
Abstract
Naïve and primed pluripotent stem cells (PSCs) reflect discrete pluripotent states that approximate the inner cell mass or the progressively lineage-restricted perigastrulation epiblast, respectively. Cells that occupy primed pluripotency have distinct epigenetic landscapes, transcriptional circuitry, and trophic requirements compared with their naïve counterparts. The existence of multiple pluripotent states has not been explored in dogs, which show promise as outbred biomedical models with more than 300 inherited diseases that also afflict humans. However, our understanding of canine embryogenesis and embryo-derived stem cells is limited. Herein, we converted leukemia inhibitory factor (LIF)-dependent and fibroblast growth factor 2 (FGF2)-dependent canine embryonic stem cells (cESCs) resembling primed PSCs toward a naïve pluripotent state using LIF and inhibitors of glycogen synthase kinase 3β and mitogen-activated protein kinase kinase 1/2 [called 2i and LIF (2iL)]. cESCs propagated in 2iL exhibited significant induction of genes associated with the naïve pluripotent state (eg, REX1, TBX3) and downregulation of primed pluripotency markers (eg, OTX2, FGF5) (P < 0.05). Differential phosphorylation of signal transducer and activator of transcription 3 (STAT3) and cell fate decisions on exposure to bone morphogenetic protein 4 (BMP4) suggested that a novel pluripotent identity has been established with 2iL. Accordingly, cESCs cultured with 2iL formed colonies at a greater efficiency than LIF-FGF2 cESCs following single-cell dissociation. Total genomic DNA methylation and histone H3 lysine 27 trimethylation signals were reduced in 2iL-treated cESCs. Our data suggest that 2iL culture conditions promote the conversion of cESCs toward an epigenetically distinct pluripotent state resembling naïve PSCs.
Introduction
P
Embryonic stem cells (ESCs) have been derived from the inner cell mass (ICM) of preimplantation murine blastocysts [2,6]. Phenotypically distinct cell lines, called epiblast stem cells (EpiSCs), can also be established from the epiblast of mouse postimplantation embryos [7,8]. Both mouse ESCs (mESCs) and EpiSCs are considered pluripotent and can contribute to developing embryos, although at temporally discrete stages of embryogenesis [9]. Because the developmental competence of mESCs and EpiSCs is associated with disparate molecular and functional characteristics, they have been designated naïve and primed PSCs [10], respectively.
Human PSCs (hPSCs) typically resemble EpiSCs with regard to flattened colony morphology and transcriptional profiles that demonstrate low-level expression of early differentiation markers [8,11]. The GRN of hPSCs/EpiSCs sensitizes them to prodifferentiation signals without inducing lineage specialization [12] and likely reflects a perigastrulation epiblast identity [13]. Conversely, naïve PSC colony architecture is characterized by a distinct compact, domed-like appearance and cells display a transcriptome similar to the ICM and early epiblast of preimplantation embryos [14].
Constraints on the developmental potency of hPSCs/EpiSCs (hereafter referred to as primed PSCs) are linked to greater genome methylation and repressive histone modifications (eg, H3K27Me3, H3K9Me2/3) that impart a more heterochromatic landscape and gene silencing [15,16]. Primed PSCs are intolerant to subculture following dissociation into single cells and as such are propagated as colony fragments [17]. In contrast, naïve PSCs show greater single-cell passage viability [18] enabling the analysis of individual cells or clonal populations [19]. Such homogeneous cell populations are advantageous to research in disease modeling and preclinical evaluation of transplantation-based regenerative therapies [20].
Derivation and maintenance of primed PSCs typically require Activin-SMAD2/3 and fibroblast growth factor 2 (FGF2) extracellular signal regulated kinase 1/2 (ERK1/2) signaling [21]. In contrast, naïve PSCs are supported by bone morphogenetic protein 4 (BMP4)-Smad1/5/8 as well as leukemia inhibitory factor (LIF)-signal transducer and activator of transcription 3 (STAT3) signaling pathways [22,23]. The transcriptional circuitry responsible for supporting the naïve pluripotent state is largely downstream of the LIF and Wnt signaling pathways. Naïve pluripotency-associated factors (eg, GBX2 and TBX3) are expressed at low to negligible levels in primed PSCs [24], with the exception of NANOG [25].
