy approaches have been suggested to antagonize inflammatory activity and neuronal damage associated with autoimmune disease of the central nervous system (CNS), for example, multiple sclerosis (MS). Intravenous MSC transplantation was reported to ameliorate experimental autoimmune encephalomyelitis (EAE), the murine model of MS, within days after transplantation. However, systemic distribution patterns and fate of MSCs after administration, especially their potential to migrate into inflammatory lesions within the CNS, remain to be elucidated. This question has of recent become particularly important, since therapeutic infusion of MSCs is now being tested in clinical trials with MS-affected patients. Here, we made use of the established EAE mouse model to investigate migration and therapeutic efficacy of murine bone marrow-derived MSCs. Applying a variety of techniques, including magnetic resonance imaging, immunohistochemistry, fluorescence in-situ hybridization, and quantitative polymerase chain reaction we found no evidence for immediate migration of infused MSC into the CNS of treated mice. Moreover, in contrast to other studies, transplanted MSCs did not ameliorate EAE. In conclusion, our data does not provide substantiation for a relevant migration of infused MSCs into the CNS of EAE mice supporting the hypothesis that potential therapeutic efficacy could be based on systemic effects. Evaluation of possible mechanisms underlying the observed discrepancies in MSC treatment outcomes between different EAE models demands further studies.
Introduction
M
Certain MHC class-II molecules are the main genetic risk factors for MS [4]. Mutations in two cytokine receptors, IL-7RA and IL-2RA, have been described as additional risk alleles for MS [5 –7] and a recently published very large genome-wide association study shows 52 single nucleotide polymorphisms besides HLA-DR as risk alleles for MS [8]. Furthermore, three environmental risk factors (Epstein-Barr virus infection [9 –11], low vitamin-D levels [12 –14], and smoking [15]) have now been firmly established. Finally, viral infections are proposed to induce the expansion of high-avidity T cells, either by molecular mimicry or by bystander activation [16].
Two major clinical manifestations of MS exist that differ in their course and their frequency. Whereas a minor fraction of patients (10%–15%) show a steady progression of disability termed primary-progressive MS (PP-MS) [17,18], 85%–90% of MS-patients suffer from relapsing-remitting MS (RR-MS) [19]. As disease progresses and disability accumulates, the majority of RR-MS patients develop secondary-progressive MS (SP-MS) [20]. Once the irreversible state of disability is manifested, the temporal acquisition of progressive invalidity is similar in PP-MS and SP-MS groups [21]. Remyelination of axons may occur during the remission phases of disease but also to a lesser extent during progressive MS periods [22,23].
Current treatment options of MS comprise interferon-β (IFN-β) formulations, glatiramer acetate, the anti-CD52 monoclonal antibody alemtuzumab, and natalizumab, a monoclonal antibody against α4-integrin. The cytotoxic drug mitoxantrone is acting nonspecifically as a DNA modulator. These therapeutics have to be injected s.c., i.m., or i.v. Fingolimod (FTY-720), teriflunomide and dimethylfumarate have been approved as orally available drugs. Fingolimod inhibits migration of lymphocytes from lymphatic organs, teriflunomide interferes with pyrimidine nucleotide synthesis, and dimethylfumarate primarily acts via influencing antigen-presenting cells in an immunomodulatory manner. Generally, available pharmaceuticals primarily act as immune modulators and immune suppressants, and only target RR-MS.
Transplantation of autologous hematopoietic stem cells (AHSCs) after intense immunosuppressive conditioning has been used as cell therapy for MS. Most if not all T cells are eradicated by this procedure followed by re-constitution of the immune system after successful engraftment of transplanted AHSCs [24 –26]. Recently, transplantation of multipotent mesenchymal stromal/stem cells (MSCs) has been suggested as a therapeutic option providing not only immunomodulatory but also reparative functions. So far, no adverse effects of autologous MSC transplantation have been reported. Controlled clinical studies will be necessary to establish the efficacy and safety of cell therapies for MS [27].
MSCs have been isolated from a variety of tissues, including bone marrow, muscle, fat,, and skin both in humans and different animals. In vitro, MSCs grow as an adherent cell layer and retain their capacity to differentiate into osteoblasts, chondroblasts, adipocytes or other connective-tissue specific cell types. Human MSCs are defined by the expression of CD44, CD73, CD90, and CD105, but are negative for CD11b, CD14, CD34, CD40, CD45, and CD80 [28]. MSCs were reported to inhibit the proliferation of activated T cells, probably by both direct cell–cell contact and paracrine factors.
MSC activation in vivo is supposedly mediated by T cells that respond to stimuli such as inflammatory cues, injury, or allogeneic tissues and cells. As a consequence, these T cells produce proinflammatory cytokines like tumor necrosis factor-α, interferon-γ (IFN-γ), IL-1α, and IL-1β thereby inducing immunomodulatory properties of MSCs [29,30].
Immunoactivated MSCs but also conditioned medium from MSCs were identified to reduce the frequency of IFN-γ and IL-2 secreting T cells [29]. The inhibitory function of MSCs in humans and nonhuman primates is mediated, at least in part, by indolamine-2,3-dioxygenase [31,32]. Inducible nitric oxide synthase is an important effector for murine MSC function in vivo [33]. Besides the production of transforming growth factor-β, hepatocyte growth factor, IL-10, and the lipid prostaglandin E2, MSCs have also been implicated in nitric oxide production that inhibits Stat-5 phosphorylation, cell-cycle progression and T-cell proliferation [34,35]. Both, the differentiation into mesodermal cell types and paracrine effects to support resident cells have been reported for MSCs to oppose tissue damage [36 –38].
In 1984, Ildstad and Sachs postulated the idea that MSCs are able to induce tolerance to allogeneic or xenogeneic grafts [39]. In 2002, the first evidence for MSCs to possess an immunomodulatory effect in vitro was demonstrated [40]. In parallel, it was published that MSC transplantation led to prolonged skin engraftment in a nonhuman primate model of allograft rejection underpinning their tolerogenic capacity [41]. Transplantation of MSCs was also reported to ameliorate EAE in mice [42]. In line with that, a beneficial impact of MSCs on repair after CNS damage was reported in several studies. Experimental models of chemical demyelination [43], stroke [44], trauma [45], Parkinson disease [46], diabetes [47], and EAE [48,49] were investigated.
It is still not clear, whether MSCs migrate to inflamed or damaged tissue or if they mediate the above effects primarily within the periphery, even though attempts have been undertaken to answer this central question [50 –52]. Access to the damaged tissue, that is, the CNS in MS and EAE, is limited by the blood-brain barrier, and therefore it is of great interest, whether MSCs exert their putative immunoinhibitory and regenerative properties in vivo by systemic effects, or if homing to the target organ is a prerequisite for the therapeutic effect of MSCs in the context of EAE and MS.
