
Abstract
Bone marrow mesenchymal stromal cells (BM-MSCs) have been demonstrated to contribute to tissue regeneration. However, chronic pathological conditions, such as diabetes and aging, can result in a decreased number and/or quality of BM-MSCs. We therefore investigated the maintenance mechanism of BM-MSCs by studying signaling through the receptor for advanced glycation end products (RAGE), which is thought to be activated under various pathological conditions. The abundance of endogenous BM-MSCs decreased in a type 2 diabetes mellitus (DM2) model, as determined by performing colony-forming unit (CFU) assays. Flow cytometric analysis revealed that the prevalence of the Lin−/ckit−/CD106+/CD44− BM population, which was previously identified as a slow-cycling BM-MSC population, also decreased. Furthermore, in a streptozotocin-induced type 1 DM model (DM1), the CFUs of fibroblasts and the prevalence of the Lin−/ckit−/CD106+/CD44− BM population also significantly decreased. BM-MSCs in RAGE knockout (KO) mice were resistant to such reduction induced by streptozotocin treatment, suggesting that chronic RAGE signaling worsened the maintenance mechanism of BM-MSCs. Using an in vitro culture condition, BM-MSCs from RAGE-KO mice showed less proliferation and expressed significantly more Nanog and Oct-4, which are key factors in multipotency, than did wild-type BM-MSCs. Furthermore, RAGE-KO BM-MSCs showed a greater capacity for differentiation into mesenchymal lineages, such as adipocytes and osteocytes. These data suggested that RAGE signaling inhibition is useful for maintaining BM-MSCs in vitro. Together, our findings indicated that perturbation of BM-MSCs in DM could be partially explained by chronic RAGE signaling and that targeting the RAGE signaling pathway is a viable approach for maintaining BM-MSCs under chronic pathological conditions.
Introduction
B
However, BM-MSCs become senescent and lose their ability to self-renew and differentiate after a long period in culture [8,9]. In general, BM-MSCs are in a quiescent state in the human body under physiological conditions [10]. Late-passage MSCs or MSCs with retinoblastoma (RB) protein knockdown showed a loss in the proliferation rate and differentiation potential, with premature senescence characteristics [11]. Considering that RB is thought to negatively control cell cycle progression, MSCs with reduced RB expression might leave the quiescent state, quickly undergo cell cycle progression, and then become exhausted in the senescent state after a few passages in culture. Based on these possibilities, the forced in vitro activation of BM-MSCs in culture may account for the loss of their properties.
Furthermore, BM-MSCs from scleroderma patients [12] and diabetic mice [13] showed abnormal characteristics and functions, which were probably related to changes in their respective niches under these pathological conditions. In addition, we previously reported that the number of BM-MSCs in Duchenne muscular dystrophy model mice gradually reduced as the pathology progressed [14]. In multiple sclerosis model mice, interferon-gamma from activated T cells changed the number of MSCs in the BM [15]. In addition, we previously demonstrated that necrotic skin-derived, high-mobility group box 1 (HMGB1) could mobilize platelet-derived growth factor receptor alpha-positive (PDGFRα+) mesenchymal cells from the BM into the circulation [16] and promote their accumulation at SDF-1α-positive inflammatory tissues [17]. These data suggested that chronic inflammatory stimuli under pathological conditions changed the abundance of BM-MSCs as well as their properties, for example, the ability of self-renewal (colony-forming capacity) and their abilities to differentiate into multiple tissue types (referred to as stemness) such as bone and fat. Understanding the molecular cue(s) that induce the loss of BM-MSC stemness in in vivo and in vitro settings enables the translation from the bench to clinical applications.
Previous data showed that in animal models of diabetes, deletion of the receptor for advanced glycation end products (RAGE) or administration of a blocking antibody against RAGE successfully attenuated the development of atherosclerosis [18,19]. In addition, it has been demonstrated that inhibiting RAGE signaling alleviated diabetic nephropathy [20,21]. RAGE is a multiligand receptor of the immunoglobulin superfamily that binds inflammatory mediators, including S100 calgranulin (S100A)8/9, HMGB1, and advanced glycation end products (AGEs). Thus, attenuating the progression of various pathological conditions by inactivating RAGE signaling has been attributed to the suppression of inflammatory signaling, including that mediated by activation of nuclear factor-kappa B (NF-κB) and p38 mitogen-activated protein kinase [22,23]. RAGE signaling also elicits oxidative stress through generation of ROS through an NADPH oxidation-dependent mechanism [18,24].
It has been reported that MSCs from diabetic mice exhibited impaired survival, proliferation, and differentiation [25,26]. However, limited reports have described the effect of AGEs on the function of MSCs [27], and the association of RAGE signaling with MSC function remains incompletely understood. Therefore, we investigated whether chronic RAGE signaling deteriorates BM-MSC function under diabetic conditions and whether the loss of RAGE signaling protects MSC functions against pathological changes, such as the depletion and impaired differentiation abilities caused by diabetic stress.
