
Abstract
The discovery of mammalian N 6-methyladenosine (m6A) methyltransferases and demethylases has enriched our knowledge of the dynamic regulation of the most prevalent posttranscriptional RNA modification, m6A methylation. This reversible methylation process of adding and removing m6A marks on RNA has been shown to have broad biological functions in fine tuning cellular processes and gene expression. Recent studies have revealed a critical role for the currently known m6A methyltransferases and demethylases in regulating the pluripotency and differentiation of stem cells. These data establish a novel dimension in epigenetic regulation at the RNA level to affect mammalian cell fate.
Introduction
S
Dynamic epigenetic modifications are known to participate in regulating both embryonic [10 –12] and adult [8,13 –16] stem cell self-renewal and differentiation. Consequently, the critical necessity of the enzymes that catalyze these modifications can be clearly demonstrated. For instance, the ablation of DNA methyltransferases Dnmt1 and Dnmt3a/3b led to severe differentiation incompetence in mouse embryonic stem cells (mESCs) [17,18]. Also, ectopic expression of some histone modification enzymes, such as myeloid/lymphoid or mixed-lineage leukemia 2 (MLL2), ubiquitously transcribed tetratricopeptide repeat X (UTX), and Jumonji domain-containing 3 (Jmjd3), severely delay or hamper the proper differentiation of embryonic stem cells (ESCs) [19,20]. Other factors, such as males absent on the first (MOF), Jmjd1a and Jmjd2c, play a pivotal role in maintaining ESC identity by modulating pluripotency transcripts [21,22]. The 5-methylcytidine (m5C) modification at the RNA level has been shown to affect posttranscriptional stem cell regulation. Dnmt2-mediated RNA m5C methylation, for example, promotes late differentiation of several organs in zebrafish [23]. The RNA m5C methyltransferase NOP2/Sun domain protein 2 (Nsun2) ensures timely anagen phase initiation and mouse spermatogenesis by controlling hair follicle stem and germ cell differentiation [24,25]. The loss of both Nsun2 and Dnmt2 expression sharply reduces transfer RNA (tRNA) m5C methylation levels and severely damages protein synthesis, leading to further proliferation and differentiation defects in mouse embryonic fibroblasts (MEFs) [26,27].
There has been considerable research in the field of epigenetic regulation in recent years. One important result is the identification of the N 6-methyladenosine (m6A) RNA posttranscriptional modification [28 –31]. “m6A” refers to the methylation of the nitrogen at position 6 of the adenosine base (Fig. 1) [32]. m6A methyltransferases and demethylases, also known as m6A “writers” and “erasers” [28,31,33 –35], catalyze a reversible m6A modification to RNAs (Fig. 1). Their regulatory role in delineating cell status and affecting cellular differentiation in mammals has only lately been discovered [36 –44] and reveals a novel dimension in epigenetic regulation at the posttranscriptional stage. Methyltransferase-like 3 (METTL3) is the most well-studied m6A writer and has been implicated in the fine-tuning of ESC pluripotency and differentiation [36 –38,40,41]. Fat mass and obesity-associated protein (FTO) is an m6A eraser that has a pronounced effect on the differentiation of preadipocytes [42 –44]. The emerging understanding of the effect of m6A methyltransferases and demethylases on mammalian stem cell pluripotency and differentiation has attracted great attention to these proteins in the search for further insights into stem cell regulation. The exact mechanisms underlying these effects are still relatively unknown, however.