In this study, we induced EAE in C57BL/6J mice by active immunization with myelin oligodendrocyte glycoprotein (MOG)-derived MOG(35–55) peptide. In this well-established model, onset of clinical symptoms/deficits occurs between 9 and 12 days after immunization. Subsequently, mice develop a maximally severe disease bout (acute phase). Remission is only partial, and mice retain a moderate, chronic deficit [2,53]. Typically, myelin-specific CD4+ Th1 but also Th17 cells are considered to mediate EAE initiation. Pathological hallmarks, for example, CD4+ T-cell infiltration of the CNS, axonal loss, and neuronal damage, are shared between MS and EAE.
By applying a therapeutic treatment regimen, we observed that transplanted MSC were not detectable in the CNS of EAE-induced mice at acute disease stage. Moreover, MSCs did not mediate beneficial effects on the EAE course, neither in the therapeutic nor in a preventive transplantation setting.
Materials and Methods
Ethics statement
All animal experiments were performed in accordance with the guidelines of animal welfare after approval (G30/08, G81/13) by the local authorities (Behörde für Soziales, Gesundheit und Verbraucherschutz Hamburg).
Mice
C57BL/6J mice were purchased from the Jackson Laboratory and bred by the animal facility of the University Medical Center Hamburg-Eppendorf. SJL/JHanHsd mice were purchased from Harlan Laboratories (Indianapolis, IN).
Isolation of murine MSCs
C57BL/6J-MSCs were isolated from 6- to 7-weeks-old male mice by flushing the bone marrow out of the tibias and femurs. Cells were cultured in 75-cm2 flasks in mouse MSC medium [DMEM/Ham's F-12 1:1 (Biochrom, Berlin, Germany), 20% preselected fetal calf serum (FCS; Lonza, Basel, Switzerland), 2 mM
Differentiation of MSCs
Routinely, MSCs were characterized by their unique capability to differentiate into cells of the mesodermal lineage [54]. For adipogenic and osteogenic differentiation MSCs were seeded in 24-well plates at 4 × 104 cells/well. After incubation overnight (o.n.), medium was replaced by mouse MSC medium supplemented with 0.1 μM dexamethasone (Sigma-Aldrich, St. Louis, MO), 0.5 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich), 0.1 mM indomethacin (Sigma-Aldrich), and 10 μg/mL insulin (Sigma-Aldrich) to induce adipogenic differentiation. Every 2 to 3 days medium was replaced for 10 μg/mL insulin in mouse MSC medium for o.n. culture. After 3 to 4 weeks MSCs were washed with phosphate-buffered saline (PBS; Gibco), fixed with 10% formalin (10 min), washed with 50% ethanol, and stained for 30 min with Sudan Red B (Fluka Sigma-Aldrich, Buchs, Switzerland). Cells were washed with 50% ethanol before staining for 5 min with Mayer's hemalum solution (Merck, Darmstadt, Germany). Tap water was used for the first washing step (1 min) of cells, which were subsequently washed three times with aqua dest. Cells were embedded in paraffin oil (Carl Roth, Karlsruhe, Germany) and subjected to microscopy. Osteogenic differentiation was induced by culturing MSCs in mouse MSC medium supplemented with 0.1 μM dexamethasone, 50 μM
Cell-surface marker expression of MSCs
At sub-confluence, MSCs were washed with PBS, detached by addition of 0.05% trypsin/EDTA, and Fc-receptors were blocked for 10 min at 4°C with anti-mouse CD16/32 antibody (clone 93) from BioLegend (San Diego, CA). Cells were washed in PBS and stained for 30–60 min at 4°C with anti-CD34-FITC (clone RAM34), anti-CD90.2-FITC (clone 53-2.1), anti-Ly-6A/E-FITC (anti-Sca-1, clone E13-161.7), anti-CD117-PE (clone 2B8), or rat IgG2a,κ-FITC (clone R35-95), all from BD Pharmingen (San Diego, CA). Anti-CD73-PE (clone eBioTY/11.8), anti-CD105-PE (clone MJ7/18), rat IgG1κ-PE (clone eBRG1), rat IgG2a,κ-PE (clone eBR2a), and rat IgG2b,κ-PE (clone eB149/10H5) were from eBioscience (San Diego, CA). After washing, data were acquired on an LSR Fortessa (405, 488, 561, 640-nm lasers) cell analyzer (BD Biosciences, San Jose, CA) and processed with BD FACSDiva [55] and FlowJo (Tree Star, Ashland, OR) software.
Irradiation of MSCs
Where indicated, cell-culture flasks containing MSCs or Superparamagnetic iron oxide (SPIO)-labeled MSCs were X-radiation irradiated at 30 Gray using an RS225 Research System (Gulmay Medical, Camberley, United Kingdom).
T-cell proliferation assays
T cells derived from spleens of 6-weeks-old C57BL/6J mice were seeded in 96-well flat-bottom plates (Greiner Bio-One) at 2 × 105 cells/well. Cells were cultured in 200 μL of complete mouse medium [50 μM β-mercaptoethanol (Gibco), 10% FCS, 1%
Quantitative polymerase chain reaction for detection of Y chromosomes
Genomic DNA was prepared from male MSCs and from peripheral blood of male C57BL/6J mice by using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The QIAamp DNA Micro Kit (Qiagen) was used to isolate genomic DNA from cervical spinal cord tissue obtained by collecting five slices (thickness: 30 μm) per EAE-induced mouse. DNA was used for quantitative polymerase chain reaction (qPCR) amplification as described [56]. Briefly, 50 ng of DNA, SYBR Premix Ex Taq II (Takara Bio, Saint-Germain-en-Laye, France), and ROX reference dye II (Takara) were incubated with 200 nM primers (MWG-Biotech, Ebersberg, Germany) for Y-chromosome (SRY) detection. Each qPCR reaction was performed in triplicates on an Mx3000P qPCR system (Stratagene, La Jolla, CA) and analyzed with MxPro software (Stratagene). The relative presence of Y chromosomes in MSCs and peripheral blood controls and in CNS sections was normalized using a control PCR against sequences located on mouse chromosome 11 (control chr11). Sequences of used primers are provided in Supplementary Table S1 (Supplementary Data are available online at
Labeling of murine MSCs by ferucarbotran (Resovist®)
For labeling of MSCs 1 day before transplantation into EAE-induced mice, cells were incubated o.n. with fresh mouse MSC medium supplemented with SPIO [Resovist, (Bayer Healthcare Pharmaceuticals, Berlin, Germany) containing 540 mg/mL ferucarbotran; 27.9 mg Fe/mL or 496 μmol Fe/mL] at 1:100 (v/v). To quantify total iron load of SPIO-labeled MSCs after incubation and repeated washing with PBS, the iron concentration in MSC suspensions used for transplantation was analyzed by atomic absorption spectrometry (AAS) using an Atomic Absorption Spectrophotometer (PerkinElmer) and AA WinLab software.