Reversible transitions between transient cell states are inherent to cultured naïve or primed PSCs, a population phenomenon termed metastability [26]. Indeed, measureable fluctuations in pluripotency factor abundance can be observed at the single-cell level [27,28]. Spontaneous reversion of primed PSCs toward a stable naïve pluripotency is not favored within basal culture conditions, but may be achieved through transgene overexpression [29]. Nevertheless, pharmacological blockade of pathways that function to destabilize the pluripotent ground state imposes fewer limitations on downstream application of naïve PSCs [30,31]. Seminal work by Bao et al. showed that rodent EpiSCs can transition to naïve pluripotency [30] by partial chemical inhibition of glycogen synthase kinase 3β (GSK3β) and mitogen-activated protein kinase 1/2 (MEK1/2) (designated 2i) [32]. Such pluripotent state resetting requires global epigenomic erasure, a process through which epigenetic specification marks are removed [16].
Early embryonic development in the domestic dog (Canis lupus familiaris) is poorly understood because canine-assisted reproductive technologies have traditionally exhibited limited success [33,34]. Notwithstanding the lag in embryo biotechnology, a number of canine embryonic stem cell (cESC) lines have been established [35 –39] with their derivations and characterizations recently reviewed [40,41]. Our research group has generated multiple cESC lines from the ICM of explanted or antisera-dissected canine embryos [39]. Although primed human ESCs (hESCs) typically do not rely on LIF signaling [42], cESC lines established by others [38] and our laboratory [43] require both LIF and FGF2 for long-term propagation in culture. The dual requirement for both LIF and FGF2 suggests, either on a single-cell or population level, that the culture may demonstrate a mixture of characteristics between naïve and primed PSCs. Interestingly, explant-derived cESCs exhibit a neural lineage default in minimal media, which is consistent with primed EpiSCs [44,45]. Although considerable heterogeneity has been reported with regard to cell surface marker and transcription factor profiles between canine PSC lines derived by different laboratories [40], none of these studies thoroughly investigated the possibility that multiple pluripotent states may be adopted by canine PSCs.
Dogs are a logical model for preclinical development of cell-based or tissue replacement therapies, with longevity and body size more comparable to humans than rodent systems [46,47]. However, pluripotency and function of canine PSC lines must be rigorously assessed at the molecular level to be considered a viable cellular source for regenerative interventions. Recent evidence of metastable pluripotent states in mammals [30,31,48] provides the rationale to postulate that culture conditions preserving canine ground state pluripotency may be defined, but there is no direct molecular evidence that multiple pluripotent states can be adopted by canine PSCs. To date, no canine PSC lines have been described that approximate the native epiblast before implantation, and the molecular networks behind canine preimplantation development are largely unknown.
The primary aim of this study was to generate naïve-like cESCs independent of ectopic transgene expression for long-term maintenance in culture. Herein we report the induction of a distinct state of canine pluripotency through modulation of environmental cues and intracellular signal transduction using 2i and LIF (2iL). This pluripotent state shares defining features with embryo-derived and induced naïve PSCs, including increased expression of genes associated with the preimplantation mammalian blastocyst, augmented STAT3 signaling, expanded clonogenic potential, and decreased global repressive epigenetic modifications.
Materials and Methods
Embryonic stem cell and feeder layer culture
cESCs were derived at the Ontario Veterinary College (OVC, University of Guelph, Ontario) from explanted blastocysts (OVC.EX) and characterized in an earlier report [39]. Mouse embryonic fibroblast (MEF) monolayer preparation and culture of cESCs were conducted as previously described [43], with some modifications. Briefly, E12.5 DR4 MEFs were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and GlutaMAX. Feeder layers were mitotically arrested with γ-irradiation (8,000 rad) and seeded at a density of 105 cells/cm2. cESCs were seeded onto growth-arrested MEFs and cultured in serum-free basal media: KnockOut DMEM/Ham's F12, 15% KnockOut Serum Replacement (KOSR), 1× GlutaMAX, 1× nonessential amino acids, recombinant insulin-like growth factor (R3IGF1; Sigma-Aldrich), and 0.1 mM 2-mercaptoethanol. Basal medium was supplemented with 10 ng/mL human recombinant LIF and 4 ng/mL recombinant human FGF2 (LIF-FGF2) for maintenance of control cESCs; or 10 ng/mL human recombinant LIF, 0.5 μM MEK inhibitor PD0325901 (PD; Selleck Chemicals), and 3 μM GSK3β inhibitor CHIR99021 (CHIR; BioVision) for establishment and culture of 2iL cESCs.
