
Abstract
The role of extracellular vesicles (EVs) in mediating the immunosuppressory properties of mesenchymal stem cells (MSCs) has recently attracted remarkable scientific interest. The aim of this work was to analyze the transport mechanisms of membrane and cytoplasmic components between T lymphocytes and adipose tissue-derived MSCs (AD-MSCs), by focusing on the role of distinct populations of EVs, direct cell–cell contacts, and the soluble mediators per se in modulating T lymphocyte function. We found that neither murine thymocytes and human primary T cells nor Jurkat lymphoblastoid cells incorporated appreciable amounts of MSC-derived microvesicles (MVs) or exosomes (EXOs). Moreover, these particles had no effect on the proliferation and IFN-γ production of in vitro-stimulated primary T cells. In contrast, AD-MSCs incorporated large amounts of membrane components from T cells as an intensive uptake of EXOs and MVs could be observed. Interestingly, we found a bidirectional exchange of cytoplasmic components between human AD-MSCs and primary T lymphocytes, mediated by tunneling nanotubes (TNTs) derived exclusively from the T cells. In contrast, TNTs couldn't be observed between AD-MSCs and the Jurkat cells. Our results reveal a novel and efficient way of intercellular communication between MSCs and T cells, and may help a better understanding of the immunomodulatory function of MSCs.
Introduction
M
According to our present knowledge, MSCs exert their immunomodulatory function on T lymphocytes by reducing the proliferation of activated T cells, promoting their differentiation toward regulatory T cells, and altering the cytokine profile released by the different T cell subsets [8,9]. MSCs have been shown to modulate the function of immunologically competent cells by the secretion of various cytokines and soluble factors creating an immunosuppressive environment [10,11]. While the effects of the soluble factors secreted by the MSCs are well-established, several studies have indicated that cell–cell contacts also play a critical role in mediating the suppression of T cell function by MSCs. In these studies, inhibitory cell surface molecules, such as B7-H1 and B7-H4, were found to suppress T cell activation and proliferation [12,13]. The cell contact-dependent immunoregulation of MSCs was also shown to involve the FasL/FAS-mediated T cell apoptosis [14] as well as the Notch signaling [15]. The induction of regulatory antigen-presenting cells by MSCs may also occur in a contact-dependent manner by STAT3 signaling, resulting in the suppression of T cell activation [16].
It is likely that membrane components—receptors and antigens—and cytoplasmic components—nucleic acids and proteins—are transmitted between MSCs and T cells, and play an important role in modifying the function of T cells. Therefore, it would be important to clarify the exact mechanisms responsible for the membrane-mediated cross talk between MSCs and T lymphocytes, as no comprehensive study has been performed yet in this regard.
Recently, the role of extracellular vesicles (EVs) in the directed transfer of membrane proteins and cytoplasmic components between different cell types has been extensively investigated [17,18]. EVs are secreted subcellular structures surrounded by a phospholipid bilayer and include exosomes (EXOs), microvesicles (MVs), and other EV subsets (such as apoptotic bodies) released by various cell types [19]. The International Society for Extracelluar Vesicles has recently recommended minimal criteria to classify the EVs, depending on their size, morphology, mechanism of cellular release, and other features, including their biochemical, biophysical, and functional parameters [20]. The best characterized categories of EVs are the EXOs and the MVs. The difference between these two categories is based on their size and the way of cellular release: EXOs are in the range of 50–100 nm and generated by the exocytosis of multivesicular bodies, while MVs are in the range of 100–1,000 nm and released directly from the plasma membrane. In the past few years, several research groups revealed the therapeutic potential of MSC-derived EVs, since these particles reflect the phenotype and specific functions of their cellular counterparts [21,22]. The immunomodulatory properties of MSC-derived EVs have been less well investigated so far, although this field began to attract considerable scientific and clinical interest.
The primary aim of this work was to investigate the transfer mechanisms of membrane and cytoplasmatic components between adipose tissue-derived MSCs (AD-MSCs) and T lymphocytes, and explore the immunomodulatory potential of AD-MSC-derived MVs and EXOs on in vitro-stimulated peripheral T lymphocytes and Jurkat cells. We examined the transport mechanisms between human or murine stromal cells and various T cells, including mouse thymocytes, human peripheral T lymphocytes, and Jurkat cells, by using membrane and cytoplasm-labeling dyes.
To study the efficacy of membrane constituent transfer, we established two model systems. On the one hand, the AD-MSCs were labeled with PKH67 or DiI fluorescencent membrane-labeling dyes and then cocultured with unlabeled thymocytes, T lymphocytes, or Jurkat cells. On the other hand, distinct populations of MSC-derived EVs were labeled directly and thereafter cocultured with the unlabeled T cells as acceptor cells. Since the two EV subpopulations, MVs and EXOs, are dissimilar in point of their physiological function, composition, and mechanisms of cellular release [23], we tested the influence of each vesicle fraction in separate experiments. The uptake of MSC-derived EXOs by acceptor T cells has not been convincingly confirmed by any membrane-labeling techniques yet, and there are no data about the uptake of homogeneous, typical MSC-derived MVs in vitro. In a reverse experimental lineup, the capability of AD-MSCs to accumulate membrane components during coculture with T cells or through microvesicular and exosomal uptake was also investigated.
For testing the cytoplasmatic material transfer, the donor cells were labeled with calcein AM and the fluorescent staining of the unlabeled acceptor cells was followed either after coculture of the different cell types or incubation of the acceptor cells with donor cell-derived EVs. The immunomodulatory potential of AD-MSC-derived MVs and EXOs was investigated using primary T cells stimulated with Concanavalin A (ConA) or anti-CD3/CD28 antibody-coated beads, and proliferation inhibition was also tested in Jurkat cells. We studied the effects of MSC-derived vesicle subsets, vesicle-free culture supernatants, and coculture conditions on T cell proliferation and interferon gamma (IFN-γ) production. Besides the allogeneic human and murine experimental set-ups, we also examined xenogeneic models, due to the extensive use of human MSCs or MSC-derived EVs in mouse disease models [24 –26]. The better understanding of the transport mechanisms between human MSCs and T cells may provide important new information for cell-free therapeutic approaches using MSC-derived EVs.
Materials and Methods
Cell isolation and culture
All animal studies were reviewed and approved by the MTA Research Centre for Natural Sciences Institutional Animal Care and Use Committee based on the ARRIVE guidelines. All studies using primary human cells and cell line were performed with the permission of Medical Research Council, Committee of Human Reproduction (24083-3/2013/HER).