Generation of MSC cytospins
MSCs were cultured to subconfluence, plated at 2 × 105 cells per 25-cm2 flask (day 0) and incubated with fresh mouse MSC medium. Depending on the experiment, medium was supplemented with 1:100 (v/v) ferucarbotran (day 1), whereas corresponding controls were kept without ferucarbotran. At day 2, MSCs were washed 5× with PBS, and fresh medium was added. At day 3, MSCs were spun on SuperFrost/Plus glass slides (Karl Hecht) at 160 g and 30,000 or 50,000 cells/spot, using a Rotofix 32A centrifuge and cyto-chambers (four-fold, 30 mm2) from Hettich (Tuttlingen, Germany), to achieve even distribution of cells.
Quantification of MSC proliferation after SPIO-labeling
MSCs were cultured to subconfluency and plated at 5 × 104, 1 × 105 or 2 × 105 cells per 25-cm2 flask (day 0). At day 1, ferucarbotran was added to the cell-culture medium. After o.n. incubation Resovist-treated and control cells were washed five times with PBS, and fresh mouse MSC medium was added (day 2). Cell numbers were quantified after Trypan Blue (Sigma-Aldrich) staining to discriminate live/dead cells at the following days: day 4 (2 × 105), day 7 (5 × 104, 1 × 105), and day 9 (5 × 104).
Electron microscopy
Since Resovist was temporarily not available, these experiments were performed with a different SPIO ferumoxide (11.2 mg Fe/mL, Endorem®; Guerbet, Roissy, France). Cells were incubated with ferumoxide o.n. at 1:100 (v/v). MSCs were washed 5× with PBS, incubated for another 24 h and detached with trypsin/EDTA. MSCs were fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich) and 1% glutaraldehyde (Science Services, Munich, Germany). Subsequently, cells were spun down and resuspended in 20 μL 2% Agarose (Gibco). Agarose embedded MSCs were cut into 1-mm3 cubes and rinsed three times in 0.1 M cacodylate buffer (pH 7.2–7.4). They were fixed in 1% osmium tetroxide (Science Services) in cacodylate buffer for 20 min on ice. Blocks were washed extensively in cacodylate buffer and dehydrated in an ascending ethanol series. Finally they were rinsed two times in propylene oxide (Sigma-Aldrich) and embedded in Epon (Carl Roth). Ultrathin sections were prepared and analyzed with a TEM 902 (Carl Zeiss, Jena, Germany).
Induction and scoring of EAE
For EAE experiments, female C57BL/6J mice were housed in “individually ventilated cages (IVC)” racks at least 1 week before the active induction of EAE. For immunization, mice were injected subcutaneously at one site of each flank with 200 μg of MOG(35–55) peptide (NeoMPS, San Diego, CA) in incomplete Freund's adjuvant (BD Difco Diagnostics, Sparks, MD) supplemented with 4 mg/mL mycobacterium tuberculosis H37 Ra (BD Difco). Additionally, mice were injected intravenously with 300 ng of pertussis toxin (Calbiochem Merck, Darmstadt, Germany) on the day of immunization and 48 h later. Body weight and clinical score were monitored in a blinded way on a 0 to 5 scale with classifications of disease severity: 0 = healthy, 1 = limp tail, 2 = ataxia and/or paresis of hind limbs, 3 = paraplegia, 4 = paraplegia with forelimb weakness, 5 = moribund or dead.
Transplantation of MSCs
MSCs were grown to sub-confluence and washed 2× with PBS. In case of SPIO-labeled MSCs cells were washed additional 3× with PBS. Cells were detached by trypsin/EDTA. For intravenous transplantation, 1 × 106 cells were resuspended in 200 μL DMEM/Ham's F-12 1:1 and 20 IU/mL heparin-sodium (Braun, Melsungen, Germany). Immediately before injection, cells were passed through 70-μm cell strainers (BD Falcon). MSCs were transplanted at disease onset (day 11 after immunization) or at days 3 and 8 in the prevention setting.
Preparation of mouse tissue
Mice in deep anesthesia were transcardially perfused with cold PBS followed by 4% cold PFA in PBS. Preparation of brain, spinal cord, spleen, liver, and lungs was followed by postfixation of the tissue in 4% PFA in PBS (30 min, 4°C). The tissue was impregnated 2–3 days in 30% sucrose in PBS for cryoprotection. Cervical, thoracic, and lumbar spinal cord in addition to cerebellum and forebrain were separated. The tissue was embedded in Tissue Freezing Medium (Jung; Leica Biosystems, Nussloch, Germany) and frozen in n-pentane (Carl Roth). Tissue was stored at −80°C.
Detection of ferucarbotran-labeled murine MSCs by magnetic resonance imaging in vitro
All MR-studies were performed on a small-animal magnetic resonance imaging (MRI) system at 7.0 T (ClinScan; Bruker, Ettlingen, Germany) using a birdcage design mouse body coil with an inner diameter of 40 mm. To test whether SPIO-labeled MSCs are detectable and can be quantified by MRI, we performed a preliminary experiment: SPIO-labeled MSCs derived from C57BL/6J mice were locally injected into gelatin-filled 50-mL conical bottom tubes (Greiner Bio-One). Injection of cells was allowed by holes drilled into the tube walls. This approach was adopted from previous studies [57,58]. Tubes were placed centrally in the coil and images were acquired using a T2*-weighted (T2*w) three-dimensional (3D) spoiled gradient echo (GRE) sequence with a flip angle of 25° and an isotropic voxel size of 80 μm (3D GRE, imaging parameters shown in Supplementary Table S2). Signal events were enumerated to determine distinct cells and total cell numbers per injection site. In detail, scans were analyzed by using the ImageJ [National Institutes of Health (NIH), Bethesda, MD] program, which sequentially counts all positive signals of all scan images belonging to one injection. Each injection site was covered by ∼35 subsequent images. After the automated dot count, dots that appeared on subsequent slides at the same positions (indicating scans of the same cell/dot picked up by more than one scan) were manually subtracted. Injections of 500 or more cells were not exactly quantifiable as MSC/dots were forming clusters that did not allow clear single cell discrimination. A detailed dot quantification was done for the injection of 50 cells.
Detection of ferucarbotran-labeled MSCs by MRI after transplantation into EAE-induced mice
Mice were anesthetized by isoflurane inhalation anesthesia (2% isoflurane, 98% oxygen, flow rate 500 mL/min). General anesthesia was achieved after 10 min and mice were subjected to MRI imaging for 30 min. Respiration triggering was performed using a pressure pad (SA Instruments, Stony Brook, NY), mice were warmed during measurement. High-resolution T2*w images of the brain stem and the upper spine, sensitive to SPIO presence, were acquired in sagittal and transversal direction. Additionally, sagittal T2-weighted images were acquired for anatomical reference. MRI-sequence parameters are listed in Supplementary Table S2.
Histological staining
Seven-micrometers slices were generated using a cryostat and mounted on glass microslides (SuperFrost/Plus). Sections were subjected to hematoxylin and eosin (HE) staining following standard protocols. For iron detection (Turnbull staining) slides were incubated in ammonium sulfide (Merck) for 30–45 min, rinsed 2× in aqua dest. and stained for 20 min with ferricyan potassium solution (Sigma-Aldrich). After washing (aqua dest., 2×) sections were counterstained by nuclear fast red (5–7 min). Slides were washed with aqua dest. and subsequently dehydrated using ascending alcohol series and xylene. For analysis of iron incorporation by MSCs in cell-culture experiments MSCs were incubated with ferucarbotran or ferumoxide o.n. at 1:100 (v/v). Thereafter, cells were washed five times with PBS and incubated for another 24 and 6 h, respectively. After repeated washing with PBS cells were detached with trypsin/EDTA and transferred to glass microslides (SuperFrost/Plus) for Turnbull staining.