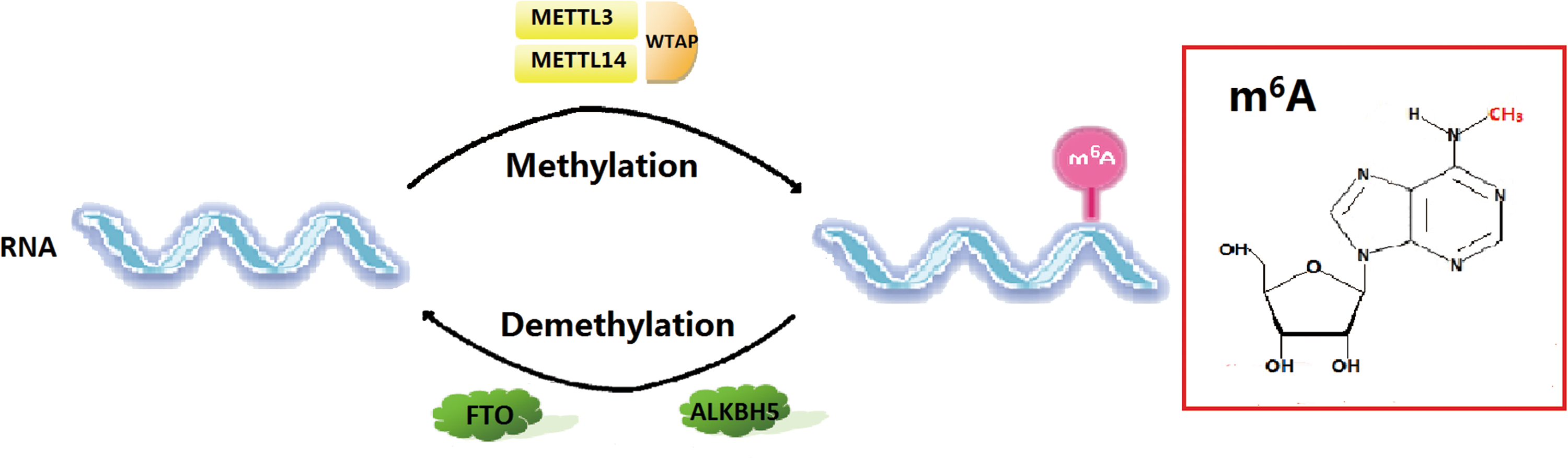
Reversible m6A methylation in RNAs. The m6A methyltransferases (METTL3, METTL14, and WTAP) and demethylases (FTO and ALKBH5) regulate the dynamic deposition of m6A marks on RNAs. The internal base modification m6A is highlighted in the red box. ALKBH5, AlkB Homolog 5; FTO, fat mass and obesity-associated protein; m6A, N
6-methyladenosine; METTL3, methyltransferase-like 3; WTAP, Wilms’ tumor 1-associating protein. Color images available online at
In this review, we briefly summarize the general features of m6A RNA modification and highlight the effect of m6A methyltransferases and demethylases on stem cell pluripotency and differentiation through the regulation of m6A levels.
General Features of m6A RNA Modification
There are many forms of nucleoside modification required to transform precursor RNA transcripts to fulfill the multiple functions of RNA [45]. Over a hundred posttranscriptional modifications have been identified in a wide range of RNAs, including messenger RNAs (mRNAs), tRNAs, ribosomal RNAs (rRNAs), small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs) [46 –51]. m6A modification is the most prevalent of these chemical modifications in mRNA and lncRNA in eukaryotes [32,45,52]. Previous studies have revealed that m6A modification occurs in 0.1%–0.4% of all adenosine residues in total cellular RNAs [32,52 –54]. A comprehensive analysis of m6A localization at the transcriptome level found that 46.0% of mRNA had one m6A peak, 37.3% had two, 11.2% had three, and 5.5% had more than three [55]. Furthermore, m6A is highly conserved among a variety of eukaryotic species as diverse as mammals, yeast, plants, and flies [39,53,55 –60]. It has even been identified in viruses [61,62]. There is much that is unknown about m6A, even though it is so ubiquitous and was first discovered in the 1970s [32,49,50,52,63,64]. This is due, in large part, to the shortage of available detection techniques [55,65]. There are two primary reasons for the technical difficulty in detecting m6A. First, N 6-methylated adenosine possesses the same Watson–Crick base pairing ability and efficiency as unmethylated adenosine [66]. Second, m6A is inert to chemical modifications that are applied in an attempt to facilitate the detection of m6A [30,55,65].