AD-MSCs were obtained from a 5-year-old male donor after hip surgical intervention and from a healthy 30-year-old female donor after liposuction. Mouse AD-MSCs were isolated from the abdominal and inguinal fat of two young adult female and male (10–12 weeks of age) C57Bl/6 mice. Both the human and mouse MSCs were used for further experiments between passage 2–6. Fat tissues were treated with 0.1% w/v collagenase type IV (Sigma–Aldrich) at 37°C for 30 min. Digestion was stopped by adding a growth medium (DMEM-F12 1:1 supplemented with 10% v/v FBS, 2 mM
MV and EXO isolation from cell culture supernatants
MV and EXO preparations were isolated by the combination of differential centrifugation and gravity-driven filtration. After collection of the culture medium, cells were removed by centrifugation at 300 g for 10 min, and thereafter the cell debris was removed by 2,000 g centrifugation for 10 min. The supernatants were filtered by gravity through a 0.8 μm syringe filter unit to completely remove apoptotic bodies (Millipore) and centrifuged at 12,500 g for 20 min at room temperature (RT) to get pure MV isolates. After removing the residual MVs by centrifugation at 20,500 g for 40 min, the supernatant was filtered by gravity through a 0.22 μm syringe filter unit (Millipore) and finally was ultracentrifuged in a Beckman L7 ultracentrifuge using 70.1 TI type rotor (Beckman Coulter) at 100,000 g for 70 min at 4°C to get the homogenous EXO isolate. After washing the samples with PBS, the EV preparations were resuspended in 1 × PBS buffer and used without freezing for further experiments.
Dynamic light scattering analysis
DLS measurements were achieved as described previously [27]. In details, we used an ALV goniometer with a MellesGriot diode-pumped solid-state laser (CVI MellesGriot) at 457.5 nm wavelength. The intensity of the scattered light was measured at 90° and the autocorrelation function was calculated using a PC-based data acquisition system developed in the Institute of Biophysics and Radiation Biology, Semmelweis University, Budapest, Hungary. We used the maximum entropy method to determine the particle size distributions. The diameter of the particles was calculated using sphere approximation, since the intensity of the scattered light is strongly dependent on the size of the particles and particles of larger size might be overemphasized.
Tunable resistive pulse sensing
TRPS analysis was carried out by using a qNano device (IZON Science) as described previously to determine the size distribution and concentration of EV isolates [28]. Calibration was performed by CPC200B and CPC400 beads (with 203 and 340 nm mode diameters, respectively). The concentrations of calibrating beads were known and 1:1,000 dilutions were used. Consequently, at least 500 events were registered with a linear particle rate in time using NP100 and NP400 nanopore membranes stretched between 45 and 47 mm. Voltage was set to 0.2–0.34 V to achieve a stable average current (126–130 nA) with a low average RMS noise.
Transmission electron microscopy
To analyze the morphology and the size of MV preparations, pellets were fixed with 4% paraformaldehyde at RT for 60 min, postfixed in 0.5% osmium tetroxide for 60 min at RT, and en bloc stained with 1% uranyl-acetate in H2O for 30 min at RT. Thereafter samples were rinsed with distilled water, dehydrated through an ascending ethanol series, embedded in LR white according to the manufacturer's protocol, and cured for 24 h at 60°C. Ultrathin sections were stained with 4% uranyl acetate in 50% methanol for 8 min and Reynold's lead citrate for 3 min. The fixed (4% paraformaldehyde) and resuspended EXO preparations were deposited onto Formvar–carbon-coated EM grids. Samples were contrasted first in a solution of uranyl oxalate and afterward were contrasted and embedded in a mixture of 4% uranyl acetate and 2% methylcellulose in a ratio of 100 μL/900 μL, respectively. The samples were finally air-dried and analyzed. Samples were analyzed in JEOL JEM 1011 transmission electron microscope operating at 60 kV. Images were collected using Olympus Morada 11 megapixel camera and iTEM software (Olympus). All reagents and materials used for electron microscopy were purchased from Sigma-Aldrich.
Flow cytometry of cells and EVs
Annexin V Alexa Fluor-488 (V13241) conjugate was purchased from Life Technologies. The anti-human CD3 (561808), CD63 (556020), CD73 (550257), and CD90 (555596) antibodies conjugated with PE, CD44 antibody conjugated with FITC, IFN-γ conjugated with BV421 (562988), and FITC/PE-conjugated CD4/8 Simultest antibodies (342407) were purchased from BD Biosciences, as well as the FITC-conjugated anti-mouse Sca-1 (553335) and PE-conjugated CD3 (561799), CD4 (557308), CD44 (553134), CD63 (564222), and CD90 (553014) antibodies. Cells and resuspended MV isolates were incubated with the antibodies for 30 min at 37°C/RT (cells/MVs, respectively). After washing all samples, the cells were resuspended in 0.5% BSA/PBS, while the MV pellets were resuspended in 1 × PBS. For annexin V labeling, annexin binding buffer was used at RT for washing and labeling the samples as described by the manufacturer's instructions. The same method was used for labeling the samples with the corresponding isotype controls (all purchased from BD Biosciences): mouse IgG1-PE (345816), IgG2b-FITC (555742), IgG1-BV421 (562438), and IgG1/IgG2a FITC/PE simultest (342409), and rat IgG2a-FITC (554688), IgG2a-PE (554689), and IgG2b-PE (553989). To verify the vesicular content of the MV preparations, Triton X-100 was added to the samples at a final concentration of 0.05% as described previously [27]. As a result of the Triton X-100 treatment, the fluorescent events disappeared suggesting the vesicular nature of the isolates.