For double staining of iron and ionized calcium binding adaptor molecule 1 (Iba1; Wako Chemicals, Neuss, Germany) the Ventana Benchmark XT (Ventana, Tuscon, Arizona) was used. Antigen retrieval for Iba1 detection was performed by boiling for 60 min in 10 mM citrate buffer, pH 6.0. Sections were incubated with primary antibody for 1 h followed by incubation with anti-rabbit secondary antibody or Histofine Simple Stain MAX PO Universal immunoperoxidase polymer (Nichirei Biosciences, Wedel, Germany). Antibody binding was visualized with the ultraview universal DAB detection kit from Ventana (Tuscon, Arizona). Subsequently, sections were stained for iron as described above with one change: Iba1-iron double stainings were not counterstained with nuclear fast red to not interfere with brownish DAB staining for Iba1.
Images were acquired using a Leica DMD108 digital microscope (Leica, Wetzlar, Germany). Photographs were taken with 4× (overview) and 20× (close up) magnification. In case of ferumoxide-marked MSCs images were acquired on a CKX41 inverted microscope (Olympus, Hamburg, Germany).
Y-Chromosome FISH
For Y-chromosome detection, we modified a published protocol [59]. Briefly, 15-μm tissue sections where taken using a cryostat and air dried for 30 min. Subsequently, tissue sections were pretreated in 10 mM sodium citrate buffer pH 6.0 for 10 min at 80°C. After cooling to room temperature, tissue was stained with Sudan black (1% in 30% ethanol/H2O), washed and incubated for at least 1 h in 50% formamide/2 × saline sodium citrate (SSC) buffer (pH 7.0) (v/v). Mouse Y-chromosome-specific probe (Empire Genomics, Buffalo, NY) was diluted in sample buffer and applied to the tissue section, sealed with a cover slip and rubber cement, preincubated for 2 h at 45°C, denatured for 5 min at 85°C and incubated for at least 16 h at 37°C for hybridization. After this, excessive probe was removed by washing the tissue twice for 15 min at 37°C in 2× SSC and subsequently the tissue was stringently washed twice for 5 min at 60°C in 0.1× SSC. Following an additional washing step in 2× SSC at room temperature for 5 min, the tissue section was mounted in DAPI fluoromount-G (SouthernBiotech, Birmingham, AL) and air dried overnight. Pictures were taken with a Zeiss Axio Imager.M2 microscope (Zeiss, Jena, Germany).
Cytospins were prepared as described above and Y-chromosome FISH detection was done as described for spinal cord tissue. For data acquisition of cytospins we used a Leica TCS SP5 confocal microscope and Leica application suite software (LAS-AF-lite).
Statistical analysis
MSC-mediated suppression of T-cell proliferation was assessed by one-way ANOVA followed by Bonferroni post hoc analysis using Prism 5.02 software (GraphPad Software, La Jolla, CA). EAE disease courses were analyzed by two-way ANOVA and Bonferroni multiple testing correction (post-hoc analysis). Data are shown as mean values ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Proliferation of MSCs after labeling by ferucarbotran incorporation was analyzed by unpaired t-test and shown as mean values ± SD.
Results
Confirmation of MSC characteristics for mouse bone marrow-derived cells
To characterize plastic-adherent MSCs according to the ISCT criteria [60], we first analyzed their potential to differentiate into cells of mesodermal origin (Supplementary Fig. S1). Cells readily displayed adipogenic differentiation marked by the accumulation of fat. Expectedly, osteogenic differentiation led to large depositions of calcium. Chondroblasts were identified by their secretion of proteoglycans. In addition, we confirmed the expression of consensus stromal surface markers CD73, CD90.2, CD105, and Ly-6A/E (Sca-1), and the absence of CD34 and CD117 (Supplementary Fig. S2) [60].
No impact of SPIO particles on MSC vitality and properties
It was previously shown that human and rat MSCs can efficiently be labeled by incubation with SPIO particles in o. n. cell cultures, which allowed detection by MRI [57,58,61,62]. We here observed efficient incorporation of Resovist in C57BL/6J-derived MSC as confirmed by Turnbull staining of cytospins (Supplementary Fig. S3). Complete marking of MSCs was also observed by microscopy. Notably, similar to ferumoxide-labeled MSCs, also ferucarbotran-marked cells appeared to be rather unequally loaded with iron particles, which is in agreement with Ittrich et al. [57]. As determined by AAS, iron concentrations in 1:100 (v/v) ferucarbotran-labeled MSC suspensions were 171.45 mg Fe/L compared to 0.34 mg Fe/L in nontreated control MSCs. About 1 × 106 MSCs were transplanted in a total volume of 200 μL, that is, applied cell suspensions contained 34.29 μg Fe and 0.068 μg Fe, respectively. In vitro, marking of MSCs by Resovist incorporation did not impair their proliferative capacity as reflected by absolute cell numbers counted at days 7 and 9 in proliferation assays (Supplementary Fig. S4).
In accordance with published data, we also confirmed that an SPIO dilution of 1:100 (v/v) ferumoxide in cell-culture medium resulted in efficient labeling of C57BL/6J- and SJL/JHanHsd-derived MSCs (Supplementary Fig. S5). Electron microscopy validated MSC incorporation of ferumoxide particles. Iron particles were not cross-linked to the extracellular cell matrix, but instead accumulations of particles could readily be found in intracellular vesicle-like structures (Supplementary Fig. S6).
SPIO-labeled MSCs suppress T-cell proliferation in vitro as efficient as control MSCs
We evaluated MSC functionality in vitro following previous studies [42]. Proliferation of splenocytes from syngeneic donor mice was induced by stimulation with anti-CD3 antibody. Both coculture with MSCs that allowed direct cell-cell contact and transwell-culture with MSCs physically separated from splenocytes significantly reduced splenocyte proliferation, regardless of MSC irradiation (Fig. 1A, C). Notably, SPIO-labeled MSCs reduced splenocyte proliferation with comparable efficiency (Fig. 1B, D) indicating that the uptake of SPIO particles did not interfere with MSC function in vitro. As expected, in the absence of anti-CD3 antibody there was no stimulation of splenocytes (Supplementary Fig. S7).

MSCs reduced T-cell proliferation in vitro. Splenocytes were activated with anti-CD3 antibody. Coculture with MSCs
No impact of transplanted MSCs on acute-phase EAE
We next assessed the efficacy of MSC infused after EAE onset (therapeutic setting). The passage numbers of MSCs used for transplantation were P6 and P10, and cells of each passage were used for SPIO-labeling. Cells of the two passages were not pooled but transplanted in parallel. Since chromosomal instability was previously reported for murine MSC [56], we confirmed presence of the Y chromosome by qPCR (not shown) before labeling MSCs with SPIO particles.