Accumulating evidence indicates that m6A occurs within a consensus sequence Pu[G/A]Pu[G>A]m6AC[U>A>C] [39,55,57,63,67 –70]. Two groups independently developed a novel approach to investigate m6A modification at a transcriptome-wide level by combining highly specific m6A-antibody-mediated RNA immunoprecipitation with high-throughput sequencing [55,57]. m6A peaks are identified within ∼7,000 human gene coding and ∼300 noncoding transcripts with a preferred distribution near stop codons, in 3′-untranslated regions (3′ UTRs), and within long external exons [55,57]. The specificity of the distribution of m6A modifications has been supported by further research [29,36,39,71 –73]. However, Ke et al. demonstrated in their experiment that m6A deposition rises sharply within 150–400 nucleotides of the beginning of the last exon, but does not appear to be concentrated around the stop codon [74]. They discovered that when the stop codon is not in the last exon, there is no obvious spatial overlap between m6A peak and the stop codon. The correlation of m6A sites with such distinct landmarks provides evolutionary insight in uncovering the functions of m6A in RNA metabolism. Recently, Linder et al. applied an ultraviolet cross-linking and immunoprecipitation method combined with reverse transcription to map m6A at single-nucleolar resolution [75]. This will greatly facilitate functional studies of m6A in the future.
Immunoblot analysis has shown that m6A is widely distributed and occurs in many tissues. There is a greater concentration in the brain, liver, and kidney, however, [55]. m6A modification has also been demonstrated to be gene- and cell-type specific. This is supported by the finding that m6A is preferably expressed in ESCs, induced pluripotent stem cells (iPSCs), and neural stem cells (NSCs). The transcription factors instrumental in defining the identity of these specific cells, such as Nanog, had abundant m6A modifications [36,38,39,41]. This implies a strong relevance of m6A to stem cell regulation. Moreover, it has been shown that m6A modification affects the circadian clock [76]. It has wide ranging effects on RNA metabolism, including RNA stability [36,37,77,78], splicing [43,57,70,79], nuclear export [80,81], mRNA-protein interactions [72,80,82], and mRNA-miRNA interactions [39,55,83,84]. Thus regulation of m6A RNA modification is complex and dynamic with a profound influence on gene expression and cellular processes as a consequence [30,31,33,85].
Purification of HeLa nuclear extracts revealed a methyltransferase complex that catalyzes the formation of m6A [68]. A 70-kDa Adomet-binding subunit MT-A70, or METTL3, was discovered in a wide variety of human tissues and identified as a crucial subunit of this multicomponent methyltransferase complex [79]. METTL14, a homolog of METTL3, was shown to work synergistically with METTL3 in m6A methylation [37,60]. Wilms’ tumor 1-associating protein (WTAP) was identified as a third factor by its effect on m6A cellular deposition by interacting with the METTL3-METTL14 complex [60,70]. The discovery of two mammalian m6A demethylases in the alkylated DNA repair protein B (AlkB) family, FTO and AlkB Homolog 5 (ALKBH5) [80,86], is also of great importance in understanding the regulation of m6A. These two factors work together with m6A methyltransferases to dynamically regulate m6A within RNAs.
m6A Methyltransferases
m6A RNA modification is performed by a multicomponent methyltransferase complex. Investigating the structure and mechanism of this methyltransferase complex is one of the high research priorities for gaining further insight into the biological function of ubiquitous m6A modification. Many studies have been directed at purifying this catalytic subunit, yet only the three subunits mentioned above have been identified to date: METTL3, METTL14, and WTAP. METTL3 and METTL14 exhibit a methyltransferase activity, whereas WTAP serves as a regulatory subunit [33,34,60,68,70,77,79,87].