EXOs were absorbed to the surface of 4 μm aldehyde/sulfate latex beads (Life Technologies). Five microliters of latex bead was incubated together with freshly isolated EXOs resuspended in 50 μL PBS for 15 min at room temperature. Then, 110 μL PBS was added to each sample to reach a final dilution of three-fold. After 30 min of incubation at RT, a suitable amount of 1 M glycine was added (100 mM final concentration) and samples were incubated for 30 min at RT. Next, an equal volume of 2% (w/v) bovine serum albumin was added and samples were blocked for 2 h at RT. After blocking, the samples were diluted in PBS to a final volume of 1.5 mL and centrifuged at 2,000 g for 5 min at RT. After washing, the bead pellet was resuspended in PBS for staining as described above. EV preparations were analyzed by using an Attune flow cytometer (Life Technologies). The instrument settings and the vesicular gate were set as described previously [29] using Megamix beads (BioCytex) and 1 μm Silica Beads Fluo-Green (Kisker) (Supplementary Fig. S4; Supplementary Data are available online at
Labeling the cells and EVs with fluorescent dyes and conditions of cocultures
Labeling with the PKH67 Fluorescent Cell Linker Kit (Sigma-Aldrich) was performed according to manufacturer's instruction. Cell staining was stopped by adding an equivalent volume of FBS. To stop the staining of EVs, 1% BSA/PBS was added. The calcein assay (Sigma-Aldrich) was performed only in case of living cells; calcein AM (250 nM) was diluted in the growth medium. Cells were incubated in the dye solution for 30 min at 37°C. The mouse thymocytes, T cells, and Jurkat cells were treated with verapamil (at a final concentration of 10 μM for 30 min at 37°C) before coculturing. After labeling with either the PKH67 kit or calcein AM, the cells were washed twice with 1 × PBS. To label the cells with Vybrant DiI cell-labeling solution (Life Technologies), cells were washed with 1 × PBS and then incubated with the prewarmed dye solution (5 μg/mL DiI in 1 × PBS) for 5 min at 37°C. Then cells were washed thrice with the culture medium to remove excess dye. The labeled and unlabeled cells were cocultured in a 1:20 ratio (MSCs to mouse thymocytes, human T cells, and Jurkat cells, respectively). During coculture, we used a ratio of 6 × 104 MVs/cell and 6 × 105 EXOs/cell on average, respectively, but we tested other ratios as well.
Confocal laser scanning microscopy
For confocal laser scanning microscopy measurements, μ-Slide eight-well glass bottom imaging chambers (Ibidi) were coated with human plasma fibronectin (Millipore). Fibronectin was used at 10 μg/cm2 concentration. For T lymphocyte sorting, peripheral blood mononuclear cells (PBMCs) were labeled with PE-conjugated anti-CD3 antibody as described previously and sorted by FACSAria flow cytometer (BD Biosciences). MSCs were labeled with calcein AM, while human T cells were stained with DiI as described previously, and the reverse lineup was also tested. Cells were seeded onto the imaging chambers and cocultured for 4, 12, and 24 h, respectively. After the incubation, the coculture was analyzed with an Olympus FluoView 500 Laser Scanning Confocal Microscope with 60 × oil immersion objective. The imaging chambers were incubated in a heating and incubation system (Ibidi) during the whole process at 37°C with 5% CO2 tension. Images were analyzed with FluoView Application Software (Ver. 05.00.110).
Analysis of the immunomodulatory potential of AD-MSCs, AD-MSC-derived EVs, and MSC-conditioned medium on in vitro-stimulated T lymphocytes and Jurkat cells
To determine the immunomodulatory potential of MVs and EXOs on in vitro-stimulated T cells, 8 × 104 purified PBMCs were seeded in each well of a 96-well plate. To stimulate the T cells, Dynabeads Human T-Activator CD3/CD28 (Life Technologies) was used by adding 2 μL beads to each well to obtain a bead-to-cell ratio of 1:1. Alternatively, T cells were also stimulated with ConA (Sigma-Aldrich) at a final concentration of 2 μg/mL. The EVs were incubated with the cells for 4 days at different ratios: 3 × 104 MVs/cell, 6 × 104 MVs/cell, and 12 × 104 MVs/cell or 3 × 105 EXOs/cell, 6 × 105 EXOs/cell, and 12 × 105 EXOs/cell on average, respectively. Nonstimulated and stimulated T cells, as well as stimulated T cells cultured in MSC-conditioned medium at different dilutions were also tested in all experiments. As positive controls, AD-MSC-activated T cell cocultures were analyzed using different cell-to-cell ratios: 1:5, 1:10, 1:20, and 1:50. The effect of AD-MSC-EVs on the proliferation of Jurkat cells was tested without using either Dynabeads or ConA, and the same EV–cell ratio was used as in case of experiments with activated T cells. Jurkat cells (5 × 104) were seeded in each well of a 96-well plate. For the quantification of cell proliferation, resazurin (Sigma–Aldrich) was used at a final concentration of 0.1 mg/mL. After 1 h of incubation at 37°C with 5% CO2 tension, supernatants were transferred to a 96-well optical plate, and resorufin fluorescence was read at 540/579 nm.
Intracellular IFN-γ assay
For intracellular IFN-γ measurements, purified PBMCs were stimulated with Dynabeads or ConA for 24 h in the presence of MVs or EXOs. During the last 6 h of the experiments, cells were incubated in the presence of monensin (Sigma-Aldrich) at a final concentration of 8 μg/106 cells. PBMCs were labeled with anti-CD4/anti-CD8 antibodies, fixed and permeabilized using Fix&Perm cell permeabilization reagents (Life Technologies), and stained with BV421-labeled anti-IFN-γ antibody. Flow cytometry measurements were performed by measuring IFN-γ expression on gated CD4- or CD8-positive cells. Similar to the proliferation assays, the effect of both the MSC-conditioned medium and the T cell-AD-MSC cocultures was tested.
Carboxyfluorescein succinimidyl ester cell proliferation assay
For quantification of T cell proliferation, the CellTrace carboxyfluorescein succinimidyl ester (CFSE) Cell Proliferation Kit (Life Technologies) was used. 1 × 107 human peripheral T lymphocytes were washed with 1× PBS and resuspended in 1 mL of 5% (w/v) FCS/PBS. CFSE stock solution was added to the cell suspension in a final concentration of 1 μM. After 5 min of incubation at room temperature in the dark, cells were washed with 10-fold volume of 5% (w/v) FCS/PBS, thrice at RT. After centrifugation at 300 g for 5 min, the cells were seeded onto 96-well plates and stimulated as mentioned above. Cell proliferation was analyzed after 4 days by flow cytometry. The proliferation was assessed as the percentage of viable T cells undergoing four cell divisions and the inhibition rate of lymphocyte proliferation was calculated as follows: (percentage of T cell proliferation under different coculture conditions/percentage of untreated, activated T cell proliferation) × 100.
Prostaglandin E2 immunoassay
EXOs and MVs were isolated from the supernatant of human peripheral T cells activated with Dynabeads human T-Activator CD3/CD28. The isolated MVs and EXOs were added to the human AD-MSC culture at different ratios: 1.5 × 104 MVs/cell, 3 × 104 MVs/cell, and 6 × 104 MVs/cell or 1.5 × 105 EXOs/cell, 3 × 105 EXOs/cell, and 6 × 105 EXOs/cell on average, respectively. After 48 h of incubation, the PGE2 content of the MSC supernatant was analyzed using the Prostaglandin E2 Parameter Assay Kit (R&D System), according to the manufacturer's instructions.