Mice showed marked signs of continuous clinical EAE aggravation (EAE onset) from day 7 after EAE induction. SPIO-labeled and control MSCs were transplanted at day 11, and recipient mice were subjected to MRI analysis immediately, 24 h, and 48 h later (SPIO-MSC-transplanted: 10, 9, and 7 mice per day, respectively, of 11 mice; MSC-transplanted: 5 mice per day of 11 mice; DMEM/Ham's F-12 controls: 5 mice per day of 8 mice). In addition, tissue was prepared from transplanted mice (day 13: 3 mice per cohort after MRI). EAE scores are shown in Fig. 2A. No significant differences between groups were identified. Supplementary Table S3 summarizes the evenly distributed scores of EAE-affected mice across groups that were used for organ preparation to exclude biased treatment outcome, that is, an MSC migration potentially affected by full-blown versus mild EAE status of recipient mice.
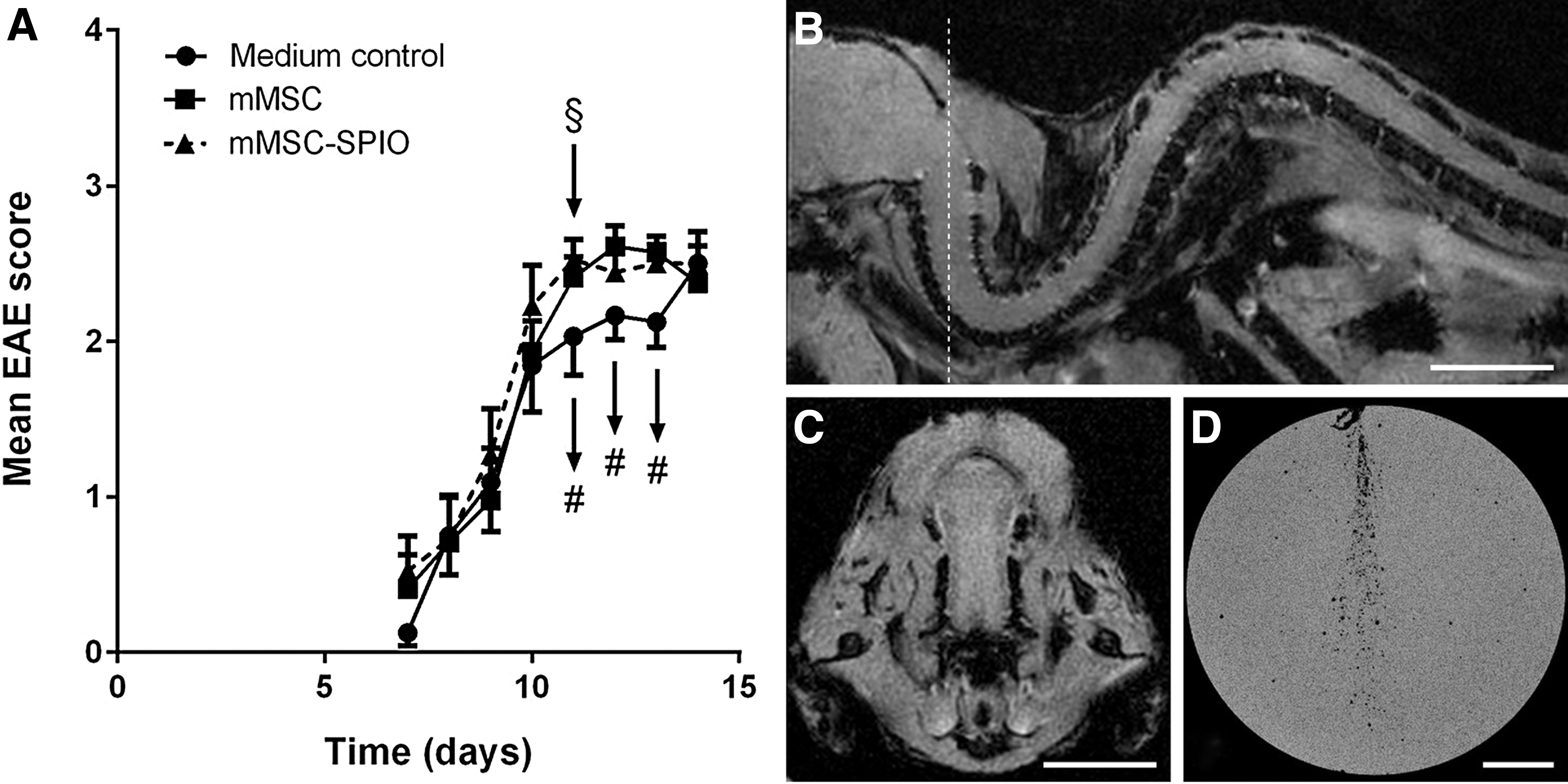
Analysis of MSC-SPIO-transplanted EAE-induced mice by MRI. From day 7 after immunization the disease course of EAE-induced mice was assessed on a daily basis. Mice were transplanted with medium, MSCs, or ferucarbotran-labeled MSCs (§) at day 11 after immunization ( = day 0) and subjected to MRI analysis (#) immediately after injection of cells, 24 h later, and 48 h after injection to identify MSC migration into the CNS, the site of inflammation
No evidence for migration of transplanted MSCs into the CNS of EAE-induced mice
By MRI, no Resovist-labeled MSCs were detectable in the CNS of transplanted mice at any time point under investigation (Fig. 2B, C). We focused on the analysis of CNS tissue up to 48 h after MSC transplantation, since clinical amelioration has been reported to appear as an immediate effect. If this effect necessitates the migration of MSCs to sites of inflammation, MSCs must be detectable in the CNS of EAE-induced recipient mice. Moreover, long-term engraftment of MSC was not observed in previous studies [4].
Sagittal MRI images demonstrated the sensitivity of MRI sequences (Supplementary Fig. S8). Notably, in a preliminary experiment using a gelatin-filled tube to inject defined numbers of Resovist-labeled MSCs, it was possible to enumerate and to detect 49 positive signals by MRI (50 injected cells, Fig. 2D). To further confirm MRI results, tissues of mice, which were sacrificed 48 h after transplantation, were additionally subjected to qPCR, Y-FISH, and immunohistochemistry (IHC). HE stains displayed inflammatory lesions within cervical spinal cord sections of EAE-induced mice treated with medium (control), MSCs or Resovist-marked MSCs (Fig. 3A). Turnbull staining identified isolated iron particles within some inflammatory infiltrates of the cervical spinal cords (Fig. 3A) and more evenly distributed in lung- and liver sections of MSC-SPIO-transplanted mice (Fig. 3B). Next, to verify the presence of iron-loaded male MSCs within the CNS, cervical spinal cord sections were stained for the presence of Y chromosomes. The Y chromosome was readily detectable in male control mouse CNS sections and clearly confined to nuclei (Supplementary Fig. S9A). In contrast, we could not detect positive signals within spinal cord sections of the female, EAE-induced mice transplanted with male MSCs or SPIO-labeled MSCs sections. (Supplementary Fig. S9B). As a further control, MSCs were labeled by Resovist incorporation and used for Y-chromosome detection in vitro. As expected, ferucarbotran-treated MSCs revealed the same positive Y-chromosome signal (Supplementary Fig. S10) ruling out an impact of the ferucarbotran-labeling on FISH analysis.
