
Abstract
This study was focused on characterizing the differentiation of bone marrow-derived mesenchymal stem cells (MSCs) into corneal-like cells. Mouse MSCs were isolated from the bone marrow, grown in cell culture for 3 weeks, and purified using a magnetic activated cell sorter. Purified MSCs were cultured with an extract prepared from excised corneas and in the presence or absence of insulin-like growth factor-I (IGF-I). Analysis by quantitative real-time polymerase chain reaction showed that the expression of corneal specific markers, such as cytokeratin 12 (K12), keratocan, and lumican, was already induced after a 3-day cultivation and gradually increased during the 10-day incubation of MSCs with the extract. The presence of IGF-I significantly increased differentiation. Immunofluorescence analysis of differentiated MSCs showed positive results for the K12 protein. The morphology of the differentiated cells and the expression of cell surface markers CD45, CD11b, CD73, CD44, and CD105 were comparable in the control and differentiated MSCs. Proliferative activity was even higher in differentiated cells than in untreated MSCs. Both untreated and differentiated MSCs inhibited the production of interleukin-2 and interferon-γ in spleen cells stimulated with Concanavalin A. The results thus show that MSCs cultured in the presence of corneal extract and IGF-I efficiently differentiate into corneal-like cells. The differentiated cells possess characteristics of corneal epithelial cells and keratocytes, while at the same time maintaining MSC properties.
Introduction
S
MSCs represent a population of multipotent stem cells that can be obtained relatively easily from various sources. They can be isolated in a sufficient amount from bone marrow or adipose tissue and are able to differentiate into a number of various cell types, including those that form bone, cartilage, muscle, fat, and other connective tissues [9], or can even transdifferentiate into other cell types, including corneal epithelial cells [7,10]. Furthermore, MSCs possess potent immunosuppressive and immunoregulatory properties [11,12] and are a source of numerous growth and trophic factors [13,14]. All these properties contribute to their therapeutic potential and make them promising candidate for cell populations for ocular surface regeneration. Indeed, numerous studies have demonstrated the ability of MSCs to treat damaged ocular surface and LSCD [7,15,16].
Although the ability of MSCs to differentiate into corneal cells is still a matter of debate [17], many authors clearly demonstrated the expression of markers of corneal epithelial cells or keratocytes in differentiated MSCs under selective conditions. For example, Du et al. [18] used reduced-serum medium supplemented with ascorbate and insulin for differentiation, Park et al. [19] cultured MSCs in keratocyte-conditioned medium, and the medium from LSC cultures was used by Gu et al. [10]. In other studies, the coculture of MSCs with corneal epithelial cells or with corneal stromal cells induced the expression of corneal epithelial cell-associated markers [20,21].
In our previous study, we found that insulin-like growth factor-I (IGF-I) supports the differentiation of LSCs into corneal-like cells [22]. In the present study, we tested whether mouse bone marrow-derived MSCs have the potential to differentiate into corneal epithelial cells using the extract from the cornea, and whether the differentiation process is increased in the presence of IGF-I. We also evaluated the characteristics of MSCs differentiated with corneal extract and IGF-I and compared them to the untreated MSCs.
Materials and Methods
Animals
Mice of both sexes of the inbred strain BALB/c at the age of 2–4 months were used in the experiments. The animals were obtained from the breeding unit of the Institute of Molecular Genetics, Prague. The use of animals was approved by the local Animal Ethics Committee of the Institute of Experimental Medicine, Prague. The animals were treated in accordance with the Principles of Laboratory Animal Care.
Isolation, culture, and purification of MSCs
MSCs were isolated from the bone marrow of BALB/c mice. The bone marrow was flushed out from the femurs and tibias, and a single-cell suspension was prepared with a tissue homogenizer. The cells were seeded at a concentration of 4 × 106 cells/mL in Dulbecco's modified Eagle's medium (DMEM; Sigma) containing 10% fetal calf serum (Gibco BRL), antibiotics (100 U/mL of penicillin and 100 μg/mL of streptomycin), and 10 mM HEPES buffer (after this referred to as complete DMEM) in 75-cm2 tissue culture flasks (Trasadingen). After 72-h cultivation, the nonadherent cells were removed by washing and the remaining adherent cells were cultured with a regular exchange of medium and held to optimal cell concentration for an additional 2–3 weeks at 37°C in an atmosphere of 5% CO2. The adherent cells were harvested by 5-min incubation with 1 mL of 0.5% trypsin and gently scrapped. The cell suspension was incubated for 15 min with CD11b MicroBeads and CD45 MicroBeads (Miltenyi Biotec) according to the manufacturer's instructions and immunodepleted CD11b+ and CD45+ cells using a magnetic activated cell sorter (AutoMACS; Miltenyi Biotec). The remaining CD11b− and CD45− cells were evaluated in terms of their purity and differentiation potential.