Statistical analysis
Statistical comparisons were performed using Student's t-test. The values are presented as the mean ± standard deviation and P values <0.005 were considered significant.
Results
Isolation and characterization of distinct MV and EXO populations
EV subpopulations were isolated from the supernatant of human and mouse AD-MSCs, mouse thymocytes, human peripheral T lymphocytes, and Jurkat cells. Before vesicle isolation, fetal bovine serum of the growth medium was replaced by KnockOut Serum Replacement to avoid contamination with EVs present in FBS. For EV production, cells were grown in the log phase; therefore mouse thymocytes and human peripheral T cells were stimulated with ConA. The different EV preparations were isolated by the combination of differential centrifugation and gravity-driven filtration (see Materials and Methods and Supplementary Fig. S1A).
Before coculture experiments, the freshly isolated vesicle subpopulations were characterized by using several methods to determine the specific features of these particles. To investigate the size distribution and morphology of EVs, all isolates were characterized by dynamic light scattering (DLS), tunable resistive pulse sensing (TRPS), transmission electron microscopy (TEM), and flow cytometric analysis, respectively (Fig. 1). According to the DLS and TRPS measurements, the size distribution of the MV and EXO isolates obtained from the different cell types were similar. Isolated EXOs were within the size range of 50–100 nm, while MVs were between 100 and 1,000 nm in diameter. In addition, the TRPS analysis revealed the exact concentrations of these isolates, which are shown as an example in the histograms in case of each EV subpopulation derived from human AD-MSCs (Fig. 1A). Based on the exact vesicle concentration of each EV isolate, we could use the same dose of either MV or EXO particles in each coculture experiment. The human AD-MSCs were used as a benchmark for the standardization of EV concentrations, and we consistently isolated EVs from the supernatant of 5 × 106 MSCs after incubation for 48 h. The number of acceptor cells was always 1 × 105.

Size distribution, morphological characterization, and flow cytometric analysis of human AD-MSC-derived microvesicles and exosomes.
The MV preparations had a mean diameter of 400–500 nm in all cases, and the MVs isolated from the supernatant of each cell type were found to have similar shape, size, and electron density on the score of the electron microscopic pictures. We used different methods to analyze the distinct EV subsets. In the case of MVs, ultrathin sections of MV pellets were analyzed, while the fixed and resuspended EXO isolates were deposited onto Formvar—carbon-coated EM grids, as detailed in Materials and Methods. The EXOs formed the most uniform vesicle population according to the TRPS and electron microscopic analysis, with a mean diameter of 80–100 nm. EV subpopulations were also characterized after labeling the particles with Alexa Fluor 488-conjugated annexin V, anti-CD63, as well as with host cell type-specific, fluorophore-conjugated antibodies, using flow cytometry. All EV isolates were found to be highly positive for annexin V, which marks phosphatidyl serine externalization, and for the tetraspanin CD63 [30]. Moreover, all MV isolates were carrying antigens typical of the host cells, for example, CD73 and CD90 in case of the human MSC-derived MVs (Fig. 1B). In contrast, the MSC-derived EXOs were negative for the GPI-anchored ectonuclease CD73, proving the different origin of these particles compared to MVs.
To demonstrate the functional nature of these particles, human AD-MSCs were cocultured with different concentrations of EXOs and MVs derived from human peripheral T cells activated with CD3/28 microbeads. After 48 h, the PGE2 level of the MSC supernatant was analyzed using Prostaglandin E2 Immunoassay (Fig. 1C). According to our results, both the MVs and EXOs derived from activated T cells induced a robust production of PGE2, one of the most prominent immunosuppressive compound produced by MSCs, in stem cells in a dose-dependent manner, proving the functionality of these particles.
Membrane-mediated and cytoplasmic material transfer from AD-MSCs to mouse thymocytes, human T cells, and Jurkat cells
First, we investigated the efficacy of membrane transfer from human or murine MSCs as donor cells to T cells as acceptors, including mouse thymocytes, human T lymphocytes, and Jurkat cells, respectively. We labeled MSCs with the PKH67 fluorescence membrane labeling kit, and after the MSCs fully attached to the plastic surface, unlabeled mouse thymocytes, human T cells, or Jurkat cells were added to the cultures at a donor–acceptor ratio of 1:20. After 24 h of coculture, the acceptor cells were analyzed by flow cytometry (Supplementary Fig. S2, 3rd row histograms). In the following experiments, we isolated human or murine MSC-derived MVs and EXOs, labeled the particles with the PKH67 membrane labeling kit, and cocultured these particles with the unlabeled thymocytes, T cells, or Jurkat cells, respectively. During coculture, we tested several ratios of MV–cell and EXO–cell. After 24 h of incubation, the acceptor cells were analyzed by flow cytometry (Supplementary Fig. S2, 1st and 2nd row histograms).
The results documented in Supplementary Fig. S2 show that after 24 h of coculture, neither the mouse thymocytes and human T cells nor the Jurkat cells accumulated considerable amounts of membrane components from MSCs, despite the long-term close cell–cell contacts. Similarly, after the PKH67-labeled MV or EXO treatment, negligible membrane transfer was observed toward the acceptor cells, despite even the highest dose (1.2 × 106 EXOs/cell and 1.2 × 105 MVs/cell) of EV treatment (Supplementary Fig. S2 represents the positivity in case of 6 × 104 MVs/cell and 6 × 105 EXOs/cell ratios). We concluded from these experiments that, the mouse thymocytes, human peripheral T cells, and even the Jurkat cells are not capable to incorporate considerable amount of membrane components from vesicles derived from MSCs. In addition, we did not find any differences between the allogeneic and xenogeneic models.
To investigate the phenomenon of cytoplasmic transfer between these cells, we labeled the mouse or human MSCs with calcein-AM dye and thereafter the transfer of the fluorescent calcein was followed (see Supplementary Fig. S2, 4th row histograms). Interestingly, after 24 h of coculture, the only cell type that gained calcein from MSCs was the human T lymphocyte, and we observed notable differences between allogeneic and xenogeneic models, suggesting an active recognition of a cell surface component on MSCs by human T lymphocytes. While approximately 92% of human T lymphocytes were positive for calcein after 24 h of incubation with human MSCs, only 32% of the human T cells gained calcein from mouse MSCs. In contrast, the thymocytes and Jurkat cells did not accumulate calcein from either human or mouse MSCs. It is important to note that calcein is a substrate of the MDR1 or MRP membrane transporter proteins, potentially expressed in peripheral T cells and also in Jurkat cells. To block any loss of the dye, the acceptor cells were treated with the inhibitor verapamil, at a final concentration of 10 μM for 30 min before the cocultures.