LIF-FGF2 cESC colonies were mechanically isolated, dissected into small clumps, and transferred onto fresh MEFs every 4–6 days. 2iL cESC colonies were picked from culture, dissociated enzymatically with TrypLE™ Express, and split onto new feeder layers. To remove feeder cells before experiments, cESCs were transferred to Geltrex®-coated dishes and cultured with 70% MEF-conditioned medium balanced with nonconditioned basal media. Culture medium was exchanged daily and incubators were maintained at 37°C, 5% CO2, and ambient oxygen tensions.
Unless otherwise stated, all reagents for cell culture were from Thermo Fisher Scientific. Additional bioactive molecules described in this work include Activin A (20 ng/mL), BMP4 (20 ng/mL), Noggin (50 ng/mL), and SB431542 (10 μM; Sigma-Aldrich).
Quantitative reverse transcription PCR
Cells were harvested directly into TRIzol® Reagent (Thermo Fisher Scientific) and RNA was purified using the PureLink® Total RNA Purification Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Total RNA was reverse transcribed using the SuperScript® III First Strand cDNA synthesis kit (Thermo Fisher Scientific). Quality and quantity of cDNA were determined through spectrometry using a NanoDrop ND-2000 (NanoDrop Technologies). Primer pairs were designed using NCBI Primer BLAST software for each target gene, spanning an exon–exon junction where possible. Specificity of each primer set was determined if a single amplicon of the correct size was detected on a 2% agarose gel following electrophoresis.
Expression of orthologous genes associated with murine and human naïve pluripotency (REX1, KLF4, TBX3, GBX2, LIN28, PECAM1, STAT3), and primed pluripotency (FGF5, BRACHYURY, CER1, OTX2, SOCS3, ACVR2B) was examined. For primer sequences and amplicon sizes, see Supplementary Table S1 (Supplementary Data are available online at
Immunocytochemical fluorescence microscopy
cESCs cultured on Geltrex-coated four-well multidishes or Lab-Tek™ Chamber Slides (Thermo Fisher Scientific) were fixed with 4% formaldehyde in PBS for 15 min. Cells were permeabilized with 0.1% Triton X-100 before epitope blocking with 10% goat, donkey, or rabbit serum (Sigma-Aldrich). The following primary antibodies were incubated at 4°C overnight: anti-OCT4 (1:250), anti-OTX2 (1:250), anti-GATA2 (1:200), anti-GATA4 (1:200), anti-TUJ1 (1:300), anti-H3H27Me3 (1:250), anti-CDH1 (5 μg/mL; deposited to the DSHB by Gumbiner, BM), anti-CDX2 (1:250), anti-pSMAD2 (1:150), and anti-SOX17 (1:200). For information regarding primary antibodies, see Supplementary Table S2.
AlexaFluor-conjugated secondary antibodies (Thermo Fisher Scientific) were diluted 1:250 in 5% serum and cells were counterstained with NucBlue® specialized DAPI formulation (Thermo Fisher Scientific). The primary antibody was replaced with dimethyl sulfoxide and incubated with the AlexaFluor secondary antibody to control for antibody specificity. Fluorophore-conjugated anti-SSEA4 (eBioscience) was diluted in PBS and incubated for 90 min with nonpermeabilized cells.
Cells were imaged on a Leica DMI 6000B with Orca Flash digital camera (Hamamatsu Photonics) using Leica Application Suite Advanced Fluorescence software. H3K27Me3 immunolabeled cells were observed with a Zeiss LSM 510 Duo Vario confocal microscope (Zeiss Canada, Inc.). Serial optical sections were taken in a total depth of 50–80 μM. More than 30 images were assessed per treatment in three independent experiments. Nuclei were categorized based on the presence of well-delineated H3K27Me3 foci throughout the input stack. For representative images, extended depth of field composites was generated using ZProjector plugin on ImageJ (National Institutes of Health). Brightness or contrast was set using secondary-only control and equivalent imaging parameters were applied to all other images.