The aim of the present study was to assess the effects of RAGE deficiency on BM-MSCs in a chronic diabetes model and under in vitro culture conditions. We demonstrated that loss of the colony-forming capacity of BM-MSCs in diabetic mice was ameliorated in RAGE knockout (RAGE-KO) mice. We also found that RAGE-KO BM-MSCs have great capacity for multidifferentiation into different mesenchymal lineages in vitro. Our findings provide a possible molecular target for inhibiting the reduction in BM-MSC abundance in diabetes models, and our results represent an advanced strategy for preparing MSCs with maintained stemness in long-term culture for clinical applications.
Materials and Methods
Mice
All animals were handled according to approved protocols and the guidelines of the Animal Committee of Osaka University. All experimental mice were housed in a cage with 12-h light–12-h dark cycles. Solid food and water were supplied ad libitum. The temperature and humidity in the animal room were maintained at ∼23°C and ∼55%, respectively. C57BL/6, Leprdb +/+ Dock7m-J (db/db) mice were purchased from CLEA Japan. RAGE-null (RAGE-KO) mice were kindly provided by Hiroshi Yamamoto (Kanazawa University, Japan).
Streptozotocin treatment
Type 1 diabetes mellitus (DM1) was induced in 4–5-week-old WT or RAGE-KO mice by intraperitoneal (i.p.) injection with streptozotocin (STZ; 100 mg/kg; Sigma) each day for two consecutive days. STZ was dissolved in a citrate acid buffer solution (0.05 M, pH 4.5) just before injection. Blood glucose levels were examined 7 weeks after the first STZ injection using a glucotest (Sanwa Kagaku Kenkyusho Co., Ltd.) after obtaining blood from the tail vein. Control mice were injected with buffer alone.
Isolation of bone marrow cells
Isolation of bone marrow cells (BMCs) was performed as previously described [16,28]. Briefly, under sterile conditions, BMCs were isolated by flushing the femurs and tibiae with phosphate-buffered saline containing 2% fetal bovine serum (FBS/PBS), and the resulting suspension was then filtered through 40-μm cell strainers. The filtrate was centrifuged at 4°C for 10 min at 1,500 rpm, and the pellet was used for fluorescence-activated cell sorting (FACS) analysis and primary cell culture.
Primary cell culture
Isolated primary BMCs were cultured in α-MEM (Life Technologies) containing 20% FBS and antibiotics at 37°C under a hypoxic condition (5% O2). BMCs adhering to the plastic dishes were considered to be BM-MSCs. BM-MSCs at passage 0 were grown for ∼1 week (until colonies were visible) and then harvested by incubation with 0.25% trypsin/EDTA at 37°C for 10 min. The harvested BM-MSCs at passage 0 were replated and maintained to 70%–80% confluence up through passage 3 (P3). The medium was changed every 3 days.
Adipocyte, osteocyte, and chondrocyte lineage differentiation assay
Adipocyte and osteocyte differentiation assays were performed with culture-expanded BM-MSCs at P3. BM-MSCs were seeded at 5 × 104 cells/well in a 12-well plate or 1 × 105/well in a 6-well plate on the day before inducing adipocyte or osteocyte differentiation. The Mesenchymal Stem Cell Adipogenesis Kit and Mesenchymal Stem Cell Osteogenesis Kit (Chemicon International) were used for adipocyte and osteocyte differentiation. To verify differentiation into the adipocyte lineage, Oil Red O staining was performed using the Lipid Assay Kit (Cosmo Bio Co. Ltd.), according to the manufacturer's instructions. Accumulation of Oil Red O was recorded by reading the absorbance at 540 nm on a microplate reader. To determine osteocyte differentiation, alkaline phosphatase (ALP) staining was performed using the ALP Staining Kit (Cosmo Bio Co., Ltd.), according to manufacturer's instructions. The cells stained with Oil Red O or ALP were visualized by light microscopy using a DMI4000B microscope (Leica).
FACS analysis of BMCs
Freshly isolated BMCs (2 × 106 cells) were suspended in 200 μL of staining buffer (2% FBS/PBS), followed by incubation with a purified rat anti-mouse CD16/CD32 antibody (Mouse BD Fc Block™; BD Pharmingen) for 10 min at 4°C. The cells were incubated with the following fluorophore-conjugated antibodies for 30 min at 4°C: anti-mouse/human CD44 PE-Cy7 (clone IM7; BioLegend), anti-mouse CD106 PE [clone 429 (MVCAM.A); BioLegend], anti-mouse lineage cocktail with isotype control APC (BD Pharmingen), anti-mouse CD117 (c-kit) FITC (clone 2B8; BioLegend), anti-mouse c-kit APC (clone 2B8; eBioscience), and anti-mouse/rat Ki67 FITC (clone SolA15; eBioscience). As matching isotype controls, fluorescence-conjugated anti-mouse IgG antibodies obtained from either BioLegend or eBioscience were used in each experiment. Stained cells were then analyzed on a BD FACSCanto™ II flow cytometer (BD Biosciences). Gates were defined based on staining with isotype-matched negative control antibodies. FACS data were analyzed using FlowJo software, version 6.3.3 (Tree Star, Inc.).
RNA extraction and real-time polymerase chain reaction
Total RNA was extracted using ISOGEN (Nippon Gene), according to the manufacturer's instructions. cDNA synthesis was performed using 2 μg of total RNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems). Real-time polymerase chain reaction (PCR) was performed with SYBR Premix EX Taq (TaKaRa), using oligonucleotide primers. The sequences of the oligonucleotide primers were designed with the Universal Probe Library Assay Design Center (Roche). The sequences of the oligonucleotide primers used in this study are listed in Table 1. Quantitative data were obtained in triplicate within a single experiment on a 384-well plate, based on the standard curve method, using CFX Manager software (BioRad). All data were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression, which served as an internal control. Data were expressed as the fold increase relative to GAPDH expression.