Phenotypic characterization of MSCs by flow cytometry
Untreated and differentiated MSCs were washed in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin and incubated for 30 min on ice with the following anti-mouse monoclonal antibodies (mAbs): allophycocyanine (APC)-labeled anti-CD44 (clone IM7; BD PharMingen), phycoerythrin (PE)-labeled anti-CD105 (clone MJ7/18; eBioscience), APC-labeled anti-CD11b (clone M1/70; BioLegend), fluorescein isothiocyanate-labeled anti-CD45 (clone 30-F11; BioLegend), or PE-labeled anti-CD73 (clone: TY/11.8; eBioscience). Dead cells were stained using Hoechst 33258 fluorescent dye (Invitrogen) added to the samples 10 min before flow cytometry analysis. Data were collected using an LSR II cytometer (BD Biosciences) and analyzed using a FlowJo software (Tree Star).
Differentiation of MSCs to adipocytes and osteoblasts
MSCs were cultured for 2–3 weeks and separated by magnetic cell sorting. The cells were cultured in a complete DMEM supplemented with specific adipogenic (containing 0.1 mM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, 0.1 mM indomethacine, and 0.5 mg/mL of insulin) or osteogenic (0.1 mM dexamethasone, 0.1 mM L-ascorbic acid, and 10 mM β-glycerophosphate disodium salt pentahydrate) reagents [23]. Cell differentiation was confirmed by staining with Oil Red O or Alizarin Red S.
Preparing the corneal extract
The corneas were harvested and cut into small pieces in serum-free DMEM (one cornea/125 μL of medium) postmortem. The samples were frozen at −80°C and thawed/frozen in three cycles for 10 min each. The extracts were filtered through a 0.22 μm filter and stored at −80°C until used.
Differentiation of MSCs
MSCs were cultured for 3, 7, or 10 days in complete DMEM with extract from the corneas and in the absence or presence of IGF-I (20 ng/mL; PeproTech). The concentration of the extract in the culture medium was 20% and increased to 40% during the culturing and exchange of the medium. The culture medium was exchanged every 2–3 days.
Detecting gene expression
The expression of genes for K12, keratocan, and lumican in cultured MSCs was detected using a quantitative real-time polymerase chain reaction (qPCR). The following primers were used for amplification: K12 (sense: GTGAGTCCGCTGGTGGTAAC, antisense: CATCAGCACAGCAGGAAGTG), keratocan (sense: TCCCCCATCAACTTATTTTAGC, antisense: AGTTTGGGGTTGCCATTACA), lumican (sense: GGATGGCAATCCTCTCACTC, antisense: TCATTTGCTACACGTAGACACTCAT), and GAPDH (sense: AGAACATCATCCCTGCATCC, antisense: ACATTGGGGGTAGGAACAC). Untreated or differentiated cells were transferred into Eppendorf tubes containing 500 μL of TRI Reagent (Molecular Research Center), and the total RNA was extracted according to the manufacturer's instructions. One microgram of RNA was treated with deoxyribonuclease I (Promega) and used for subsequent reverse transcription. The first-strand cDNA was synthesized using random hexamers (Promega) in a total reaction volume of 25 μL using M-MLV Reverse Transcriptase (Promega). qPCR was performed in a StepOnePlus real-time PCR system (Applied Biosystems) as previously described [22]. The PCR parameters included denaturation at 95°C for 3 min, 40 cycles at 95°C for 20 s, annealing at 60°C for 30 s, and elongation at 72°C for 30 s. Fluorescence data were collected at each cycle after an elongation step at 80°C for 5 s and were analyzed using StepOne Software version 2.2.3 (Applied Biosystems). Each individual experiment was done in triplicate. A relative quantification model was applied to calculate the expression of the target gene in comparison to GAPDH used as an endogenous control.
Determining metabolic cell activity
The metabolic activity of living cells was determined by the WST-1 assay. The assay is based on the ability of living cells to cleave tetrazolium salts by mitochondrial dehydrogenases into water soluble formazan, which is then measured by spectrophotometry. MSCs (2 × 105 cells/mL) were cultured in complete DMEM with or without extract from the corneas and IGF-I in 24-well tissue culture plates (Corning) for 7 days at 37°C in an atmosphere of 5% CO2. WST-1 reagent (Roche) (10 μL/100 μL of the medium) was added to each well and the plates were incubated for another 4 h to form formazan [24]. Formazan
Immunostaining with anti-K12 antibody
Corneal cells (prepared by trypsinization of corneal tissue) and untreated or differentiated MSCs (3.7 × 105 cells/mL) were fixed for 20 min with 4% paraformaldehyde and permeabilized for 10 min with 0.1% Triton X-100. The samples were incubated with goat polyclonal anti-K12 antibody (Santa Cruz Biotechnology) for 1 h at room temperature and then with a secondary donkey anti-goat IgG antibody conjugated with Alexa Flour 594 (Invitrogen). The cells were rinsed with PBS containing 0.05% TWEEN and fixed on glass slides with Mowiol 4-88 (Calbiochem) in the presence of the nuclear dye 4′,6-diamidino-2-phenylindole (DAPI). Visualization of the fluorescent label was performed using a fluorescent microscope (Leica).