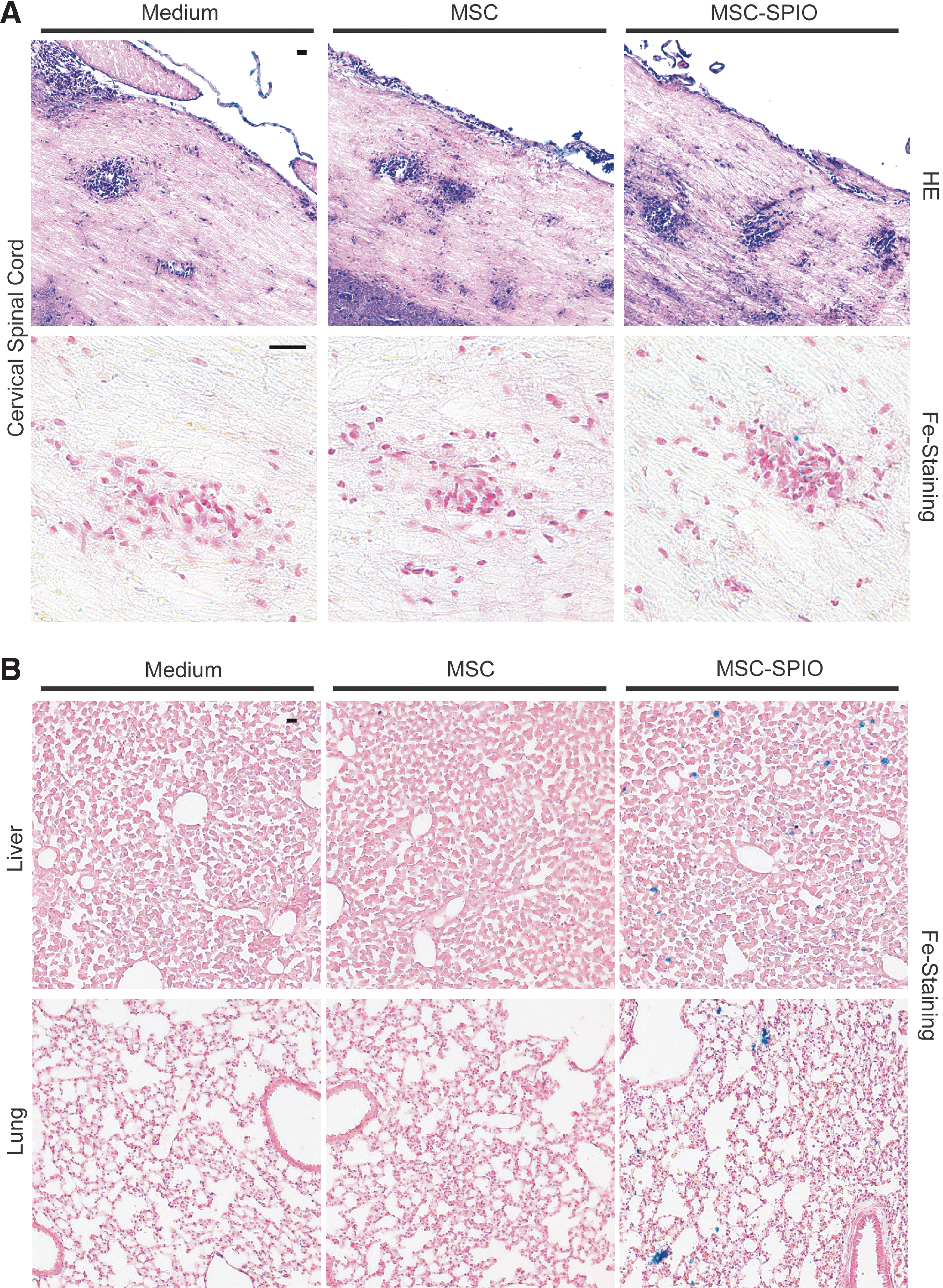
SPIO particles were detectable in tissue sections of EAE-affected mice upon MSC-SPIO transplantation. At acute EAE-disease phase, cellular infiltration of the CNS was observed. In case of MSC-SPIO transplantation, SPIO particles were detectable within the parenchyma of cervical spinal cords
In summary, results indicate that control and ferucarbotran-labeled male MSCs transplanted into female recipient mice should be traceable by Y-FISH. However, as we could not detect Y-chromosome-specific signals in the CNS of recipient mice, we conclude that transplanted MSCs did not reach the spinal cord.
To corroborate the absence of MSCs in CNS inflammatory lesions, CNS-tissue sections of all three (medium-, MSC-, or MSC-SPIO-transplanted) EAE-affected mouse groups were analyzed by qPCR for the presence of infused male cells. In contrast to positive controls, that is, genomic DNA from male C57BL/6J MSCs expanded by in vitro cell culture and from blood of male C57BL/6J mice, no Y chromosome was detectable in cervical spinal cord sections. Based on our qPCR results, the calculated detection limit was one male in more than 30,000 female cells (not shown).
MSCs are found in liver and lungs early after transplantation
To characterize in more detail the iron particles observed within various tissues of MSC-SPIO-transplanted mice, for example, CNS, liver, and lung (Fig. 3 and Supplementary Fig. S8), we probed sections for the detection of iron and co-stained to identify Iba1+ macrophages/microglia by IHC. In cervical spinal cord sections of MSC-SPIO-transplanted mice very few iron particles were found that colocalized with microglia and/or CNS-infiltrating macrophages 48 h after infusion (Fig. 4A). In contrast, in liver sections colocalization of iron with macrophages/Kupffer cells was detected at comparatively high frequencies (Fig. 4A). Together these data indicate that the presence of SPIO particles in CNS-sections was due to phagocytic activity of monocyte-derived cells. To verify homing of intact cells to the liver, we repeated IHC at an earlier time point, that is, 24 h post MSC infusion. As shown in Fig 4B, at that time point intact and freshly engulfed MSC can be detected in the liver. In line with the IHC data, consecutive MRI scans showed that the profound accumulation of iron in spleens and livers of MSC-SPIO-transplanted mice already occurred at the day of transplantation and remained stable for 3 days.