ALP, alkaline phosphatase.
Statistical analyses and microscopy
Values are expressed as the mean ± standard error of the mean (SEM). Statistical analyses were performed by conducting Student's t-test. For all statistical tests, statistical significance was evaluated at P < 0.05.
Results
Reduction of the number of BM-MSCs and an increased proportion of proliferating BM-MSCs in DM2 model mice
To determine the influence of diabetes on BM-MSCs, the numbers of BM-MSCs in db/db mice and WT mice were analyzed. In db/db mice, the number of colony-forming cells in the BM showed a tendency to decrease compared with that observed in WT mice (Fig. 1A), although this change was not significant. FACS analysis revealed that the prevalence of the Lin−/ckit−/CD106+/CD44− population (CD44− cells), which was previously shown to be a quiescent MSC population [14], was significantly lower in db/db mice compared with that observed in WT mice (Fig. 1B). In addition, the percentage of Ki67+ cells in the CD44− cell population increased in db/db mice (Fig. 1C), suggesting that diabetic conditions promoted the proliferation of CD44− cells. These data suggested that proliferation of the quiescent BM-MSC population was driven by diabetic conditions; however, chronic pathological conditions consequently reduced the total number and frequency of BM-MSCs.
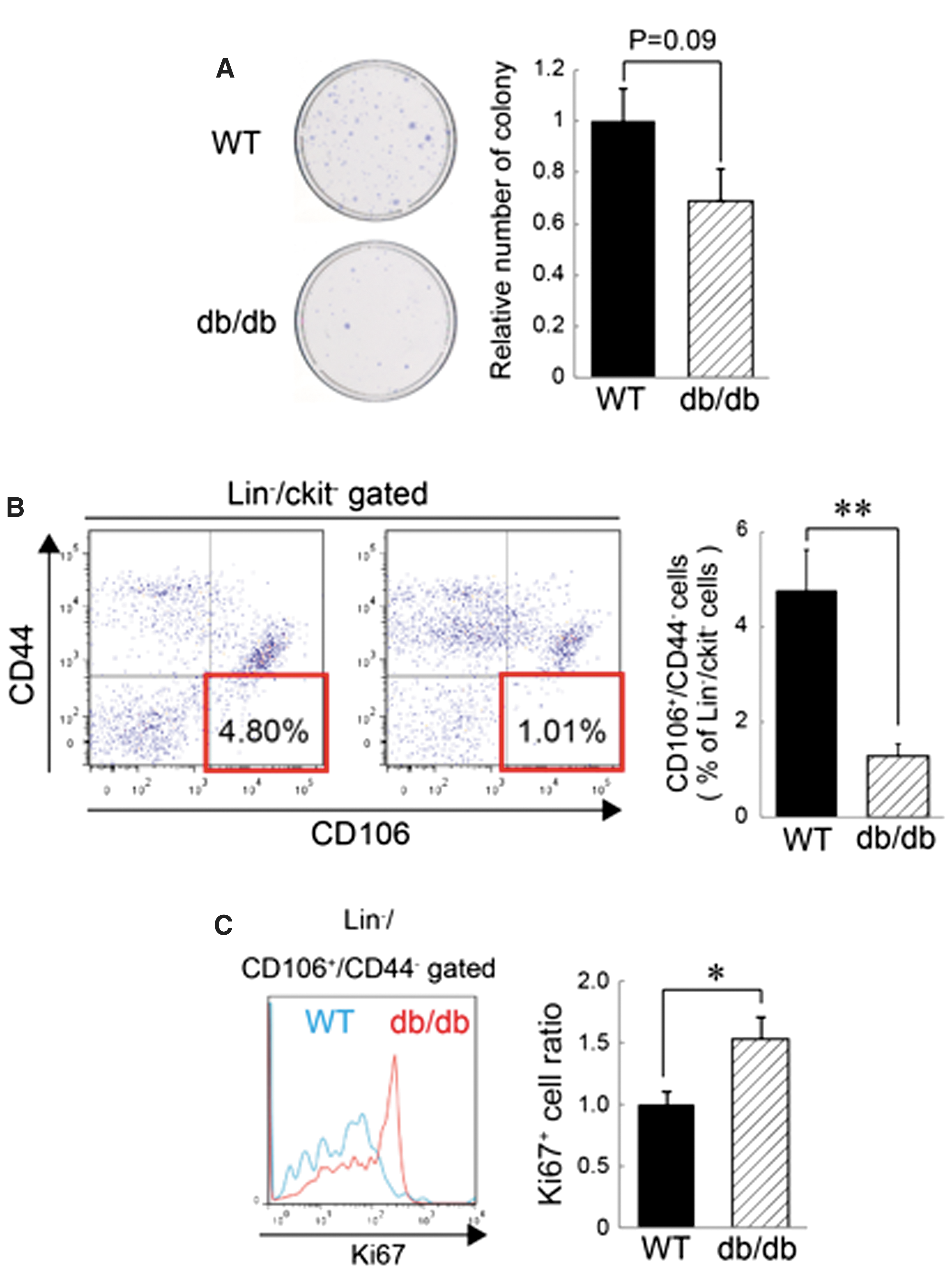
Reduction of the number and frequency of BM-MSCs in type 2 diabetes mellitus (DM2) model mice.