Immunostaining with phalloidin
Untreated or differentiated MSCs (2.5 × 105 cells/mL) were fixed for 20 min with 4% paraformaldehyde and permeabilized for 10 min with 0.1% Triton X-100. The samples were then incubated with Phalloidin conjugated with Alexa Fluor 568 (Invitrogen) for 1 h at room temperature. Cell nuclei were stained with DAPI for 1 min and samples were mounted with VECTASHIELD. Visualization of the fluorescent label was performed using a fluorescent microscope (Leica).
Comparing the immunosuppressive properties of untreated and differentiated MSCs
Spleen cells (0.6 × 106/mL) from BALB/c mice were stimulated with Concanavalin A (ConA; Sigma-Aldrich), as described previously [25]. Cells were cultured in a volume of 0.4 mL of complete DMEM in 48-well tissue culture plates (Corning) alone or were stimulated with 1 μg/mL of ConA. Untreated or differentiated MSCs were added to these cultures at a lymphocyte/MSC ratio of 8:1. Supernatants were harvested after a 24-h incubation for interleukin-2 (IL-2) determination and after a 48-h incubation period for interferon-γ (IFN-γ) determination. The concentrations of cytokines in the supernatants were determined by ELISA using cytokine-specific capture and detection mAbs purchased from BD Pharmingen and following the manufacturer's instructions.
Statistical analysis
The statistical significance of differences between individual groups was calculated using the Student's t-test. A value of P < 0.05 was considered statistically significant.
Results
Characterization of MSCs
The purified cells had a uniform spindle-shaped morphology. The purity and phenotypic markers of MACS-separated MSCs were evaluated by flow cytometry. The results showed that MSCs were positive for CD44 and CD105, but negative for CD11b and CD45 (Fig. 1A). In addition, the MSCs were characterized by their ability to undergo specific adipogenic (Fig. 1B) and osteogenic differentiation (Fig. 1C). These observations showed that the adherent MACS-separated bone marrow-derived cells possess the phenotype and differentiation characteristics of MSCs.
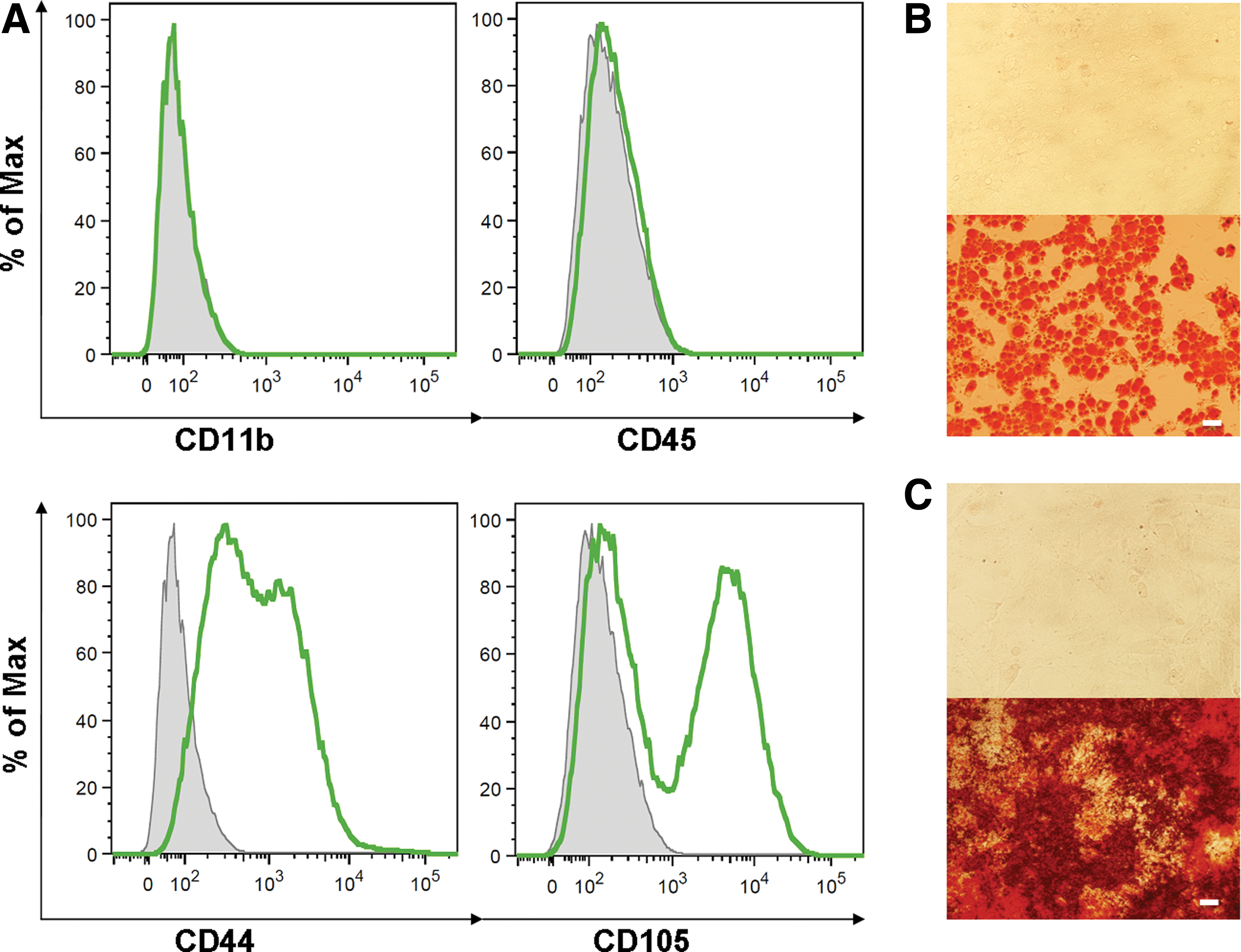
Characterization of untreated bone marrow-derived MSCs.