Membrane-mediated and cytoplasmic material transfer from mouse thymocytes, human T lymphocytes, and Jurkat cells to MSCs
To investigate the cytoplasmic or membrane-attached transfer of cell components to MSCs from mouse thymocytes, human T lymphocytes, or Jurkat cells, we repeated the same experiments in a reverse lineup (Supplementary Fig. S3). First, we directly labeled all donor cells with the PKH67 fluorescent membrane labeling kit, and then cocultured them with unlabeled MSCs at a 1:20 ratio (MSCs–T cells). After 24 h of coculture, we analyzed the acceptor cells by flow cytometry. As in the previous experiments, we isolated MVs and EXOs from the supernatants of these cells as well, labeled the particles with the PKH67 membrane labeling kit, and cocultured the vesicles with the unlabeled mouse or human MSCs. We examined several EV–cell ratios as well and found that the PKH67 positivity of acceptor MSCs is proportional to the concentration of added EVs (the dose-dependent increase of MSC positivity for PKH67 dye is shown in Supplementary Fig. S1C, D). Supplementary Figs. S2, S3 and Table 1 represent the positivity using the ratio of 6 × 104 MVs/cell and 6 × 105 EXOs/cell on average, respectively.
MV, microvesicle; ADSC, adipose tissue-derived mesenchymal stem cell.
As shown in Supplementary Fig. S3 (3rd row histograms), after 24 h of cell-to-cell coculture, both the mouse and the human MSCs accumulated a large amount of membrane components. Similarly, after the fluorescently labeled MV or EXO treatment, most of the acceptor MSCs were highly positive for the fluorescent membrane labeling dye (Supplementary Fig. S3, 1st and 2nd raw histograms). However, we found significant differences between the allogeneic and the xenogeneic models in the case of membrane transfer from mouse thymocytes to human MSCs. The xenogeneic membrane transfers were significantly less efficient either in the case of MVs or EXOs, although in the long-term cocultures, this difference was less pronounced (Supplementary Fig. S3, 1st column histograms), compared to the allogeneic model (Supplementary Fig. S3, 4th column histograms).
When mouse thymocytes, human T cells, or Jurkat cells were labeled with calcein AM, only 20% of the mouse MSCs gained calcein from mouse thymocytes, and there was no demonstrable transport of cytoplasmic calcein toward human MSCs. Both the human and the mouse MSCs gained similar amounts of calcein from human T cells, while there was not any observable transport of calcein from Jurkat cells to MSCs (Supplementary Fig. S3, 4th row histograms). During these experiments, in case of labeling T cells and Jurkat cells, verapamil was used again to avoid the loss of fluorescent calcein by potential membrane transporter extrusion. The summarized results of the experiments assessing membrane and cytoplasmic material transfer between these cells are shown in Table 1. The expression level, which was lower than 10% based on flow cytometric measurements, or the weak positivity, which could not be confirmed by fluorescent microscopy, is marked with minus. The positivity between 10%–33% is marked with one plus, the level between 33%–66% is marked with double plus, and the triple plus indicates the strong positivity for PKH67 dye or calcein.
We supposed that MVs might play a role in the cytoplasmic calcein transport from T cells toward MSCs since both in coculture models and by using T lymphocyte-derived MV preparates, most of the MSCs gained membrane components from T cells. Therefore, after labeling the cells with calcein AM, we isolated and analyzed the MVs by flow cytometry. All MV isolates were highly positive for calcein, independent from the host cell type. However, even after 24 h of coincubation with the MVs, there was no demonstrable calcein signal in MSCs. According to these results, we had to presume that the amount of calcein dye delivered by MVs is not sufficient to reproduce the high positivity measured after cell–cell coculture and another mechanism must be responsible for the large-scale cytoplasmic transport, which definitely requires close cell–cell contacts.
Detection of tunneling nanotubes between MSCs and T lymphocytes
To clarify the mechanism responsible for the intensive cytoplasmic calcein transfer between human MSCs and T lymphocytes, we analyzed the cocultured cells with confocal laser scanning microscopy and looked for the presence of tunneling nanotubes (TNTs).
When the MSCs were labeled with CellTracker CM-DiI fluorescent dye and human T cells were labeled with calcein AM, no detectable TNT formations originated from MSCs after 24 h of incubation (Fig. 2B). However, in a reverse set-up, when T cells were labeled with CellTracker CM-DiI and MSCs were labeled with calcein AM, we found plenty of TNTs between MSCs and T cells. These results indicate that the nanotubes originated from the T cells in all cases (Fig. 2A). The TNTs were formed after 2–3 h of coculture and existed for several hours. Acceptor cells were checked for calcein transfer at three different time points: after 4, 12, and 24 h of coculture. After 4 h, only 1% of the acceptor cells were positive for calcein. However, as a result of a 12-h long incubation, 40%–50% of the acceptor cells became positive, and after 24 h, almost all cells gained calcein.