Western blotting
Cells were mechanically detached from 60-mm culture dishes and lysed in 200 μL RIPA buffer (150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris pH 8.0) with a protease and phosphatase inhibitor cocktail (EMD Millipore). Total protein concentration was determined by DC™ Protein Assay (BIO-RAD Technologies). Protein lysates were resolved on 4%–12% SDS-PAGE gels (Thermo Fisher Scientific) and transferred onto Immobilon™ PVDF membranes (Thermo Fisher Scientific). Epitope blocking was achieved by incubation with 5% skim milk powder/TBS/0.1% Tween-20 for 1 h.
The following primary antibodies were used: anti-OCT4 (1:1,000), anti-SOX2, anti-NANOG (1:500), anti-OTX2 (1:1,000), anti-pSTAT3 (1:1,500), anti-STAT3 (1:1,000), anti-pERK1/2 (1:2,000), anti-ERK1/2 (1:2,000), anti-pSmad2 (1:1,000), anti-Smad2/3 (1:1,000), anti-histone H3 lysine 27 trimethylated (1:1,000), anti-histone H3 acetylated (1:1,000), anti-histone H3 (1:1,000), and β-actin horseradish peroxidase (HRP) conjugate (1:30,000).
After a 16-h primary antibody incubation period, membranes were washed three times with TBS/0.1% Tween-20. HRP-coupled secondary anti-rabbit or anti-mouse IgG antibodies (Jackson Laboratories) were diluted in TBS/0.1% Tween-20 and applied to membranes for 1 h at room temperature. Immune reactive protein epitopes were visualized by chemiluminescence on Molecular Imager® VersaDoc™ System (BIO-RAD) and Quantity One software (BIO-RAD Technologies). Densitometry was performed using ImageLab 3.0 software (BIO-RAD Technologies).
Response to transforming growth factor β signaling
LIF-FGF2 and 2iL cESCs were exposed to 20 ng/μL recombinant human BMP4 in addition to their standard culture media preparation for a period of 96 h. Cells were harvested from multiwell dishes using TRIzol reagent for RNA isolation. RNA purification and first-strand cDNA synthesis were performed for quantitative reverse transcription PCR (RT-qPCR) (as in RT-qPCR section, above). In addition, canine cells from each experimental group were fixed with formaldehyde and prepared for immunocytochemistry (as in Immunocytochemical fluorescence microscopy section, above).
To examine LIF-FGF2 cESC dependence on SMAD2/3 signaling, cultures were supplemented with either 10 μM activin receptor-like kinase (ALK) inhibitor SB431542 or 20 ng/mL Activin A for 96 h before protein lysate harvest for western blotting.
Single-cell cloning efficiency and population doubling
To examine clonogenic potential, individual LIF-FGF2 or 2iL cESC colonies were mechanically isolated and incubated in 0.05% Trypsin to dissociate into single cells. Cells were resuspended in culture media containing LIF, FGF2, LIF + FGF2, or LIF +2i and passed through a 40-μM cell strainer (Thermo Fisher Scientific). Media with no growth factor or chemical inhibitor supplementation were applied for negative controls. Five hundred cells were seeded per well of a 24-well plate (n = 6). After 7 days, the number of SSEA4+ colonies were counted. Clonogenic efficiency was calculated as a percentage of SSEA4+ colonies per number of cells seeded. For linear-phase population doubling, 100,000 cESCs were seeded onto MEF-coated 35-mm dishes in triplicate. Biological replicates were harvested at 96 and 144 h and the total cell number was determined. Population doubling time was calculated using the following formula:
Metaphase spread preparation and karyotyping
cESCs were seeded into Geltrex-coated T75 flasks and grown to ∼60% confluency before metaphase spread preparation, as previously described [39]. Approximately 16 h before spread preparations, dividing cells were synchronized with Methotrexate (10−7 M final concentration) in fresh growth media. Cells were released from replication block with 60 μL of 10 mM thymidine and incubated for 3 h at 37°C. cESCs were treated with 1.25 mM ethidium bromide for 50 min to prevent excessive chromosome compaction and 10 μg/mL colcemid for 30 min to induce metaphase arrest.