Differentiation of MSCs
The MSCs were cultured in the absence or presence of the corneal extract and with or without recombinant IGF-I (20 ng/mL) for 3, 7, or 10 days. The expression of genes for cornea-associated markers was determined by qPCR. Figure 2 shows that the expression of genes for K12, keratocan, and lumican was already upregulated 3 days after the culture with the extract. Adding IGF-I to the culture medium significantly increased the expression of the tested genes.

The expression of genes for K12, keratocan, and lumican in untreated and differentiated MSCs was determined by qPCR. The cells were cultured for 3, 7, or 10 days untreated (Unt), with the extract from the corneas (Ext) and in the presence of the extract and IGF-I (Ext+IGF-I). Each bar represents mean ± SD from four to five determinations. The asterisks represent statistically significant (*P < 0.05, **P < 0.01) difference in the gene expression between MSCs treated only with the extract or with the extract and IGF-I. Freshly purified MSCs are marked as a control (C). IGF-I, insulin-like growth factor-I; qPCR, quantitative real-time polymerase chain reaction.
The differentiation potential of the MSCs was confirmed by immunostaining for the K12 protein using anti-K12 antibody. As demonstrated in Fig. 3, the K12 protein was clearly detected in the MSCs differentiated with the extract (Fig. 3B) and with the extract and IGF-I (Fig. 3C). Untreated MSCs were used as a negative control for K12 expression (Fig. 3D), while isolated corneal epithelial cells served as a positive control (Fig. 3A).
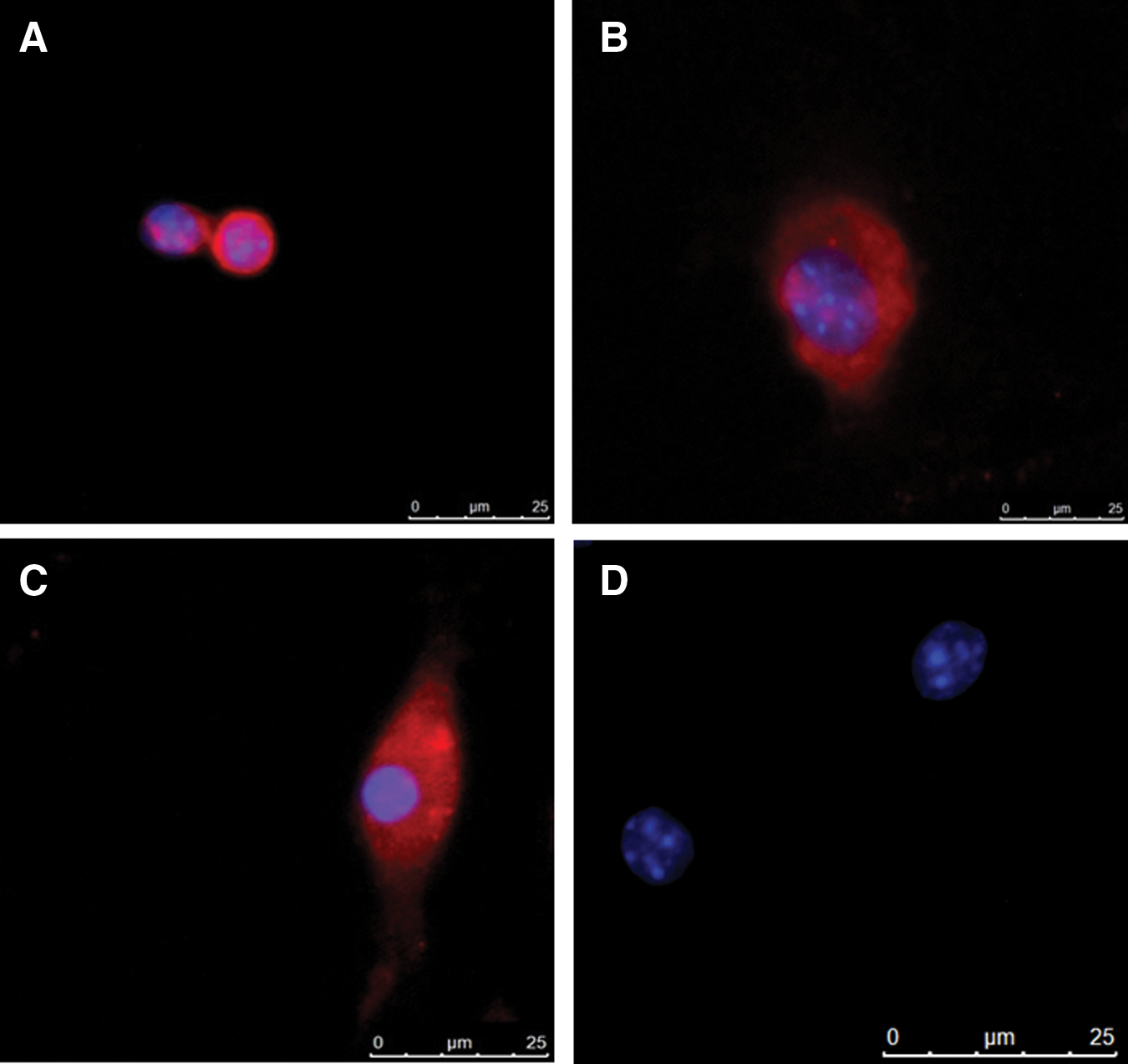
Immunostaining for K12 protein in corneal cells and untreated or differentiated MSCs. Single cell suspensions of corneal cells
Morphology, growth, and gene expression of differentiated MSCs