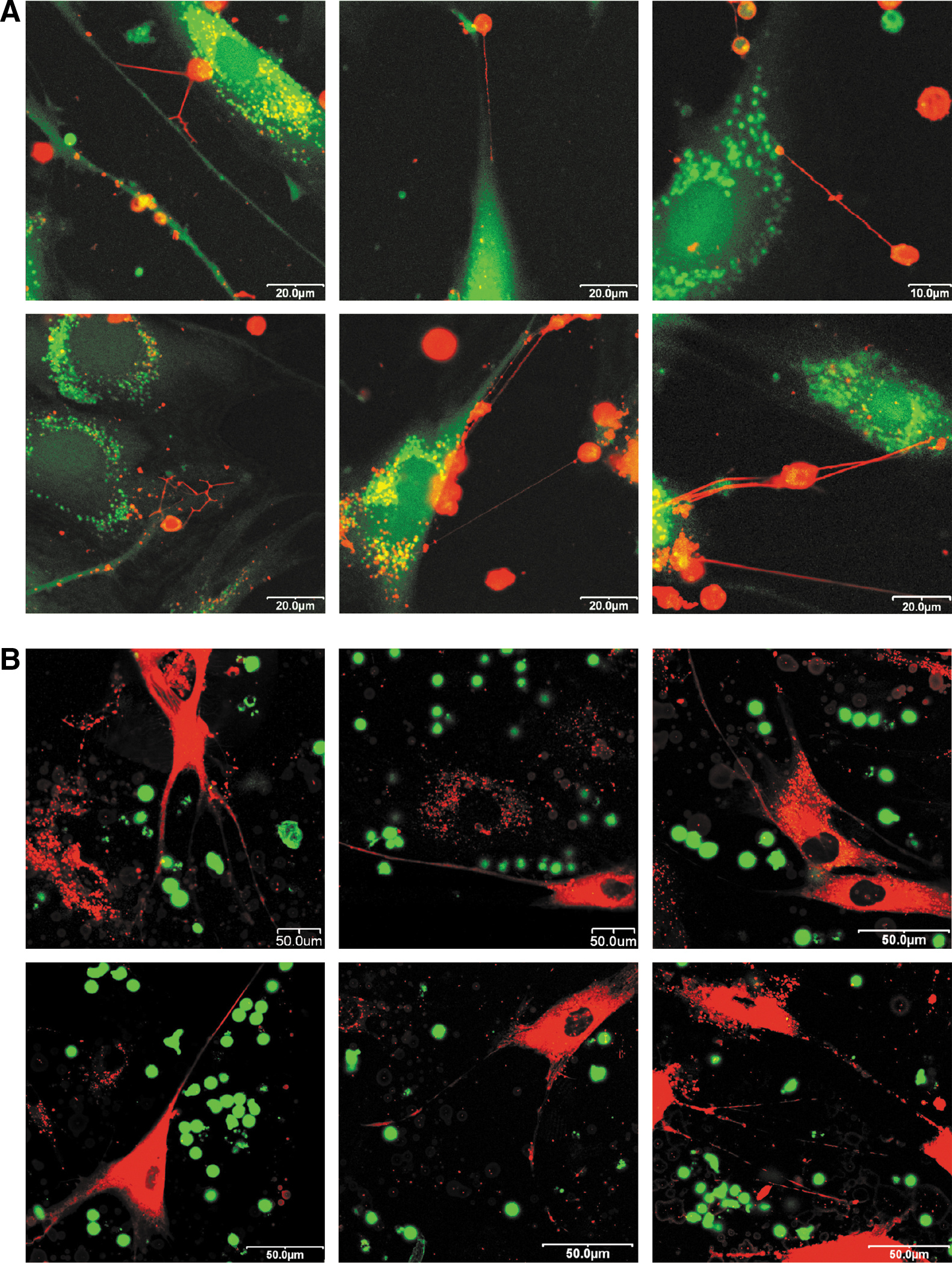
Confocal laser scanning microscopic images of human AD-MSC and human T cell coculture.
In conclusion, the TNTs formed very quickly and these tubes were always derived from T lymphocytes. Some of them reached 200 μm in length (Supplementary Fig. S4). The cytoplasmic calcein transfer between the cells required several hours. It is important to note here that Jurkat cells formed TNTs with each other, but did not form these nanotubes toward AD-MSCs under the same coculture conditions.
Immunomodulatory potential of AD-MSC-derived MVs and EXOs on in vitro-stimulated T lymphocytes
After clarifying the mechanisms responsible for the exchange of membrane components between AD-MSCs and T lymphocytes, functional assays were performed to analyze the effects of MSC-derived MVs and EXOs on the proliferation and IFN-γ production of in vitro-activated cytotoxic and helper T lymphocytes. The effect of both EV subpopulations on the proliferation of Jurkat T lymphoblastoid cells was also investigated.
To analyze the functional effects of MSC-derived EVs, in vitro-stimulated PBMCs were cocultured with different concentrations of either MVs or EXOs and the proliferation rate of lymphocyte cells was followed for 4 days. After removing all the EVs by ultracentrifugation, the functional effects of undiluted and diluted MSC supernatants were tested as well. We also investigated the immunosuppressive capacity of MSCs during cocultures using different MSC–T cell ratios. The activation of T lymphocytes was performed by using human T-Activator CD3/CD28 microbeads or ConA. All experiments were performed by using resazurin reduction assay and also by CFSE cell proliferation assay with very similar results.
Our results showed that neither the MSC-derived MVs nor the EXOs caused a significant reduction of T cell proliferation, despite using a very high EV concentration (1.2 × 105 MV/cell and 1.2 × 106 EXO/cell) (Figs. 3D, 4D, and 5B). In the reverse experimental lineup, as shown earlier, all MSCs incorporated EVs derived from T cells (Supplementary Fig. S3) and a robust increase of PGE2 production was observed (Fig. 1C). Moreover, during those experiments, only half of this EV–cell ratio was applied.

Immunomodulatory potential of AD-MSCs, AD-MSC-CM, and MSC-derived microvesicles and exosomes assessed by resazurin assay. T cells were activated with CD3/CD28 microbeads during all comparative experiments.

Immunomodulatory potential of AD-MSCs, AD-MSC-CM, and MSC-derived microvesicles and exosomes assessed by resazurin assay. T cells were activated with Concanavalin A during all comparative experiments.