RAGE-KO mice were resistant to the reduction of BM-MSCs in type 1 diabetes model mice
Next, to investigate whether RAGE signaling contributes to pathological changes of BM-MSCs in DM model mice, we induced DM1 in RAGE-KO mice by STZ injection. We confirmed the absence of RAGE expression in RAGE-KO mice by real-time PCR (Fig. 2A), indicating that RAGE expression was completely absent in this model.
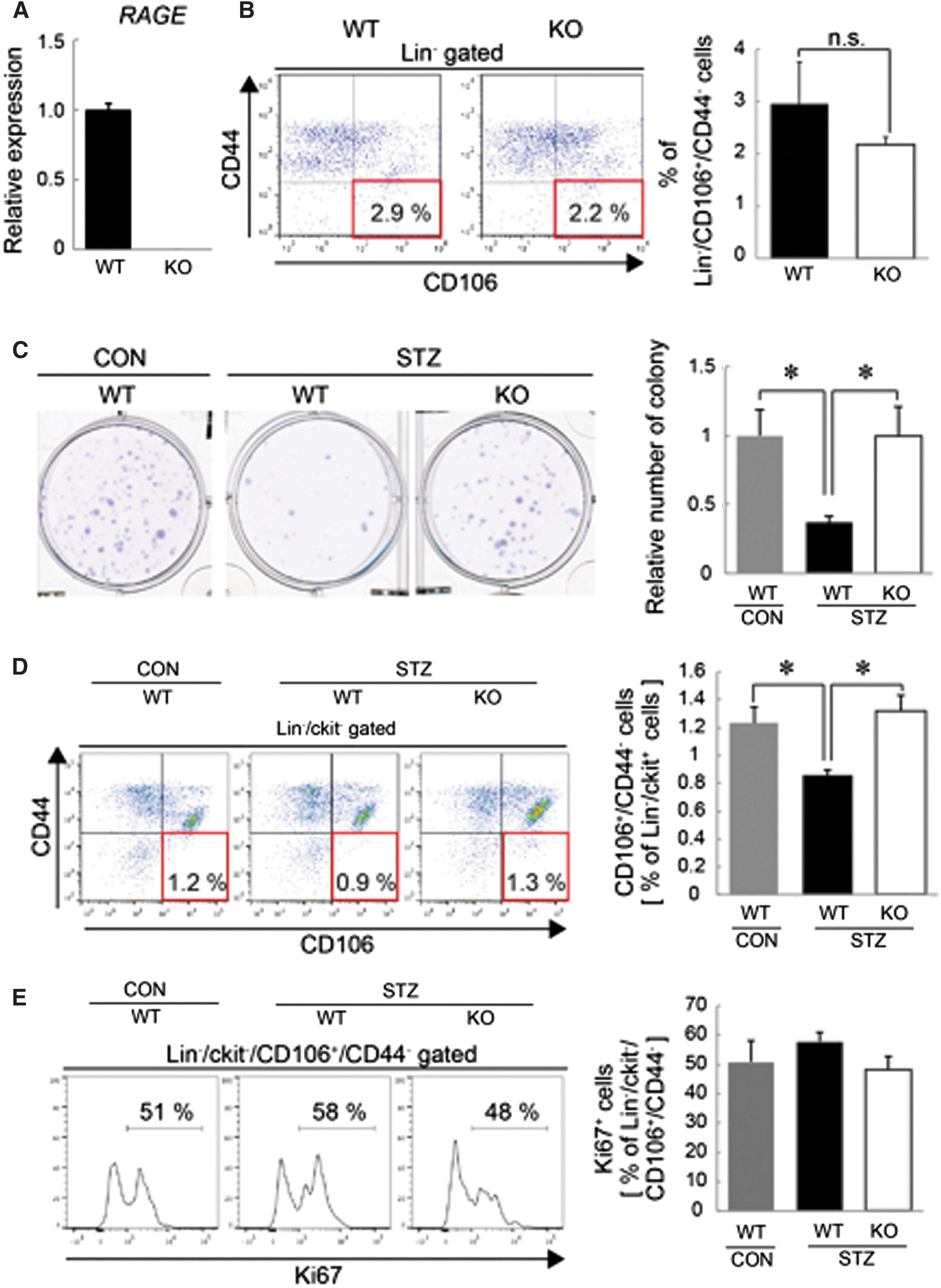
RAGE-KO mice are resistant to the reduction of BM-MSCs in an STZ-induced DM1 model.