The morphology of the untreated and differentiated MSCs is shown in Fig. 4. Both cell types had a typical fibroblast-like shape and adhered to plastic and glass surfaces. The expression of cell surface markers CD45, CD11b, CD73, CD44, and CD105 was determined by flow cytometry. The analysis revealed that both cell types had a similar expression profile (Fig. 5). Results from the WST-1 assay showed that differentiated MSCs have rather better proliferation activity than untreated cells (Fig. 6).
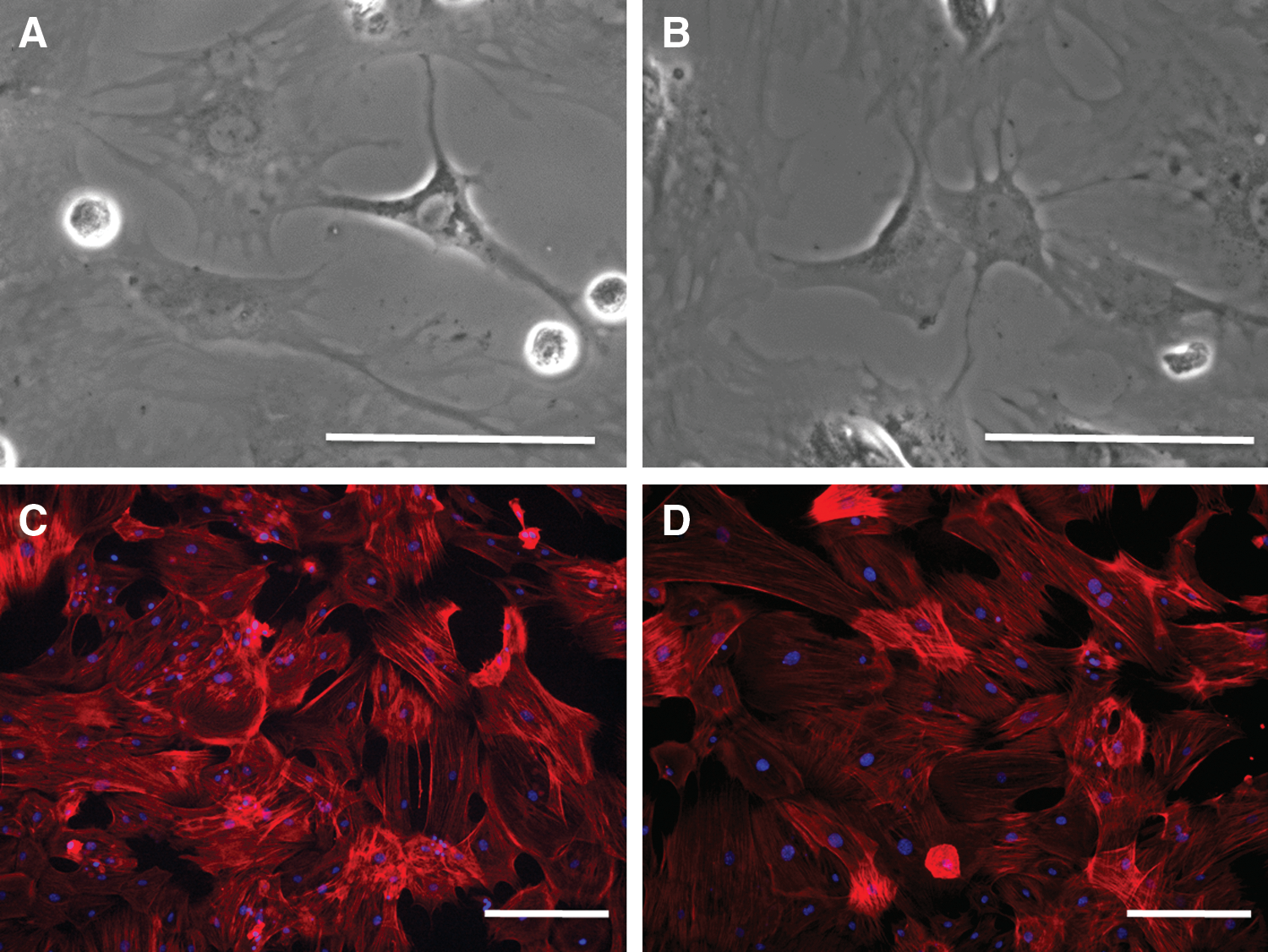
Comparison of morphology of untreated and differentiated MSCs. The growing untreated MSCs
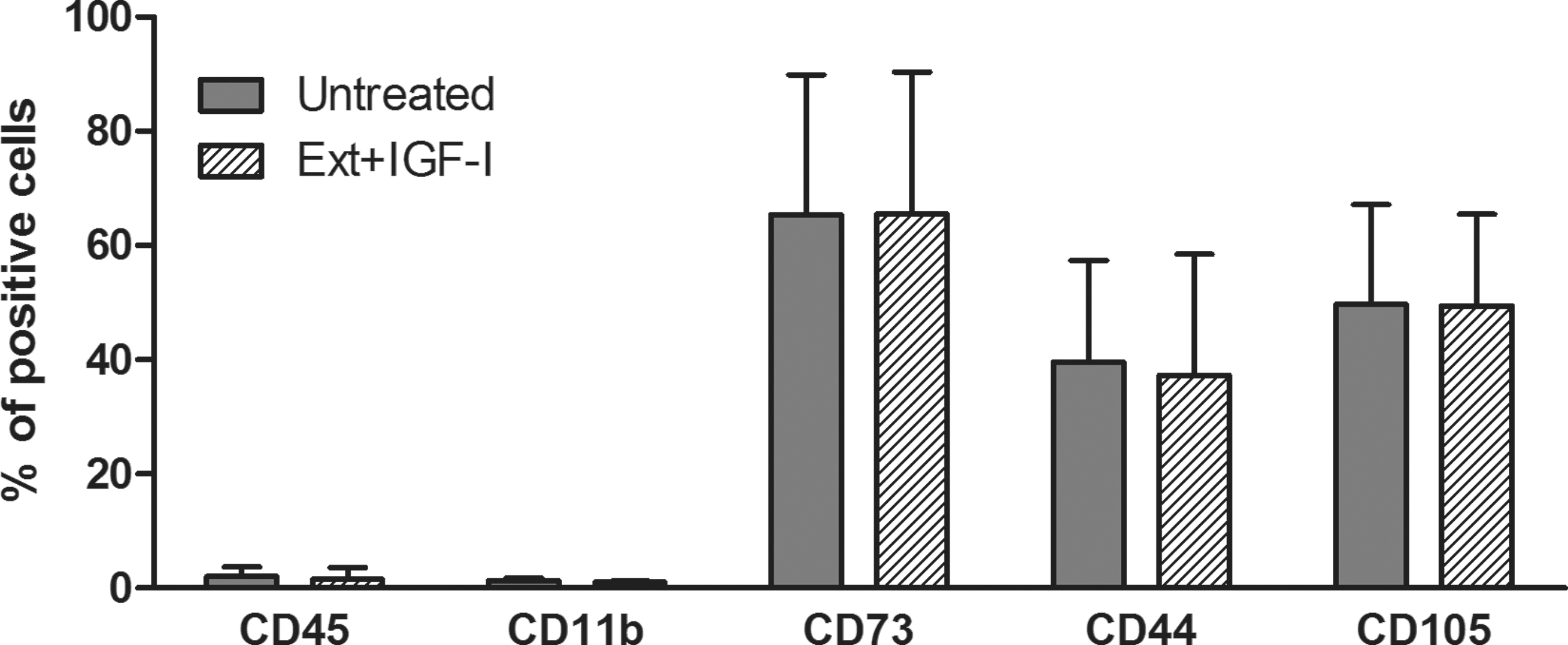
Comparison of the expression of cell surface markers in untreated and differentiated MSCs. Flow cytometry analysis of CD45, CD11b, CD73, CD44, and CD105 markers expressed in untreated (Unt) or differentiated (Ext+IGF-I) MSCs is demonstrated. Each bar represents mean ± SD from three determinations.