Cells were then dissociated, suspended in hypotonic solution (75 mM potassium chloride), and incubated at 37°C for 15 min. A few drops of ice-cold Carnoy's fixative (3:1 methanol:acetic acid) were added and mixed by gentle agitation. The cells were pelleted and resuspended in the fixative twice before dropping the cells onto fixative-soaked microscope slides to prepare metaphase spreads. At least 30 metaphase cells were examined for each treatment group. Karyotypes were assembled according to ideograms using SmartType (Digital Scientific UK Ltd).
5-Methylcytosine DNA enzyme-linked immunosorbent assay
Purified genomic DNA was extracted from triplicate populations of LIF-FGF2 cESCs and 2iL cESCs (passage 5) using the PureLink Genomic DNA kit (Thermo Fisher Scientific). Global 5-methylcytosine (5-mC) levels were determined using the 5-mC DNA enzyme-linked immunosorbent assay (ELISA) kit (Zymo Research Corp.) according to the manufacturer's instructions. A standard curve was generated using E. coli control samples provided with the kit and raw data were calculated as percent 5-mC per 200 ng genomic DNA. Output values were multiplied by a fold difference of CpG island density factor specific to sample species [49] and expressed relative to LIF-FGF2 cESCs as baseline controls.
In vitro directed differentiation
Colonies of 2iL-treated cESCs were mechanically isolated and briefly triturated in basal culture media (KO DMEM/Ham's F12, 15% KOSR). Disaggregated 2iL cESCs were transferred to suspension culture in nonadherent dishes to form embryoid bodies (EBs). Basal media were changed every other day for 7 days.
To promote neuroectoderm differentiation, 2iL-derived embryoid aggregates were gently dissociated and seeded onto laminin- and poly-L-ornithine-coated dishes in the presence of TGF-β and BMP antagonists (1 μM SB431542 and 20 ng/mL Noggin). Mesodermal fate was induced using 10 ng/mL BMP4 and 10 ng/mL FGF2 on a gelatin substratum. Endoderm differentiation was induced by chemical stimulation of β-catenin signaling (1 μM CHIR) and 20 ng/mL Activin A on Geltrex-coated dishes. After 2 weeks under differentiation conditions, adherent cultures from each experimental group were fixed with formaldehyde for immunocytochemistry.
Teratoma formation
1.2 × 106 LIF-FGF2 or 2iL cESCs (n = 3) were transplanted into Non-Obese Diabetic Severe Combined Immunodeficient (NOD/SCID) interleukin-2 receptor γ-chain null (IL2Rγ−/−) mice (Jackson Laboratory). hESC lines hES2 were injected as positive controls (n = 2). Cells were suspended in 50 μL Geltrex and kept on ice (<20 min) before intramuscular injection in rear hind leg. Injection sites were monitored every other day by palpation. Between 6 weeks (for hESC transplants) and 10 weeks (for cESC transplants) postinjection, mice were anesthetized with isofluorane and sacrificed by cervical dislocation, and the area of muscle around the injection site was excised. Samples were rinsed in PBS before overnight fixation in 4% paraformaldehyde. Fixed tissues were processed for embedding and sectioning. Whole section hematoxylin and eosin staining was performed. Animal experiments were approved as ethical by the Animal Use Subcommittee (AUS #2008–101).
Statistical analyses
Statistical analyses were performed using Microsoft Excel 2010 or GraphPad Prism software for all experiments. Results are expressed as mean ± standard error of biological replicates. Two-sample statistical significance was determined using unpaired Student's t-tests, while parametric data were analyzed by a one-way analysis of variance followed by Tukey's post-hoc test. Data were indicated as statistically significant if the P value was <0.05.
Results
LIF and small-molecule inhibitors (2i) induce compact cESC colony morphology
OVC.EX cESCs were expanded on MEFs by mechanical dissociation and expansion in LIF-FGF2 media. Consistent with earlier work on the same cESC lines [39], LIF-FGF2 cESCs exhibited flattened morphology akin to hESCs/EpiSCs (Fig. 1A-I) and did not survive passaging regimens involving enzymatic dissociation reagents or mechanical trituration to single cells.

Morphologically distinct cESCs can be isolated from culture using LIF and 2i (2iL).