First, no significant difference in the frequency of the Lin−/CD106+/CD44− BMC population was observed between WT mice and RAGE-KO mice under physiological conditions (Fig. 2B). To induce DM1, we intraperitoneally injected STZ into WT or RAGE-KO mice. The injection of STZ successfully increased blood glucose levels from 178.5 ± 5.0 to 594.5 ± 3.4 mg/dL in WT mice after 7 weeks. The average blood glucose level after STZ injection was not significantly different between WT and RAGE-KO mice (571.0 ± 18.8 mg/dL), suggesting that STZ injection induced DM1 in RAGE-KO mice as well. We then analyzed RAGE expression in cultured BM-MSCs isolated from mice 7 weeks after STZ injection and db/db mice, and showed no significant upregulation of RAGE (Supplementary Fig. S1A, B; Supplementary Data are available online at
BM-MSCs from RAGE-KO mice are resistant to proliferation under growth condition in vitro
Because the number and frequency of BM-MSCs in RAGE-KO mice were abated under the STZ-induced diabetic condition, we next investigated whether the RAGE signaling also plays a key role in the maintenance of BM-MSCs in vitro. Previous data have shown that pluripotency-related genes, such as Nanog and Oct4, are expressed in MSCs and regulate their multipotency [8,29]; therefore, the expression of these genes in WT and RAGE-KO BM-MSCs expanded in vitro at P3 was analyzed by real-time PCR. BM-MSCs derived from RAGE-KO mice expressed significantly more Nanog and Oct4 mRNA compared with WT BM-MSCs (Fig. 3A).
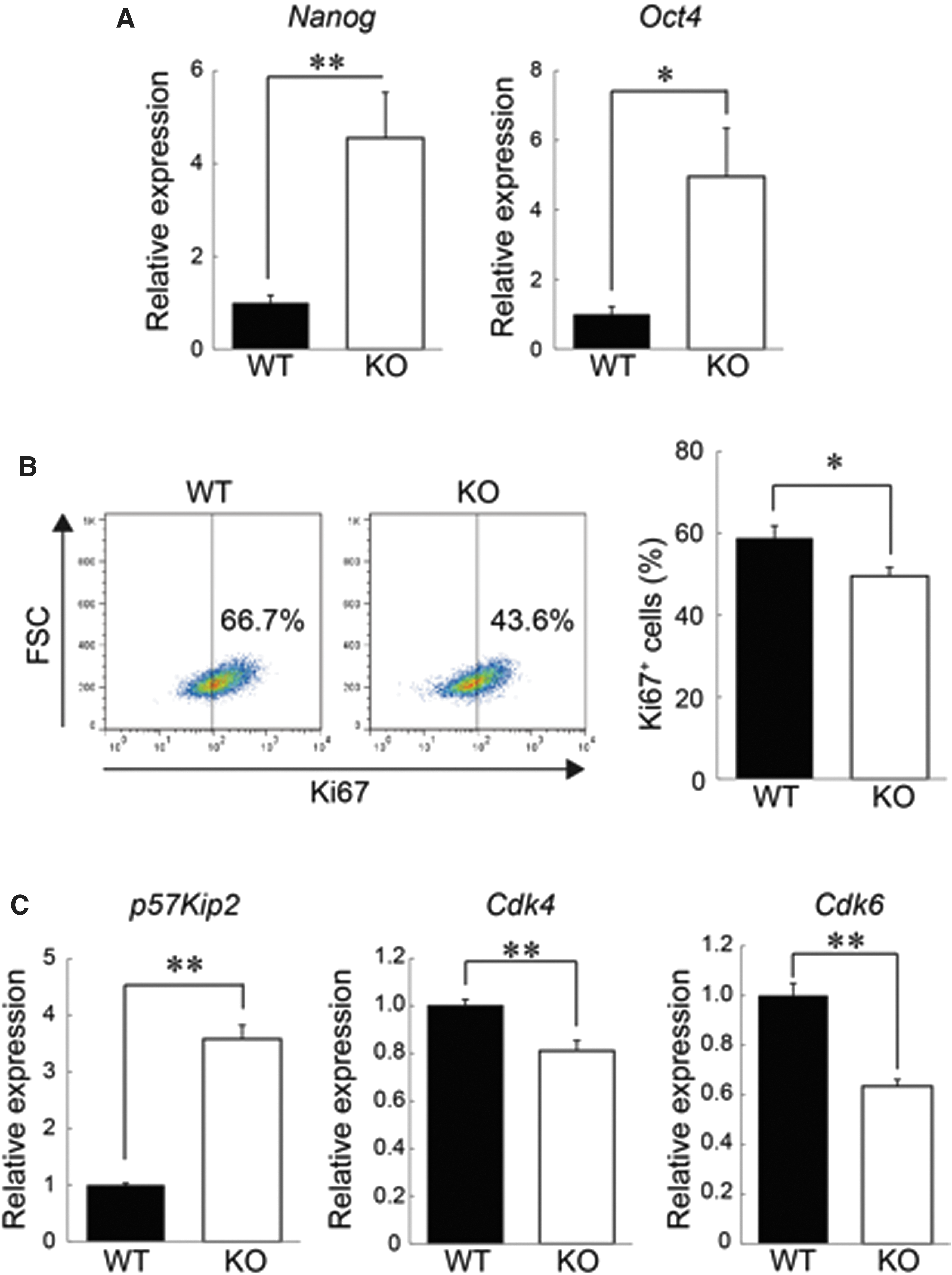
RAGE-KO BM-MSCs showed higher expression of pluripotency genes and slowed cell cycle progression.