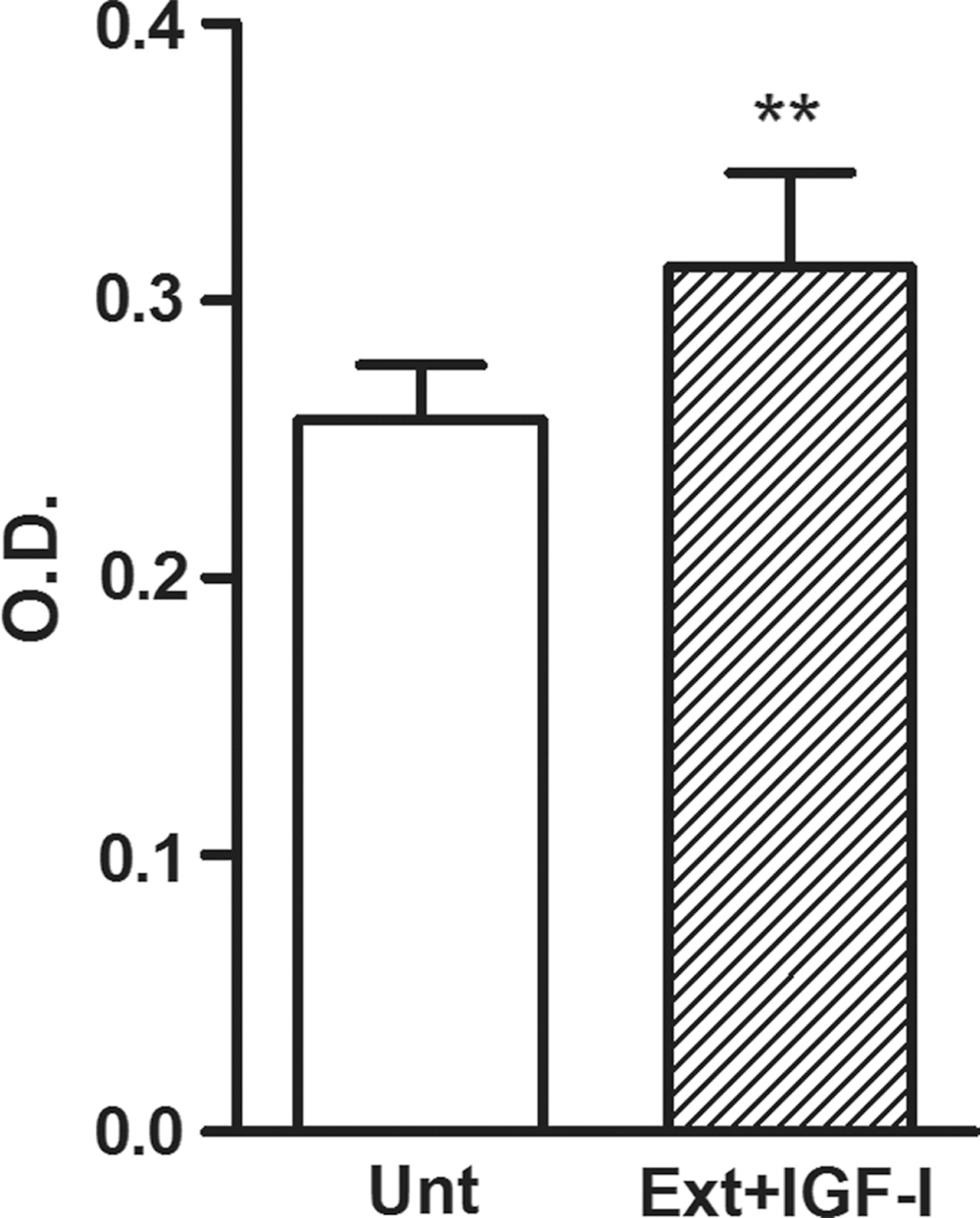
Comparison of the metabolic activity of the untreated (Unt) and differentiated (Ext+IGF-I) MSCs. WST-1 reagent was added to the cell cultures for 4 h to form formazan. The absorbance was measured using a Sunrise Remote ELISA Reader at a wavelength of 450 nm. Each bar represents mean ± SD from three determinations (**P < 0.01).
Immunosuppressive properties of untreated and differentiated MSCs
Spleen cells were stimulated with T-cell mitogen ConA in the absence or presence of untreated or differentiated MSCs (the ratio of lymphocytes to MSCs was 8:1). The production of IL-2 and IFN-γ was determined in the supernatants by ELISA. As demonstrated in Fig. 7, both cell types significantly inhibited production of tested pro-inflammatory cytokines.

Comparison of the immunosuppressive properties of untreated and differentiated MSCs. Spleen cells were cultured unstimulated or were stimulated with ConA in the presence of untreated (Unt) or differentiated (Ext+IGF-I) MSCs. The production of IL-2 and IFN-γ was determined in the supernatants after a 24 h (IL-2) or 48 h (IFN-γ) incubation period by ELISA. Each bar represents mean ± SD from three determinations (*P < 0.05, **P < 0.01, ***P < 0.001). ConA, concanavalin A; IL-2, interleukin-2; IFN-γ, interferon-γ.