LIF-FGF2 cESCs were grown to 60–80% confluency on MEFs and switched to PD-, CHIR-, and LIF-containing media (2iL) (Fig. 1B). Culture in the presence of 2iL was followed by high levels of cell death for a period of 72 h. Within 4 to 5 days, three-dimensional cell clusters emerged from a proportion of cESC colonies. By 10 to 14 days, mature, dome-shaped colonies with tight edges typical of naïve PSC morphology were observed (Fig. 1A-II). A starting population of LIF-FGF2 cESCs (3 × 105) typically yielded 15–20 colonies with compact, dome-like morphology (∼0.005%). Stable 2iL cESCs had to be mechanically isolated before passaging in bulk culture because serial enzymatic passaging did not interfere with the growth of cESC-derived neural progenitor-like cells that spontaneously developed in 2iL culture (Supplementary Fig. S1A, B).
Confocal z-scans of DAPI-stained colonies verified that 2iL-treated cells formed multilayer stacks of cells, as opposed to LIF-FGF2 cESCs that invariably grew as monolayer colonies (Supplementary Fig. S1C). On removal of 2i from the growth media, cESCs rapidly lose dome-like colony morphology with concomitant appearance of differentiated cells (Fig. 1A-III).
As genetic background has been shown to influence the ability to derive naïve PSCs [50], the procedure was repeated in three independent OVC.EX cESC lines [39] (IO3, IO7, and BE5) with preliminary characterization of morphology and expression of pluripotency markers of primary 2iL cultures. We selected one 2iL cESC line and its original LIF-FGF2 cESC line (OVC.EX.BE5) with an XY karyotype (Fig. 1C) that has been shown to readily differentiate into each of the three germ layers in vitro [39], for extended passaging and subsequent analysis. These findings suggest that a trophic environment consisting of LIF, PD, and CHIR added to KOSR-containing basal media is permissive for a scarce population, which can be subsequently enriched based on compact colony architecture.
2iL cESCs are pluripotent and exhibit transcriptional circuitry resembling an earlier developmental state
To ensure 2iL-expanded canine cells retained pluripotent characteristics, we used immunodetection of core pluripotency transcription factors. Protein analyses showed no significant difference in OCT4 or SOX2 protein between LIF-FGF2 and 2iL cESCs (Fig. 2A). Both LIF-FGF2 and 2iL cESCs exhibited prominent nuclear OCT4 staining (Fig. 2B). However, there was a significant (P < 0.05) induction of NANOG in cESCs cultured with 2iL (Fig. 2A). OTX2, a transcription factor observed in the late epiblast [51] and a critical driver of naïve-to-primed transition [52 –54], was enriched in the nucleus and perinuclear region of LIF-FGF2 cESC colonies, whereas specific intracellular OTX2 staining was not observed in 2iL cESCs (Fig. 2C). This result was confirmed by western blot analyses showing a significant increase in OTX2 from whole-cell lysates of LIF-FGF2 cESCs compared with 2iL-cultured cESCs (Fig. 2A). Furthermore, 2iL stimulated nonuniform E-Cad immunostaining in bulk cultures that was not present in LIF-FGF2 cESC populations (Supplementary Fig. S1D).