METTL3
METTL3 is a 70 kDa subunit of MT-A and is an indispensible part of this multicomponent methyltransferase complex [68,79]. A BLAST search on METTL3 revealed the presence of homologous sequences for functional consensus methylation motifs I and II [79]. The first motif is responsible for Adomet binding and the second contains catalytic domains [88]. METTL3 is located in nuclear speckles, which are enriched with mRNA processing factors. This suggests that METTL3 may be associated with RNA splicing [79]. The knockdown of cellular METTL3 led to an ∼30% decrease in m6A level in HeLa cells and ∼20% in 293FT cells [60]. Conversely, RNA probes incubated with purified METTL3 proteins from HEK293 cells exhibited a 1.5-fold increase in m6A levels [37]. Bokar first reported that the METTL3 knockdown triggered severe apoptosis in HeLa cells, which indicated that METTL3 is crucial for cell viability [53]. This observation was later confirmed by Dominissini et al. Their finding further suggested that the observed apoptosis might be linked to regulation of the p53 signaling pathway under conditions of METTL3 deficiency [57]. However, Batista et al. showed that Mettl3 depletion increased mESC proliferation [36]. The contrasting effects in somatic and stem cells imply that the function of METTL3 to regulate cell viability might depend on the cell state. METTL3 was also shown to add m6A modifications to primary miRNA to promote miRNA maturation with the help of a newly defined m6A “reader”—heterogeneous nuclear ribonucleoprotein A2B1 (HNRNPA2B1) [83,89]. Mature miRNA can enhance the binding of METTL3 to mRNA, illustrating another mechanism for miRNA to regulate mRNA and gene expression [39].
METTL3-catalyzed m6A methylation is known to be widespread in both mouse and human ESC transcripts, including key regulatory factors that define cell state and determine cell fate [36 –38,40,41]. Wang et al. first reported that the Mettl3 knockdown in mESCs led to the loss of self-renewal capability and an obvious elevation of developmental factors [37]. They argued that the increased binding of human antigen R (HuR), an RNA stabilizer protein that can recognize m6A marks [57,90], is responsible for the increased stability of developmental factors upon Mettl3 depletion [37]. However, more recent studies have demonstrated that METTL3-mediated m6A modification induces ESC differentiation rather than suppressing it [36,38,41]. Batista et al. found that silencing Mettl3 did not cause immediate lethality and it enhanced the self-renewal ability of mESCs [36]. The discrepancy in observations might be ascribed to acute versus chronic inactivation of Mettl3 or to an RNA interference (RNAi) off-target effect [36]. Chromatin-associated zinc finger protein 217 (ZFP217) modulates m6A by sequestering METTL3. Zfp217 depletion removes this inhibition to Mettl3 binding and results in impaired self-renewal and differentiation, thus further supporting the role of Mettl3 in ESCs exiting pluripotent state [38]. Mettl3 depletion during mESC differentiation results in a failure to properly repress core pluripotency factors that are enriched in Mettl3 targets [36,38,41] and a morphological abnormality [36,41].
Furthermore, Mettl3-deficient mESCs undergo partial differentiation and display aberrant lineage development [36,41]. Mettl3-knockout (KO) embryoid bodies (EBs) are retained in a rather naive and undifferentiated state when compared with those in wild-type control groups [41]. Mettl3 KO teratomas exhibit poor differentiation in all three germ layers [36,41]. The requirement of Mettl3 for differentiation was further demonstrated in Mettl3 KO mESCs. These cells displayed a compromised development of key mature structures when induced to differentiate into either cardiomyocytes or the neural lineage [36]. A sustained expression of naive markers and an inability to induce developmental markers were observed in Mettl3 KO ESCs and EBs. This contributed to the prolonged maintenance of a highly pluripotent state, vividly termed as the “hypernaive” pluripotent state [41]. In addition, diminished m6A levels in mESCs induced by Mettl3 ablation resulted in a dominance of pluripotency regulators, which not only exhibited an extended half-life but also underwent more efficient translation [38,41].
Interestingly, the depletion of Mettl3 in stem cells results in distinctive alterations in cell fate based on the cell state. This reveals a much more complex and precise control by Mettl3 to regulate proper stem cell pluripotency and differentiation [41]. Naive mESCs have high expression levels of pluripotency genes, whereas the epiblast stem cells, which are at a more advanced developmental stage, express higher levels of lineage commitment genes. Mettl3 depletion in epiblast stem cells led to further upregulation in the expression of those genes and resulted in accelerated differentiation and cell death [41]. Naive mESCs, on the other hand, experienced an obvious elevation in pluripotency genes with only a slight increment in lineage commitment genes. This resulted in the “hypernaive” state. Thus the diminished m6A abundance aggravated the existing imbalance between pluripotency and lineage commitment genes in both naive mESCs and epiblast stem cells. This resulted in an opposite, possibly pluripotent-state dependent, outcome in these two cell types at different development stages [41]. In summary, Mettl3 depletion perturbs the normal progression and fine-tuning of the balance of pluripotency and lineage commitment genes, which in turn disrupts stem cell fate and the proper pace of differentiation.