Furthermore, flow cytometry analysis revealed that RAGE-KO BM-MSCs have fewer Ki67+ cells than do WT BM-MSCs (Fig. 3B). In addition, RAGE-KO BM-MSCs showed higher expression of p57 [30,31], which is a cell cycle progression inhibitor (Fig. 3C). Instead, the expression levels of cyclin-dependent kinase (Cdk) 6 and Cdk4, which promote cell cycle progression, were lower in RAGE-KO BM-MSCs (Fig. 3C). We also analyzed the expression of Nanog and cell cycle-related genes in cultured BM-MSCs from db/db mice [type 2 diabetes mellitus (DM2) model] because the DM1 and DM2 conditions affected the number of BM-MSCs in vivo (Fig. 2C, D). The cultured BM-MSCs from db/db mice showed lower expression of Nanog and p57 (but not Cdk6 and Cdk4) than did WT BM-MSCs (Supplementary Fig. S2). Together, these data suggested that even under in vitro culture conditions, knocking out RAGE modulates the expression of the multipotency-associated genes, Nanog and Oct4, as well as the proliferative state of cultured BM-MSCs.
BM-MSCs from RAGE-KO mice sustain stemness in vitro
In addition to assessing the expression of multipotency-associated genes and the cell-cycling state of cultured BM-MSCs, their multidifferentiation abilities were also investigated in RAGE-KO BM-MSCs. As shown in Fig. 4A, more Oil Red O-stained cells were observed in RAGE-KO BM-MSCs compared with that found with WT BM-MSCs after the induction of adipocyte differentiation. The quantification of Oil Red O fluorescence confirmed the statistical significance of the increased Oil Red O staining in RAGE-KO BM-MSCs (Fig. 4A). Gene expression levels of adipocyte markers, including fatty acid-binding protein 4 (FABP-4), peroxisome proliferator-activated receptor gamma (PPARγ), and CCAAT/enhancer-binding protein alpha (C/EBPα), were higher in RAGE-KO BM-MSCs than in WT BM-MSCs (Fig. 4B). Next, BM-MSCs were differentiated into osteocytes, showing that RAGE-KO BM-MSCs were more strongly stained for ALP expression than were WT BM-MSCs (Fig. 4C). The expression levels of the osteocyte markers, ALP, osterix, osteocalcin, Runx2, BMP2, and BMP4, were higher in RAGE-KO BM-MSCs than in WT BM-MSCs (Fig. 4D). These results indicated that RAGE-KO BM-MSCs have higher differentiation potencies than WT BM-MSCs, even in culture, as indicated by increased Nanog and Oct4 expression, and maintain multipotency to differentiate into mesenchymal lineages such as adipocytes and osteocytes. Together, these data suggested that the inhibition of RAGE signaling maintains stemness under in vitro culture conditions.
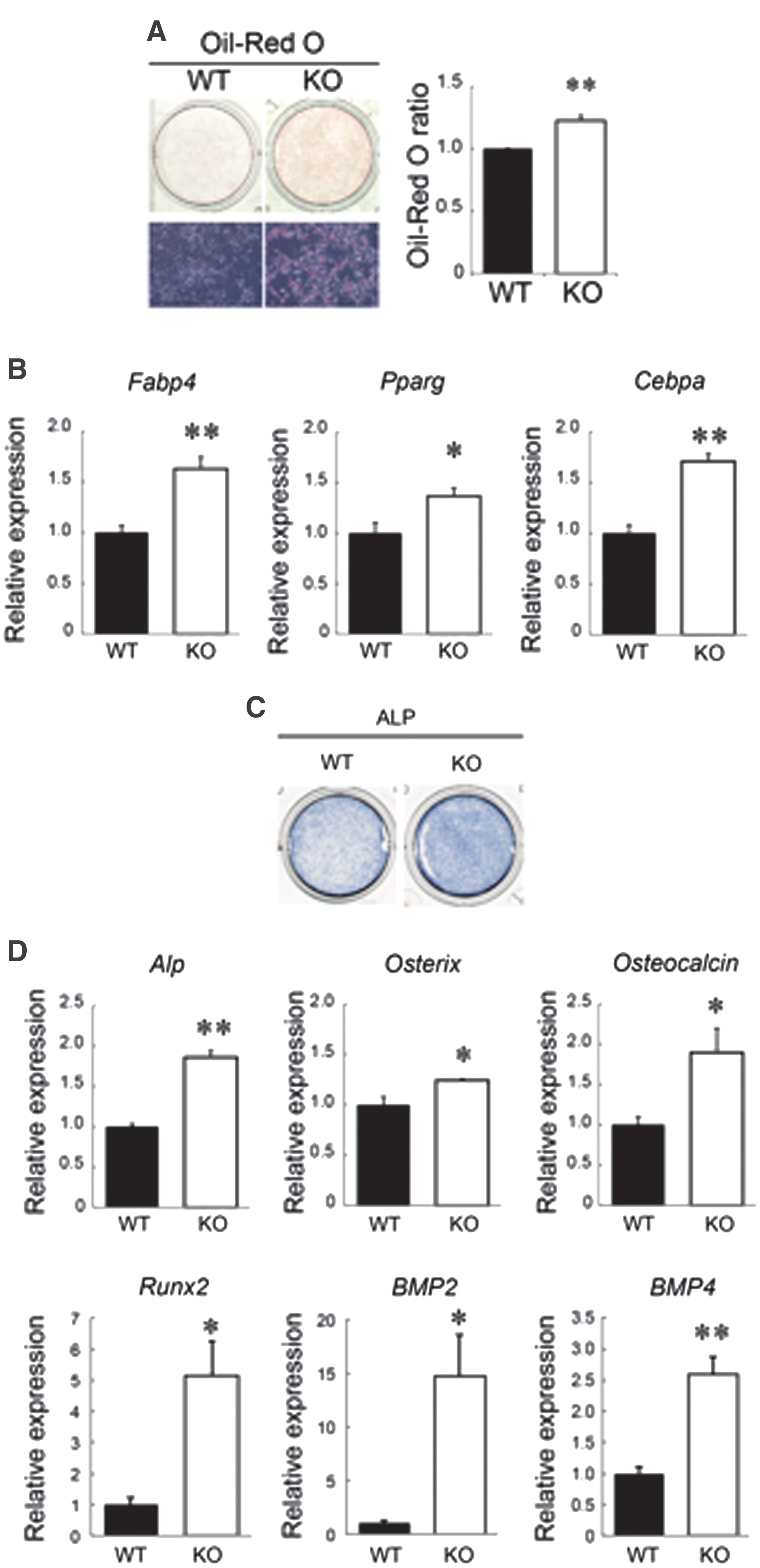
BM-MSCs derived from RAGE-KO mice sustained higher multidifferentiation potency.