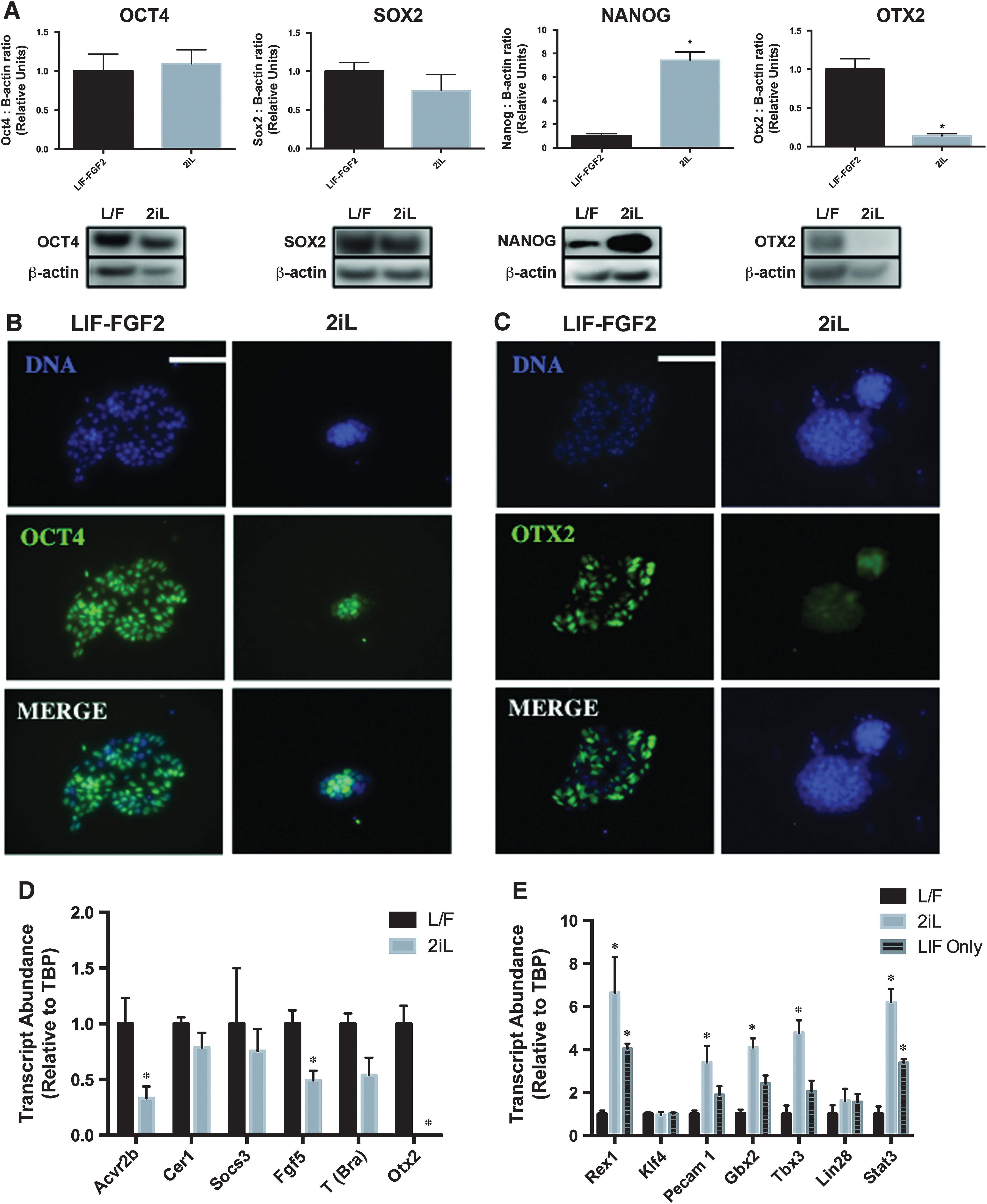
Transcription factor profiles suggest LIF-FGF2 and 2iL cESCs correlate with different epiblast stages. (
To assay in vitro pluripotential of 2iL cESCs, cells were allowed to form three-dimensional aggregates in suspension culture. 2iL cESCs formed EBs that could be directed to form putative early progenitors of endoderm, mesoderm, and ectoderm as assessed by fluorescent immunocytochemistry for expression of GATA4, GATA2, and TUJ1, respectively (Fig. 3A).