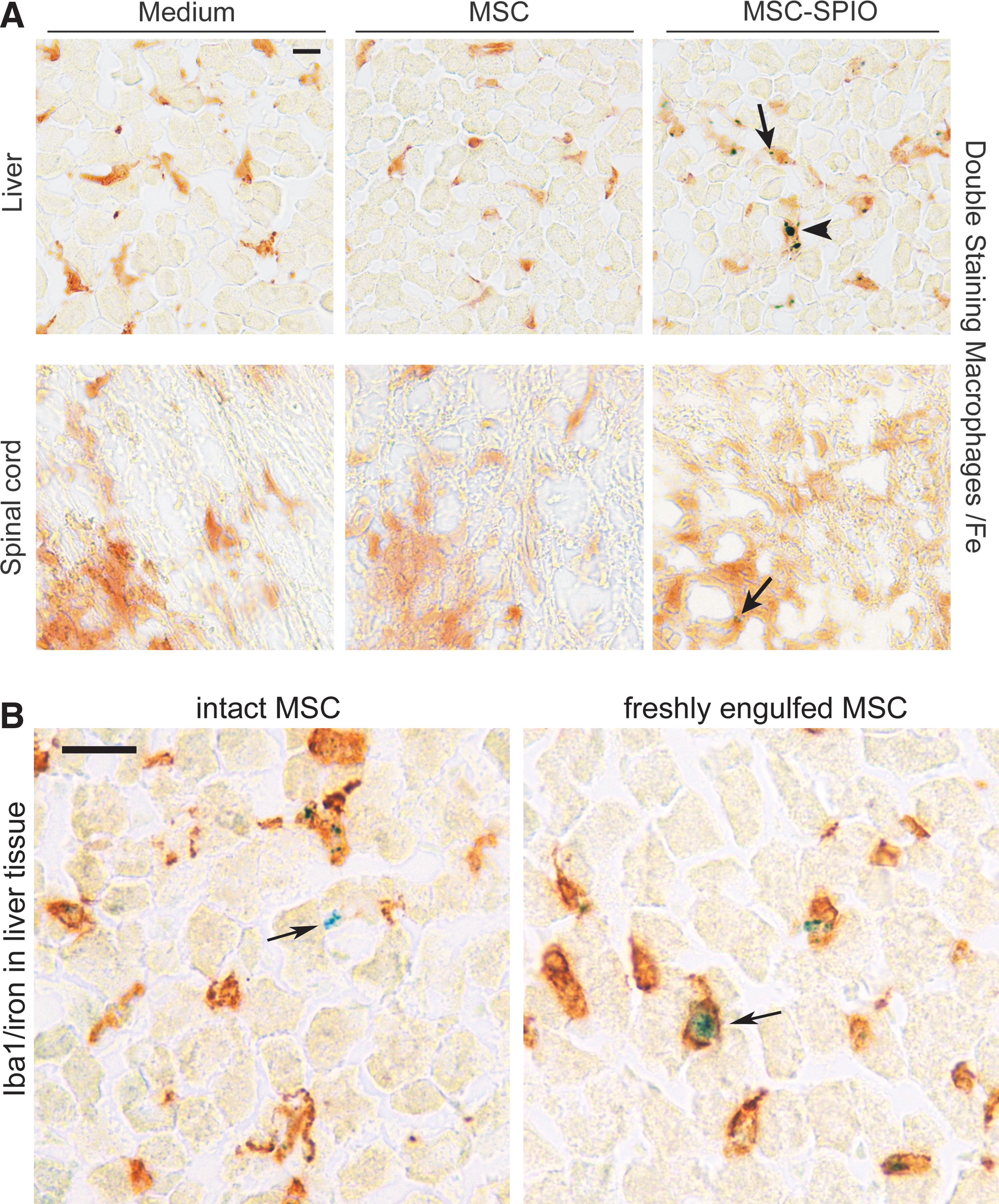
Detection of SPIO particles in the CNS and the liver of transplanted animals.
In summary, MRI data and results from FISH analysis, IHC, and qPCR do not provide evidence for the migration of MSCs into the CNS of EAE-affected mice upon transplantation at acute-disease phase. On the contrary, detection of intact MSC in the liver and iron particles in lungs, livers, and spleens indicated preferential accumulation of MSCs in these organs in agreement with previous reports [63 –67].
No amelioration of EAE after preventive transplantation of MSCs
To assess effectiveness of another therapeutic regimen, namely preventive MSC treatment of EAE [42], we transplanted MSCs at days 3 and 8 after immunization and monitored disease scores and weights of mice until day 40. We did not find significant differences, when we compared scores of MSC-transplanted mice to scores of mice transplanted with NIH3T3 mouse fibroblasts and DMEM/Ham's F-12 as control, respectively (Fig. 5). Thus, our results did not recapitulate the ameliorated EAE-disease course reported by Zappia et al. [42], notwithstanding the observed strong inhibitory effect of the used MSCs on T-cell proliferation in vitro.

Preventive treatment of C57BL/6J EAE mice did not ameliorate disease course. At day 3 and 8 after immunization mice were transplanted with MSCs, NIH3T3 fibroblasts, or medium, respectively (§). Disease course was analyzed by assessment of clinical scores
Discussion
Despite recent progress in deciphering the etiology of MS and the successful development of new drugs [68 –70] and treatment concepts [71,72], therapy options for MS patients are still limited. Fueled by encouraging outcomes of various in vitro and preclinical models, MSCs have been suggested as a novel treatment modality in autoimmune and/or neurodegenerative diseases. An ongoing phase-I/II study, MEsenchymal StEm Cells for Multiple Sclerosis (MESEMS), aims at determining the safety and efficacy of intravenously administered autologous MSCs in MS patients.
Notwithstanding these remarkable efforts to advance cell-therapeutic approaches, which could potentially address not only the inflammatory facet of MS but also neurodegenerative aspects of the disease, relatively little is known about the fate of MSCs after transplantation [73]. We here addressed this question by transplanting SPIO-labeled, sex-mismatched syngeneic MSCs in the EAE model.
It was shown that o.n. incubation of human [57] and rat-derived [58,61] MSCs resulted in efficient incorporation of ferucarbotran (Resovist) as a label. Human MSCs were also shown to engulf ferumoxide (Endorem) [57], another iron formulation for this purpose. In our study, we found high-level ferucarbotran particle uptake by C57BL/6J mouse-derived MSCs. SPIO-labeled MSCs were not compromised in their proliferative capacity and maintained their capacity to inhibit T-cell proliferation in vitro. In addition, we verified the uptake of ferumoxide particles by murine MSCs to complement earlier reports [57]. We decided to mark MSCs by SPIO-particle incorporation, since the selective marking of MSCs by covering the cell surface with ferucarbotran was reported to aggravate EAE symptoms in comparison to unlabeled MSCs [52].
To assure comprehensive detection of SPIO-labeled cells by MRI, we applied a 1:100 (v/v) dilution (280 μg/mL) of ferucarbotran for MSC marking. This was based on previous data by Peldschus et al. [61]. Those authors observed a significant improvement of MSC detection when applying a 1:50 dilution of ferucarbotran compared to 1:500 [61]. Additionally, a 1:100 (v/v) dilution of ferucarbotran still allowed the quantification of marked single cells in a preliminary in vitro proof-of-concept experiment using MSC injection into gelatin (Fig. 2D).