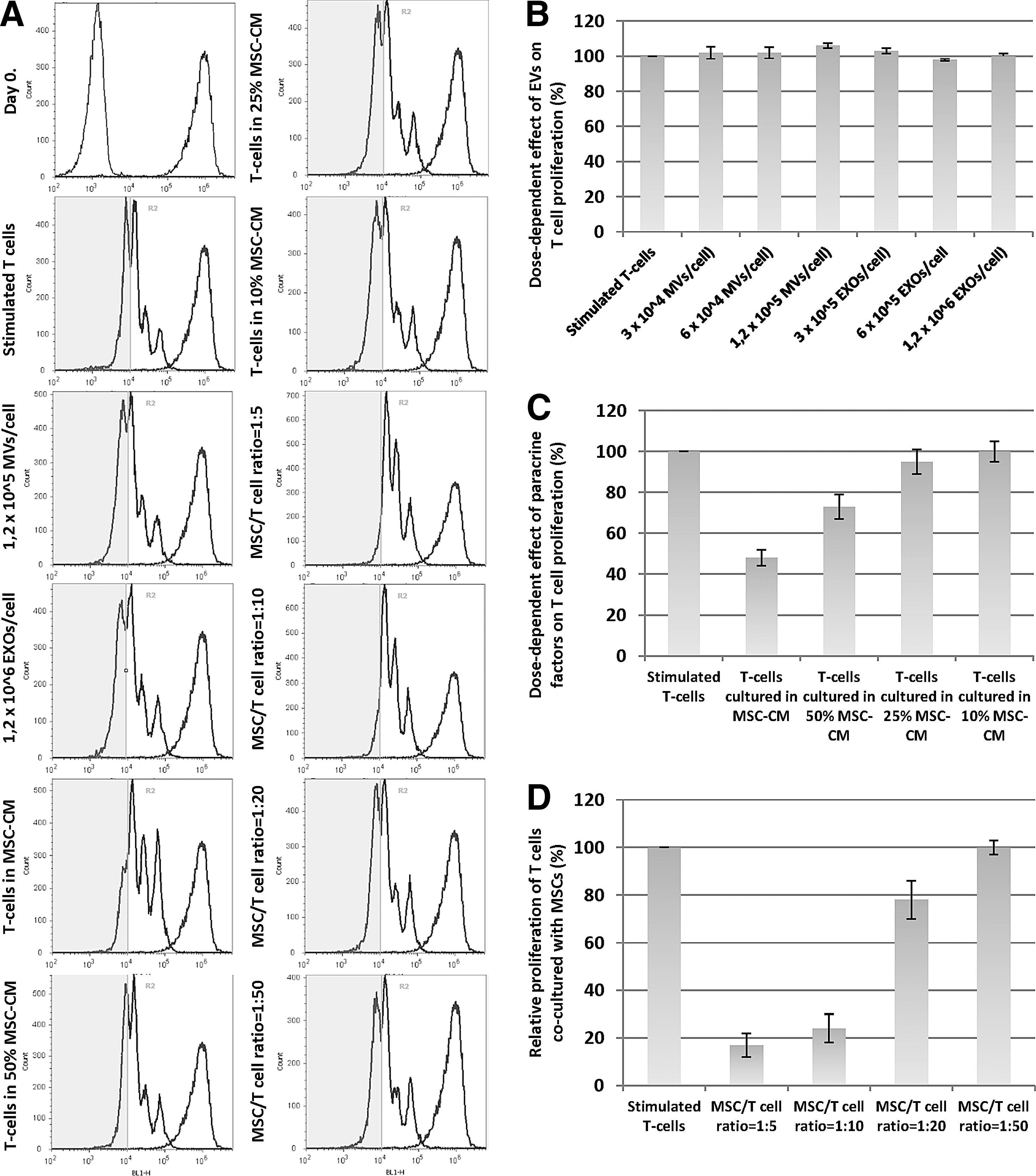
Immunomodulatory potential of AD-MSCs, AD-MSC-CM, and MSC-derived microvesicles and exosomes evaluated by CFSE cell proliferation assay.
In contrast, the activated T cells cultured in the MSC-conditioned medium (MSC-CM) had a significantly lower proliferation rate compared to the control-activated T cells. Using dilution series of MSC-CM, a lower inhibition effect could be observed (Figs. 3E, 4E, and 5C). The greatest degree of proliferation inhibition occurred during coculture conditions independent from the T cell activation method (Figs. 3A, B and 4A, B). We established different coculture conditions with different MSC–T cell ratios. At 1:50 ratio, the inhibition of T cell proliferation was negligible compared to that in other ratios, while similar inhibitory effects were found at 1:5, 1:10, and 1:20 ratios. (Figs. 3F, 4F, and 5D). At a 1:20 and 1:50 ratio, an intensive death of MSCs could be observed after 2 days. Probably the inflammatory cytokines were responsible for the cell death, because recently it has been reported that inflammatory conditions induce autophagy and promote the apoptosis of MSCs [31].
The MSC-derived EVs had no detectable effect on the production of IFN-γ by T lymphocytes (Fig. 3C). In contrast, we measured significantly lower levels of intracellular IFN-γ both in case of the presence of MSC-CMs and during coculture conditions. Furthermore, remarkable differences have been observed between the direct coculture model and the effects of MSC-CM. The decrease of IFN-γ levels produced by CD4+ lymphocytes was more significant either in the case of close cell–cell contacts or the presence of MSC-CM in culturing. After treating the T cells with ConA, there was no detectable level of intracellular IFN-γ production after 24 h.
Antiproliferative effect of AD-MSC-derived MVs and EXOs on Jurkat cells
To investigate the antiproliferative effect of EVs produced by AD-MSCs, Jurkat cells were cocultured with freshly isolated MVs and EXOs. During these experiments, a 1:10 MSC–Jurkat cell ratio was applied and the ratio of the cells and the EVs was 3 × 104 MVs/cell and 3 × 105 EXOs/cell. The functional effects of MSC-CM were also tested after removing all the EVs by ultracentrifugation. In these studies, we did not use ConA for stimulating the proliferation of Jurkat cells.
As shown in Fig. 4C, we found that during coculture of Jurkat cells with EVs, neither the MVs nor the EXOs caused the inhibition of cell proliferation. In contrast, a significant decrease of cell proliferation was observed using MSC-CM and also in AD-MSC–Jurkat cocultures. However, in contrast to previous experiments, using activated T cells, we could not find any significant difference between the antiproliferative effects of MSC-CM and the AD-MSCs in the coculture.
Discussion
Despite recent efforts to elicit physiological mechanisms responsible for the suppressory effect of MSCs on T cell function [32,33], data on contact-dependent or membrane-mediated signaling between these two cell types are incomplete. The latest findings confirmed the role of membrane-transported bioactive molecules in modulating the immune response, and these molecules have been shown to be carried by EVs [21,34,35].
It has been demonstrated that MSC-EVs carry mRNAs encoding immunomodulatory mediators [36], as well as some miRNAs enriched in MSC-derived EVs are associated with the regulation of the immune system [37]. Moreover, the proteomic profiling of EVs released by MSCs revealed the presence of numerous proteins, which are involved in inflammatory processes [38,39]. Still, although the biologically active molecules might significantly differ between MVs and EXOs due to their different biogenesis [30], their exact role in mediating the paracrine effects of MSCs has been rarely investigated in detail [40].
Recently, many research groups have focused on studying the immunomodulatory capability of MSC-derived EVs [41,42]. The various terms used in the literature to describe membrane particles released from living cells indicate that a consensus of the nomenclature still needs to be reached. Typical EXOs are often referred to as MVs, and the EXO isolates frequently contain typical MVs due to the method of isolation. Although the immunomodulatory activity of typical EXOs is well-characterized [43], the uptake of EXOs by acceptor immune cells has rarely been confirmed by membrane labeling techniques. Moreover, based on the isolation protocols used in several experimental set-ups, the MSC-EXO preparations contained typical MVs as well [44], or the isolates referred as MVs contained EXOs [36,37,45,46]. In some cases, typical EXOs were referred to as MVs [46,47]. Up to now, only a few studies have investigated the immunomodulatory effect of these particles on T lymphocytes, and there is a contradiction between the published data whether EVs derived from MSCs are as effective on T cells as the whole MSCs, regarding the immunomodulatory properties measured in vitro [44,46,48]. Moreover, it gives some reason for concern that only a small proportion of PBMCs were verified by membrane labeling technique to be susceptive to take up MSC-derived EVs and this subpopulation of PBMCs was composed of B lymphocytes [45].