More importantly, although Mettl3 heterozygous mice are viable, Mettl3 homozygous embryos have impaired development and suffer prenatal lethality [41]. Mutations in morphology were observed at postimplantation embryonic day 5.5 (E5.5–E7.5) and embryonic absorptions of Mettl3 KO embryos were detected at E8.5–E10.5 [41]. Consistent with in vitro observations, pluripotency factors were not repressed at the proper time in Mettl3 KO embryos. There was also a failure to upregulate key lineage commitment markers like Foxa2, Brachyury, and Blimp1, which mark the adequate development of endoderm, mesoderm, and primordial germ cells, respectively [41].
Notably, it was recently demonstrated that METTL3 plays a critical role in adult cell reprogramming to pluripotency [38,39]. Morphological and quantitative analyses by Chen et al. revealed that overexpression of human Myc-METTL3 successfully boosted iPSC colony numbers with an obvious increase in the mRNA level of three significant pluripotent factors: Oct4, Sox2, and Nanog [39]. On the other hand, depletion of Mettl3 with siRNA resulted in an inefficient reprogramming that could be partially rescued by the overexpression of human METTL3. These results suggest that METTL3 facilitates somatic cell reprogramming to pluripotency by upregulating m6A levels [39]. However, Aguilo et al. reported that METTL3 and m6A levels were inversely related to iPSC generation [38]. Their study showed that Zfp217 antagonizes the RNA methyltransferase activity of Mettl3 in mESCs by hampering its ability to bind to its target RNAs. The increase in m6A levels upon Zfp217 depletion destabilizes the mRNA of key pluripotency factors and leads to a sharp reduction in iPSC colonies [38]. More importantly, concomitant lentiviral infection of MEFs with Mettl3 small hairpin RNA (shRNA) partially rescues iPSC reprogramming in these cells. This suggests that m6A is a barrier for somatic cell reprogramming [38]. Interestingly, Geula et al. found that Mettl3 mainly ensures sufficient proliferation of somatic cells during the early reprogramming stage. Meanwhile, Mettl3 depletion exerts less adverse effects on iPSC generation as reprogramming proceeds into later stages after the induction of pluripotent factors [41]. Further experiments should resolve the current discrepancy and provide more understanding of the explicit underlying mechanisms.
METTL14
METTL14 shares 43% homology with METTL3 [91]. A cellular defect in METTL14 caused ∼40% and ∼35% downregulation of m6A levels in polyadenylated RNA in HeLa cells and human 293FT cells, respectively, thus exhibiting a higher methyltransferase activity than METTL3 [60]. METTL3 and METTL14 proteins form a heterodimer core complex with a stoichiometry of 1:1 [60]. METTL3 and METTL14 appear to work synergistically in this complex since it has an even larger effect on m6A levels than either METTL3 or METTL14 alone [37]. Western blot analysis also revealed a possible reciprocal stabilization of METTL3 and METTL14 [37]. RNA probes were used to show that the heterodimer complex has a preference for RNA substrates that are less structured and contain a consensus motif of GGACU [60]. Immunofluorescene analysis located the METTL3-METTL14 complex at nuclear speckles along with diverse pre-mRNA processing factors [60]. The depletion of either METTL3 or METTL14 could disrupt the nuclear speckle localization of the other [70].