The MSCs used in our study were in full conformance to MSC criteria, including expression of characteristic cell-surface markers, mesodermal lineage differentiation in vitro, and inhibition of T-cell proliferation. Importantly, SPIO labeling did not impair either growth or functional properties of MSCs. However, syngeneic transplantation of SPIO-labeled and unlabeled control MSCs into EAE-induced C57BL/6J mice did not result in beneficial effects on the course of disease, in contrast to previous reports [28]. Interestingly, and in agreement with our data, human and rat MSCs were described to lack an effect on experimental autoimmune neuritis in rats, a model of human autoimmune inflammatory neuropathies, despite proven in vitro MSC properties, that is, marker expression, differentiation, and inhibition of T-cell proliferation [74].
The reasons for the contradictory results in different models of autoimmune diseases remain to be elucidated. In this context, it is of note that we, in contrast to previous studies, performed our EAE experiments using mice housed in IVC racks. Interestingly, spontaneous EAE occurrences that were observed in TCR-transgenic SJL mice were not detectable, if mice were kept in germ-free conditions [75].
Commensal microbiota may act on T cells, for example, by microbial antigens, which mimic encephalitogenic epitopes, and/or by activation of innate immunity, to induce CNS inflammation. Moreover, transplanted MSCs were found not only to modulate T-cell but also B-cell responses in EAE, either directly or indirectly by MSC-mediated changes of T-cell effector functions [28]. Together these data indicate that the gut microbiota status of experimental model organisms may be relevant for the development of autoimmune diseases, but also the testing of cell-based therapies. It is therefore tempting to speculate that the contrasting effects of MSC transplantation on EAE were at least partially due to the different status of gut microbiota.
In any case, the divergent treatment outcomes may point toward two potential caveats in the clinical application of MSCs. First, a beneficial treatment effect of MSCs on autoimmune and/or neurodegenerative diseases may be dependent on the individual MSC donor. This notion is emphasized by the fact that divergent results were obtained within one syngeneic mouse model, for example, when comparing our data to Zappia et al. [42]. Second, it might be necessary to implement a more detailed harmonization of protocols for MSC isolation, expansion, and characterization. Isolation and propagation of MSCs as established in many laboratories has recently been demonstrated to involve clonal selection [76]. Moreover, as we have shown here, MSC preparations fulfilling all consensus criteria might be ineffective in ameliorating diseases successfully treated by MSCs in other laboratories.
Notably, functional heterogeneity might be present even within one and the same MSC preparation. Indeed, bulk MSCs and clones derived thereof were found to show different activity in a mouse model of irradiation rescue [56]. It also remains to be elucidated whether such functional heterogeneity of MSCs can be linked to phenotypic diversity.
Regardless of the clinical treatment outcome, transplanted MSCs were not detectable within the CNS of recipient mice at acute-disease stage by different high-resolution techniques, namely MRI, IHC, FISH analysis, and real-time qPCR. Given the detection of iron particles in microglia cells, it cannot be completely excluded that some of the marked MSCs entered the CNS. However, we did not detect any iron-loaded MSC by MRI at three time points early after infusion. This result is in agreement with previous reports. Indeed, therapeutic effects on EAE-disease were observed within a few days after MSC transplantation [42,50,51,77 –80], whereas MSCs that apparently migrated in the CNS of recipient mice were only detectable several weeks after transplantation [42,50,51].
Previous approaches to trace transplanted MSCs in the CNS of recipient mice made either use of MSC marking by fluorescence protein expression [42,50,51,81] or by using a xenogenic MSC source [49,79,82]. In those studies, MSCs were reported to be not only preventively [42,50,81,82], but also therapeutically [42,49,50,51,77 –79] active. Based on their analyses the authors concluded that MSCs were potentially present in the CNS at later EAE stages, that is, weeks after transplantation.
However, the observation of fast clinical improvement after MSC transplantation argues against a predominant role of CNS migration in EAE amelioration, but rather supports systemic effects of MSCs. In fact, the apparent detection of eGFP+ MSCs by immunofluorescence in lymph nodes and spleens of recipient mice underpin the involvement of MSCs in peripheral modulation of (auto)immunity [42,51]. This notion has been further supported by data from a murine demyelination model, in which the peripheral immune system is absent. MSCs were found to not exert regenerative functions and did not enter CNS lesions [83,84].
In line with our findings, trapping of MSCs in the lungs of experimental animals was shown by many groups [56,63,64,66,67]. In a comprehensive review, Kurtz [65] analyzed MSC distribution for different routes of delivery. According to that work, after intravenous transplantation MSCs were mainly found in lungs, spleen, and liver, both in healthy animals and disease models. Moreover, 48 h after intravenous injection, cells were mostly detected in the lung, liver, intestine, skin, and bone marrow [65]. Thus, our data on the distribution of transplanted MSCs in peripheral organs of recipient mice is in very good agreement with previous work.
Several studies in EAE models aimed to address the CNS-migration potential of transplanted MSCs by fluorescence imaging [42,50,81], IHC [42,49,51,79,81,82], and in vivo photon emission imaging [51]. Here, we applied very sensitive approaches, that is, MRI, Y-FISH, and qPCR, but were not able to confirm the presence of transplanted MSCs within the CNS. SPIO particles were found in the CNS, histological staining indicated their uptake by phagocytic leukocytes, which vitally contribute to inflammatory lesion formation and neuronal degeneration. We therefore suppose that possible EAE amelioration by transplanted MSC would likely be mediated by immediate systemic anti-inflammatory effects [35,85].
In summary, we did not observe beneficial treatment effects upon preventive or therapeutic MSC transplantation of EAE-affected mice. Thus, our data suggest that not all preparations of MSCs are per se therapeutically active, at least not by effects in the CNS. Different factors might contribute to the observed conflicting clinical outcomes in models of autoimmune diseases, including donor-dependent influences, heterogeneity within given MSC populations, differential composition of gut microbiota, and differences in the applied animal models. In addition, we did not find evidence for migration of transplanted MSC to the place of damage, that is, the CNS, in EAE-affected mice. In conclusion, our results contribute important information regarding the development of MSC-based cell therapies for autoimmune diseases, including MS.
Footnotes
Acknowledgments
The authors wish to thank Almut Uhde, Tanja Sonntag, and Johannes Polke for excellent technical support. We are grateful to Kristin Hartmann from the Core Facility Mouse Pathology, Dagmara Nelson, isotope lab core facility at the UMC Hamburg-Eppendorf, as well as Peter Nielsen and Rosemarie Kongi, Department of Biochemistry and Molecular Cell Biology. The authors are indebted to the UKE FACS Core Facility and the Laboratory of Radiobiology and Experimental Radiooncology, UMC Hamburg-Eppendorf, for expert technical assistance. P.A. was supported by a postdoc grant (NWF-13/12) within the Forschungsförderung Medizin (FFM) program of the medical faculty of the UMC Hamburg-Eppendorf. C.L. was supported by the “Deutsche José Carreras Leukämie-Stiftung e.V.” (DJCLS R 12/30).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.