In our comparative study, we investigated the intercellular transport mechanisms during cell–cell and cell-EV coculture conditions to make a distinction between those interactions that require close cell–cell contacts and those that can be effectively reproduced by using EVs. Importantly, we used homogeneous MV or EXO preparates without the presence of any other type of EV subsets, since the precise role of the distinct populations of MVs and EXOs still needs to be clarified in the context of membrane and cytoplasmic material transfer between MSCs and T cells. According to our results, neither murine thymocytes, human T lymphocytes, nor Jurkat cells incorporated considerable amount of membrane components from the different EV subpopulations when AD-MSCs were used as the donor cells, independent from whether allogeneic or xenogeneic experimental set-ups were used. In line with these data, neither vesicle fraction had any effect in our functional tests when the immunosuppressive effects of AD-MSC-derived EVs were assessed. Neither MVs nor EXOs reduced the proliferation rate and the IFN-γ production of the activated T lymphocytes. Upon stimulating the T cells with ConA, we obtained the same results as in case of the CD3/CD28 microbeads, regarding the proliferation rate. Similar results have been observed by Conforti et al., when MSC-derived EVs did not show a significant effect on in vitro-stimulated T cell proliferation, while a profound inhibition could be demonstrated upon coculturing of the two cell types [46].
Interestingly, using a reverse experimental lineup, AD-MSCs incorporated large amounts of membrane components independent of the source of origin, and no significant differences could be observed between the allogeneic and xenogeneic models, except that human AD-MSCs gained obviously less membrane components from murine thymocytes than murine AD-MSCs. In our subsequent functional assay, where MSCs were treated with various doses of EVs derived from activated T cells, a robust increase of secreted prostaglandin E2 was observed. T cells were specifically activated with CD3/28 microbeads by T-cell receptors. Similar to our findings, recently AD-MSCs were reported to alter their phenotype when treated with EXOs derived from cancer cell lines [49].
Assessing the potential transfer of cytoplasmic components between T cells and MSCs, we found that neither mouse thymocytes nor Jurkat cells gained calcein from AD-MSC donors; only human peripheral T lymphocytes were able to contact the cytoplasm of MSCs. Still, a significant difference could be observed between the allogeneic and xenogeneic models, suggesting an active recognition of a cell surface component on MSCs by human T lymphocytes. Interestingly, the experiments designed to reveal the role of EVs in intercellular transfer of cytoplasmic components showed that close cell–cell contacts are required for a significant cytoplasmic material transfer between AD-MSCs and T cells. To further investigate the mechanism responsible for the calcein transfer between MSCs and T lymphocytes, cocultures were analyzed with confocal laser scanning microscopy. Plenty of TNTs could be observed between these cells, which were originated from the human T cells. The role of TNTs has recently been described in the intercellular material transfer between different cell types of the immune system [50]. Interestingly, nanotubes are able to form between Jurkat cells also [51]; however, we could not observe these structures between human MSCs and Jurkat lymphoblastoid cells. TNTs have been noted between MSCs and distressed cardiomyocytes as well, and the nanotubular cross talk was suggested to stimulate the paracrine repair function of MSCs [52]. The role of TNT structures in connection with the intercellular exchange of cytoplasmic components between primary T cells and MSCs has not been described yet.
In the light of our results, we suggest that the role of MSC-derived EVs is negligible in the membrane and cytoplasmic material transfer toward T lymphocytes or Jurkat cells; thus, these vesicles are not decisive in mediating the suppressive effect of MSCs on T cells. However, an active communication exists between these two cell types through T cell-dependent mechanisms, which include vesicular or nanotube-mediated transfer of T cell components to MSCs. Receiving membrane and cytoplasmic components originated from T cells definitively alters the immunomodulatory activity of MSCs, shifting toward a more suppressive state and inducing the production of soluble factors such as prostaglandin E2. In this study, we also show that nanotube-mediated transfer of cytoplasmic components is bidirectional between these cells, therefore the MSC-derived cytoplasmic components may modulate the function of T cells.
As a summary, we conclude that the suppressive effect of AD-MSCs on T cells may be mediated by several mechanisms. The role of secreted cytokines and other soluble mediators is well-known [33] and was further confirmed by our experiments. Our results revealed that soluble factors per se mediate the antiproliferative effects of MSCs on the leukemic Jurkat cells. In our functional assays measuring proliferation and IFN-γ production of stimulated primary human T cells, we dissected the effects of soluble mediators from those of the different extracellular subsets present in cell culture supernatants. According to the recent opinion, MSCs gain their immunomodulatory phenotype in the inflammatory microenvironment [12,32]. Based on our observations, the soluble mediators of AD-MSCs collected from healthy individuals have definite suppressive effects on both the proliferation rate and IFN-γ production of activated T cells. Although the immunoregulatory function of MSCs seems to be greatly influenced by the in vivo microenvironment, an inherent suppressive potential of these cells might exist through the spontaneous release of soluble factors. Still, coculturing of the two cell types is needed to suppress the proliferation of T cells at the greatest level. Soluble mediators released by the human AD-MSCs significantly lowered the inflammatory IFN-γ production of both CD4+ and CD8+ cell types, and this effect was significantly modified when cells were cocultured. Thus, the bidirectional exchange of cytoplasmic components through TNTs and the uptake of T cell-derived EVs by MSCs should also be considered during the communication of peripheral T cells and AD-MSCs.
Finally, we have to emphasize the importance of using allogeneic models both in vitro and in vivo. Human MSCs are widely used during in vivo mice disease models [53,54], but crucial effector mechanisms differ between murine and human MSCs, which indicates a need for cautiousness when results obtained from murine or xenogeneic experimental systems are translated to the human condition [32]. In our xenogeneic model, we found that the human AD-MSCs, unlike mouse AD-MSC, did not gain cytosolic calcein from mouse thymocytes. Moreover, the uptake of thymocyte-derived MVs and EXOs was much less effective by human MSCs than by mouse MSCs. Since these phenomena are probably valid in vivo as well, it is worth considering the use of allogeneic systems in studying MSC-mediated immunosuppression in mice.
Footnotes
Acknowledgments
This research was supported by the grant KTIA_AIK_12-1-2012-0025 (Hungary). We are grateful for the help of János Matkó, Ágota Apáti, Anna Szigeti, Mónika Bátkai, and Margit Bakki. The authors are also grateful to Balázs Sarkadi for critical reading of the article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.