Studies have indicated that the effects of METTL14 depletion on ESC differentiation were similar to those of METTL3 depletion [37,41]. Mettl14 KO mESCs were incapable of timely downregulation of the pluripotency regulators necessary to prepare for an advanced developmental stage. Mettl14 KO mESCs also recapitulated the in vitro differentiation block seen in Mettl3 KO mESCs [41]. Notably, liquid chromatography-tandem mass spectrometry analysis and immunoprecipitation showed that METTL14, unlike METTL3, does not interact with ZFP217 [38]. This difference implies a functional variation between METTL14 and METTL3 that is likely caused by their structural difference.
WTAP
WTAP is the third known factor in the mammalian METTL3-METTL14 methyltransferase complex [60,70]. WTAP was first regarded as a potential splicing factor due to its nuclear speckle colocalization [92]. A reduction in WTAP resulted in perturbed cell cycle progression [93,94] and led to embryonic lethality [95]. It has been discovered that WTAP binds with the METTL3-METTL14 complex and has an indispensable role in m6A methylation [60,70,77]. In fact, the knockdown of WTAP resulted in an even more pronounced reduction in m6A levels than either METTL3 or METTL14 alone [60]. WTAP lacks catalytic domains and thus does not exhibit any methyltransferase activity of its own [60]. However, silencing WTAP led to an obvious failure of METTL3 and METTL14 accumulation in nuclear speckles. This led to the conclusion that WTAP mainly functions to enable the proper nuclear speckle colocalization of the METTL3-METTL14 heterodimer [70]. The three proteins of the METTL3-METTL14-WTAP complex share ∼36% binding sites in common and ∼43%–49% of these sites fell into intergenic regions [60]. The majority of target genes of METTL3 and WTAP proteins were associated with transcription and RNA splicing, and originated from multi-isoform genes that exhibited an increased diversity in isoform numbers upon METTL3 or WTAP depletion. This demonstrates the significant effect of m6A methylation on RNA processing [70].
Research has shown that WTAP is essential for mESC differentiation, especially the differentiation of endoderm and mesoderm [95]. Wtap-mutant mESCs failed to express the key endoderm marker Foxa2 or mesoderm marker Brachyury, exhibited aberrant morphology at the gastrulation stage, and experienced early embryonic lethality [95]. A chimera of Wtap-mutant mESCs and wild-type eight-cell-stage embryos successfully rescued the impaired differentiation and indicated that the presence of Wtap in extraembryonic tissues is essential for proper endoderm and mesoderm differentiation [95]. Another study found that Wtap and Mettl3 are extensively expressed during the early embryogenesis of zebrafish. It was also shown that the Wtap knockdown leads to a more severe embryo deformity than the Mettl3 knockdown [70], suggesting a greater role for WTAP in regulating stem cell differentiation.
m6A Demethylases
Two m6A RNA modification demethylases have been identified, FTO and ALKBH5 [80,86]. These factors provide a reverse pathway for this most abundant RNA modification mechanism and create a dynamic regulation of gene expression. The discovery of demethylases has broadened our knowledge of the diverse biological functions and molecular mechanisms of m6A RNA modification and holds great promise for clinical applications.
FTO
FTO was first detected in a fused toes (Ft) mouse mutation experiment [96]. It is widely expressed during embryonic development and is also highly expressed in the brain of adult mice, especially in the hypothalamus [96 –98]. Studies have demonstrated potential connections between FTO and several health problems and diseases associated with fat metabolism and the brain, such as obesity risk, cardiovascular disease, depression, Alzheimer's disease, and reduced cognitive ability in the elderly [99 –106].
FTO belongs to the AlkB family of non-heme Fe(II)/2-oxoglutarate -dependent dioxygenases [97,107,108]. FTO exhibits Fe(II)- and 2OG-dependent DNA demethylation activity with a preference for single-stranded DNA 3-methylthymine (3-mT) substrates. However, it also acts on 3-methyluracil (3-mU) in single-stranded RNA [97,109]. This suggested that FTO is a potential nucleic acid demethylase. A crystal structure study of FTO discovered a long loop structure that facilitated the binding of FTO to methylated strands of double-stranded DNA (dsDNA)/RNA. This may explain its substrate preference for single-stranded nucleic acids [110].