Discussion
In this study, we demonstrated that the frequency of BM-MSCs and the number of colony-forming cells in the BM decreased in both DM1 and DM2 models. These data strongly suggested that pathological serum conditions caused by prolonged hyperglycemia decrease the quantity of BM-MSCs. In diabetes, chronic hyperglycemic conditions drive glycation reactions between proteins and glucose or its derivatives, resulting in the formation of AGEs. AGEs can mediate several pathological manifestations of diabetes [32] and affect the proliferation and differentiation potential of adipose-derived MSCs [27,33]. The serum levels of several inflammatory mediators, such as S100A8/9 [20], which act through RAGE, were increased in the DM models. In addition to ligands for RAGE, we expected the upregulation of RAGE expression in DM models. However, neither the DM1 nor DM2 conditions caused a dramatic elevation of RAGE expression in cultured BM-MSCs (Supplementary Fig. S1A, B). Future analysis using freshly isolated BM-MSCs (without culturing) from DM mice will be necessary to determine the modulation of RAGE expression under DM conditions. Nevertheless, in this study, we showed that the loss of RAGE attenuated the reduction of BM-MSCs and the number of BM-derived colony-forming cells under diabetic conditions. These results supported our hypothesis that RAGE signal activation in diabetes is a key molecular determinant of BM-MSC properties. Furthermore, we found that cultured BM-MSCs derived from RAGE-KO mice expressed high levels of the multipotency-associated genes, Nanog and Oct4, and showed a high capacity for differentiation into mesenchymal lineages.
BM-MSCs are attractive candidates for tissue regeneration because they are generally accessible for isolation and have extensive expansion potential in vitro. However, the capabilities of BM-MSCs, including the multipotency, proliferation, and regeneration, gradually decline with age and disease [34,35]. Moreover, the expression of Nanog, which is an important transcription factor for maintaining the multipotency of cells, also decreased with increasing passage numbers [8] and under DM2 conditions (Supplementary Fig. S2). In this study, we observed that the number of BM-MSCs reduced in both type 1 and type 2 DM models, in agreement with previous reports [13]. Paradoxically, we found that DM model mice had more proliferative BM-MSCs than did healthy control mice. Therefore, these results lead us to conclude that systemic factors such as AGE in the serum of diabetic mice increased the proliferative state of MSCs (Ki67+) in the BM, and such BM-MSCs may eventually migrate out of the BM or undergo apoptosis after long-term exposure to pathological DM factors, which have been implicated in inducing oxidative stress through RAGE activation [18]. The number of tissue stem cells should be maintained by asymmetrical (self-renewal) or symmetrical division; however, under chronic pathological conditions, these finely tuned mechanisms were gradually disrupted [14,36]. Therefore, it is possible that the self-renewal of BM-MSCs might be impaired in DM and that consequently the number of BM-MSCs with colony-forming capacity might decrease. Additional studies are needed to identify the fate of proliferative BM-MSCs in diabetes, and it is important to know whether RAGE signaling contributes to fate choice of BM-MSCs under DM conditions.
It is noteworthy that in our study, the reduction of BM-MSCs under diabetic conditions was ameliorated in RAGE-KO mice. This finding indicates that RAGE ligands, such as S100A8/9, AGEs, and HMGB1, can disrupt MSC homeostasis in the BM. However, we cannot exclude the possibility that the maintenance of RAGE-KO BM-MSCs under diabetic conditions reflects their ameliorated conditions throughout the body as RAGE-KO mice showed amelioration of symptoms associated with cardiovascular diseases, neuropathy, loss of pain perception, and retinopathy [18,37 –40]. Therefore, exactly how the deletion of RAGE promotes maintenance of BM-MSC homeostasis remains unclear, although further mouse studies with conditionally deleted RAGE from BM-MSCs should define the contribution of RAGE signaling to BM-MSC maintenance under pathologic conditions. Transplantation of BM-MSCs can promote the repair of islets [41], diabetic nephropathy [42], and myocardial ischemic injuries [43]. In the present study, diabetic conditions such as the blood glucose level were not improved in RAGE-KO mice after STZ injection, suggesting that BM-MSCs did not transdifferentiate into pancreatic β cells in this model, even though the number and multipotency of BM-MSCs were maintained in RAGE-KO mice. As we previously reported that BM-MSCs contribute to the regeneration of peripheral organs, such as the muscle and skin [14,17,44], the primary roles and significance of BM-MSC maintenance under DM conditions are to prevent diabetes complications [45]. Further studies will elucidate the significance of BM-MSC maintenance under DM conditions and complications using RAGE-KO mice or inhibitors of RAGE signaling.