Discussion
The integrity of the cornea is ensured by a population of stem cells that reside in the limbus. When the cornea is injured, LSCs start to proliferate, differentiate, and migrate to the site of injury. To treat corneal defects in patients with unilateral LSCD, LSCs can be isolated from healthy eyes, propagated in vitro, and using an appropriate scaffold transferred to treat the damaged cornea [2,26]. However, LSC therapy is limited by the low number of cells available and harmful immunological rejection if the cells are transplanted from a genetically unrelated donor. Therefore, other sources of autologous stem cells have been tested. These include conjunctival epithelial stem cells [8], oral mucosal cells [6], dental pulp stem cells [27], hair follicle stem cells [28], or various types of MSCs [7,15,29].
One of the properties required for stem cells used in the treatment of LSCD is their capability to differentiate into corneal cells. Therefore, in the present study, we characterized mouse bone marrow-derived MSCs and tested their ability to differentiate into cells expressing cornea-associated markers, which were not detected in untreated MSCs.
MSCs isolated by a negative sorting from the population of adherent bone marrow cells were positive for CD44 and CD105 and negative for CD11b and CD45, as described earlier [30]. In addition, these cells effectively differentiated into adipocytes and osteoblasts, thus fulfilling the basic criteria for definition of MSCs [31]. Based on screening the gene expression in untreated MSCs, we identified three genes, which were not (K12, keratocan) or only weakly (lumican) expressed in unstimulated MSCs.
Previous studies have demonstrated the effects of the coculture of MSCs with corneal cells in limbal or corneal cell-conditioned medium on the differentiation of MSCs into keratocytes [18,19] or cells with markers and characteristics of corneal epithelial cells [10,20,21,32]. In the present study, we used the extract from corneas for differentiation of MSCs. We observed that already after a 3-day culture of the MSCs in the presence of the extract, the cells started to express corneal markers and their expression gradually increased. This observation is in accordance with the above studies showing the expression of corneal markers in MSCs cultured in the presence of corneal cells or in corneal cell-conditioned medium. In our previous study, we found that IGF-I plays an important role in the differentiation of LSCs into corneal epithelial cells. IGF-I, which is highly expressed in the cornea after the injury, migrates to the limbus where it binds to its receptor and triggers the differentiation process of LSCs [22]. Huang et al. [33] demonstrated that IGF-I can dose-dependently stimulate the proliferation of MSCs, upregulate the expression of CXCR4, and accelerate their migration. It has been also observed that IGF-I is secreted by MSCs after their therapeutic administration [34 –36]. Therefore, we tested whether IGF-I could also play a role in the differentiation of MSCs into corneal-like cells. Adding IGF-I into MSC cultures with extract from the corneas significantly increased the expression of genes for K12, keratocan, and lumican. IGF-I alone had no effect on the expression of these genes (data not shown).
Purified bone marrow MSCs have fibroblastic morphology. Differentiated MSCs did not change their morphology and remained in fibroblastic shape, which is comparable to previous results [20,32]. Both untreated and differentiated MSCs adhered to plastic and glass surfaces. Comparing the expression of cell surface markers did not reveal differences between untreated and differentiated cells. A similar conclusion was reached in the study where unstimulated MSCs and MSCs stimulated with a cocktail of pro-inflammatory cytokines were tested for the expression of endothelial, stromal, and adhesive markers [37]. In accordance with other studies on the proliferative and metabolic activity of differentiated cells [33,38,39], we found that MSCs differentiated with corneal extract and IGF-I have comparable or even slightly enhanced metabolic activity to untreated cells.
MSCs possess potent immunosuppressive properties and inhibit the production of various pro-inflammatory cytokines [24,40]. In the present study, we confirmed the suppressive potential of unstimulated MSCs and we showed that differentiated MSCs inhibit the production of IFN-γ and IL-2, similar to untreated MSCs.
In conclusion, we showed that IGF-I supports differentiation of mouse bone marrow-derived MSCs into cells expressing markers of corneal cells. Differentiated MSCs expressed markers of both corneal epithelial cells and keratocytes. This observation makes them a promising source of stem cells for the regeneration of damaged or diseased cornea. In addition, the differentiated cells maintain characteristics of unstimulated MSCs and suppress the production of pro-inflammatory cytokines by activated T lymphocytes.
Footnotes
Acknowledgments
This work was supported by projects 889113 and 80815 from the Grant Agency of Charles University, grant 14-12580S from the Grant Agency of the Czech Republic, and by the projects CZ.1.05/1.1.00/02.0109, CZ.2.16/3.1.00/21528, SVV 260206, UNCE 204013, NPUI LO1309, and NPUI LO1508 and also by funding from the Norwegian Financial Mechanism 2009–2014 and the Ministry of Education, Youth and Sports under project contract no. MSMT-28477/2014, project 7F14156.
Author Disclosure Statement
No competing financial interests exist.