Jia et al. reported that FTO is capable of demethylating m6A marks in nuclear RNA [86]. Overexpression of FTO significantly decreased m6A levels, while depletion of FTO by siRNA resulted in a 23% and 42% upregulation of cellular m6A levels in HeLa and 293FT cells, respectively [86]. This finding was verified in later studies using different detection techniques [43,55,73,84]. FTO was also found to be involved in heat shock response [73]. The increased binding of YTHDF2 to m6A sites of stress-inducible transcripts hampered FTO from removing m6A marks in the 5′ UTR. This consequently led to the initiation of selective mRNA translation under heat shock stress [73].
Weight loss in Fto knockdown mice has been observed in previous studies [111,112] and reveals a potential connection between FTO and fat tissue accumulation. Recently, FTO was found to downregulate m6A mRNA modification and is thus highly necessary for preadipocyte differentiation [43,44]. Research by Zhao et al. showed that the knockdown of FTO in preadipocytes significantly inhibited adipogenic differentiation [43]. FTO depletion increased m6A modification and boosted the binding ability of serine/arginine-rich splicing factor 2 (SRSF2) at its target RNA sites. This included the exonic splicing enhancer region, which shares a spatial overlap with m6A, thus promoting the inclusion of alternative exons [43,113]. FTO also affected the alternative splicing of a key differentiation regulator, Runt-related transcription factor 1 (RUNX1T1), during adipogenesis by catalyzing m6A demethylation [43]. RUNX1T1 has two isoforms, a constitutive isoform RUNX1T1-L, which hampers preadipocyte differentiation, and an isoform RUNX1T1-S without exon 6, which promotes differentiation. FTO-depleted preadipocytes exhibited a global upregulation of m6A level and a decrease in the level of RUNX1T1-L due to the increased binding of SRSF2 at the splicing site. Thus, the expression of the differentiation-promoting isoform RUNX1T1-S was severely inhibited, leading to impaired adipogenesis [43]. Zhang et al. discovered that the impaired differentiation observed in Fto demethylase-impaired preadipocytes could be rescued by a peroxisome proliferator-activated receptor γ (PPARγ) agonist. This suggests that the effect of Fto is mediated through PPARγ, which is a master regulator of adipogenesis [44].
ALKBH5
ALKBH5, like FTO, is a member of the AlkB subfamily of the Fe(II)/2-oxoglutarate-dependent dioxygenase superfamily [114]. ALKBH5 has been linked with hypoxia [114] and presumed to be an mRNA-binding protein [115]. Many members of AlkB family have been shown to exhibit a nucleic acid demethylation activity in DNA and RNA [108,116 –120]. ALKBH5 has been recently identified as a mammalian m6A demethylase [80].
ALKBH5 can efficiently demethylate m6A with enzymatic kinetics similar to FTO [80]. It decreases m6A levels in single-stranded nucleic acids much more effectively than in double-stranded or looped substrate due to its structural properties [80]. ALKBH5 is devoid of the structure necessary for binding double-stranded nucleic acids and possesses a partial region in a large loop, which has a steric clash with the complementary strand. These features cause ALKBH5 to selectively bind to single-strand methylated nucleic acids [121 –123].
ALKBH5 was found to be largely confined to nuclear speckles and play an instrumental role in modulating the modification of many mRNA-processing factors [80,114]. ALKBH5 mediates m6A mRNA demethylation and exhibits an impact on mRNA nuclear export [80]. Depletion of ALKBH5 alters the phosphorylation status of alternative splicing factor/splicing factor 2 (ASF/SF2) and promotes the interaction between mRNA and the TAP-p15 complex. This complex is responsible for mRNA export; so loss of ALKBH5 accelerates nuclear RNA export [80]. Alkbh5 has its highest expression in testes, especially in primary spermatocytes [80]. Not surprisingly, Alkbh5-deficient mice have abnormal testes, compromised spermatozoa, and halted spermatogenesis due to significant alteration in mRNA processing of important spermatogenesis-associated genes, which was triggered by increased m6A levels. In addition, mRNA in p53 signaling pathways were affected and this might account for cellular apoptosis and impaired fertility observed in Alkbh5-knockdown mice [80]. Further investigations are required to resolve the possible connection between the demethylation activity of ALKBH5 and stem cell regulation and to shed light on the more detailed mechanisms of the biological function of this newly discovered m6A demethylase.