Our series of in vitro experiments showed that RAGE-KO BM-MSCs had a higher potential for differentiation and Nanog and Oct4 expression under hypoxic culture conditions. These data suggested that RAGE signaling affected the expression levels of multipotency-related genes. Nanog and Oct4 control the self-renewal and multipotency of stem cells. Their expression levels are regulated by various signaling pathways involving STAT3, WNT, and BMP4 in embryonic stem cells [46]. RAGE signaling is reported to induce inflammation through the Jak2/STAT1 pathway [47] and to negatively regulate the proliferation of osteocytes through Wnt, PI3k, and ERK signaling [48]. Thus, RAGE-mediated signaling may directly or indirectly contribute to Nanog and Oct4 expression in BM-MSCs. As noted above, RAGE-KO mice were resistant to reduction of the BM-MSC population in a DM1 model. Moreover, in STZ-injected DM1 RAGE-KO mice, BM-MSCs tended to progress through the cell cycle at a relatively slow rate as indicated by their higher expression of p57 (Fig. 2D), which suggested that excess RAGE signaling stimulates BM-MSCs, leading to their exhaustion. In addition, significantly lower expression levels of Nanog and p57 were observed in cultured BM-MSCs from db/db mice (DM2 model) than in those from WT mice (Supplementary Fig. S2). Thus, these data partially support our hypothesis that a high glycemic condition changes the integrity of BM-MSCs in diabetes. However, we cannot conclude that pluripotency-related gene expression and the slow-cycling phenotype are associated with the multilineage differentiation ability of BM-MSCs because adipogenesis was not impaired in BM-MSCs of db/db mice even though Nanog expression in db/db BM-MSCs was significantly lower than that in normal mice (Supplementary Fig. S3). Given that db/db mice are genetically null for the leptin receptor, further studies are required to determine whether DM2 conditions disrupt the stemness of BM-MSCs in a high-fat diet-induced model or in human DM2 patient samples. Of note, cultured RAGE-KO BM-MSCs contained more slow-cycling cells compared with WT BM-MSCs in vitro, indicating that RAGE signaling influenced cell cycle distribution both in vivo and in vitro. Considering that the loss of quiescent status leads to premature senescence and impaired multilineage differentiation potential [8,9], as well as the findings from the present study, we think that inhibition of RAGE signaling could maintain BM-MSCs by modulating their proliferative state in culture.
Our data shown in Fig. 2B indicate that RAGE signaling is not involved in the maintenance of BM-MSCs under physiological conditions. However, RAGE signaling might be involved in the maintenance of BM-MSCs under specific conditions, with a significant elevation of RAGE signaling, such as under DM conditions. AGEs are reported to upregulate RAGE expression [49]. Chronic excessive stimulation with RAGE ligands, which are elevated under diabetic conditions, modulates RAGE signaling and then affects BM-MSC homeostasis. It was also reported that interleukin-6 (IL-6) is secreted from senescent-like MSCs [50]. Known RAGE ligands and some factors secreted from cultured MSCs (such as IL-6) may amplify RAGE signaling during expansion in culture as well as under DM conditions. Further studies are needed to evaluate the role of RAGE signaling on the maintenance of MSCs with molecular approaches.
Several ex vivo treatments of MSCs, such as with growth factors, hypoxic shock, and antioxidants, have been used to maintain or enhance stem cell functions. Our current findings suggest that both in vivo and in vitro inhibition of RAGE signaling prevent diabetic- or culture-associated changes from occurring in MSCs. Therefore, we believe that our data may reveal an additional viable preconditioning strategy that can maintain the properties of MSCs for expansion in clinical therapies. However, a more comprehensive approach is necessary to determine whether blocking RAGE signaling is sufficient to mediate the maintenance of BM-MSC multipotency for clinical usage. Further analyses with RAGE neutralizing antibodies and the identification of specific ligands are required. In addition, it is of great interest to know whether BM-MSCs preconditioned by treatment with RAGE neutralizing antibodies can promote significant improvements in clinical conditions. Our findings should aid in the development of novel strategies to enhance the efficiency of MSC preparation for future therapeutic applications.
Footnotes
Acknowledgments
The authors thank H. Yamamoto (Kanazawa University) and H. Goto for the gift of the RAGE-KO mice and the experimental assistance, respectively. This work was partly supported by the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development (AMED), by JSPS KAKENHI grant number JP25293244 and JP16H05369, and by Grants-in-Aid for JSPS Fellows (12J03873 and 15J03833).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.