The Involvement of m6A Readers in Stem Cell Regulation
m6A “readers” recognize m6A marks in RNA and are crucial for numerous cellular RNA processes that define cell state and safeguard proper cell development, such as mRNA stability [82], alternative splicing [124], and translational efficiency [125]. The YT521-B homology (YTH)-domain protein family, which comprises YTH domain family protein 1–3 (YTHDF1-3) and YTH domain containing protein 1–2 (YTHDC1-2), is a group of relatively well-characterized m6A readers. YTHDF1 promotes both ribosome occupancy and the translation initiation rate of m6A-modified mRNA to enhance mRNA translation efficiency [125]. YTHDF2, on the other hand, mediates mRNA degradation by recruiting m6A-modified mRNA to decay sites [82]. Wang et al. suggest that YTHDF1 and YTHDF2 might be coupled to ensure a timely response to cell development and differentiation cues and to provide the developing cell with sufficient protein productions [125]. Widespread m6A modification has been detected in many pluripotent and lineage commitment transcripts in ESCs. This suggests that m6A readers may be involved in the fine-tuning of the expression of various genes essential for cell fate transition by affecting RNA metabolism, such as transcript stability [37,41], and thereby contribute to proper and efficient stem cell pluripotency and differentiation.
Perspectives
Great progress has been made in understanding m6A RNA posttranscriptional modification in recent years. The identification of a succession of novel m6A methyltransferases [60,70] and demethylases [80,86] has revealed a new epigenetic regulatory mechanism for stem cell pluripotency and differentiation [36 –43]. However, the field is still in its infancy since studies related to this topic are small in number. Also, there have been discrepancies in some of the results. The exact regulatory functions and underlying mechanisms are largely unclear; so continuing studies are necessary to improve our understanding. For instance, Wtap was shown to affect the differentiation of mESCs into endoderm and mesoderm [95]. However, whether or not ectopic differentiation due to Wtap loss-of-function was a result of reduced m6A methylation requires more solid evidence. In addition, more in vivo studies are needed to confirm the extent to which the in vitro observations correlate with in vivo development.
Current studies have been mainly directed to the regulatory role of m6A writers and erasers in the differentiation of ESCs. However, their actions in adult stem cells, which are responsible for the development of various tissues, have been poorly studied. This critical gap in our knowledge requires more research that includes the possible association between dynamic m6A modification and adult stem cell differentiation. The preferential expression of m6A writers and erasers in certain tissues suggests an avenue of research to determine how they are associated with the development of those specific tissues and if any observed effects are accomplished by reversible m6A RNA modification. For example, FTO has been found to be highly expressed in the brain as well as both white and brown adipose tissue [97,98]. Experiments have demonstrated an Fto-dependent preadipocyte differentiation [43]. Fto-deficient mice suffered a high risk of postnatal lethality and retarded growth [98,111,112,126] and this was associated with neural regulation [98]. An upregulation of m6A was detected by immunoblotting throughout the neuronal developmental process [55]. Further studies may resolve exactly how the m6A demethylase activity of FTO is associated with neuronal development and NSC differentiation. The most recent study involving Mettl3 demonstrated that it is closely associated with the regulation of ESC pluripotency and Mettl3 KO mice were embryonic lethal [41]. This exciting finding should be followed by developing tissue-specific Mettl3 KO mice to investigate potential biological functions of Mettl3 beyond the embryonic developmental stage.
Moreover, the role of METTL3 in somatic cell reprogramming is still very perplexing and needs further study [38,39,41]. iPSCs have great importance and promise in the future of regenerative medicine and disease treatment. The exact functions and mechanisms of m6A RNA modification in cell reprogramming are therefore of high clinical value and certainly worth continued investigation.
Footnotes
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (NSFC 81371173, 81321002) to Q.Y.
Author Disclosure Statement
No competing financial interests exist.