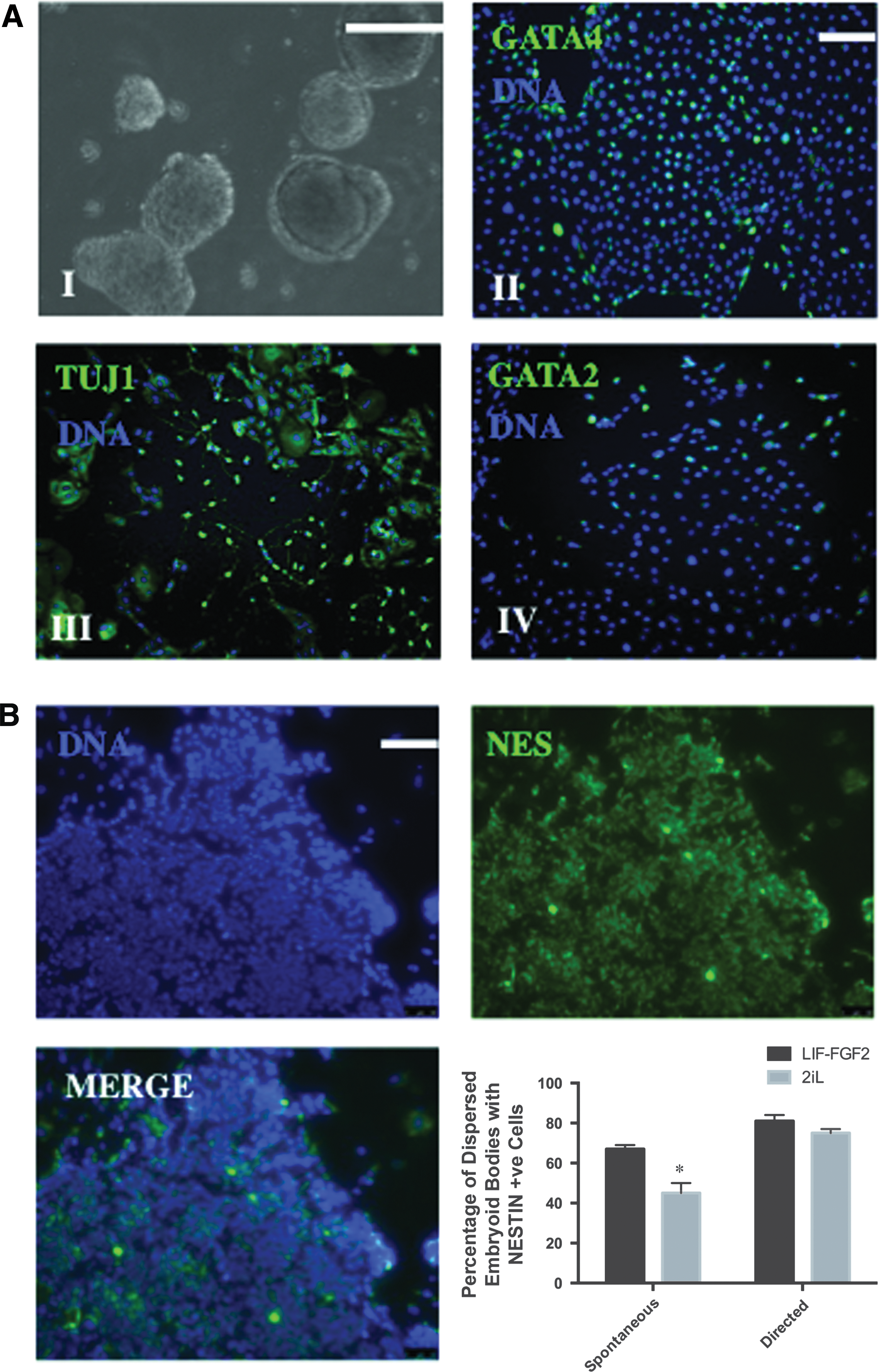
cESCs expanded with 2iL retain in vitro trilineage differentiation potential with attenuated neural differentiation bias.
The ability of LIF-FGF2 and 2iL cESCs to form NESTIN-positive neural stem-like cells under spontaneous and directed differentiation conditions was also investigated to study whether 2iL culture alters the inherent neural bias of OVC.EX cESC lines. Intriguingly, 2iL cESCs exhibited a lower efficiency of neural differentiation under spontaneous differentiation conditions (minimal media), but not under directed differentiation culture (Noggin-containing media), compared with LIF-FGF2 cESCs (Fig. 3B). These results suggest that LIF-FGF2 and 2iL cESCs have equivalent potential to form neural progenitor cells, but expansion in 2iL reduces the intrinsic neural differentiation bias of LIF-FGF2 cESCs.
We next investigated the gene expression signatures of several orthologous markers of naïve and primed pluripotency in LIF-FGF2 and 2iL cESCs to determine whether these cells bear transcriptional similarities to typical mouse or hESCs. RT-qPCR analysis revealed that naïve-like cESCs stabilized in 2iL exhibit significant (P < 0.05) reductions in the relative transcript abundance of late epiblast markers, including FGF5, OTX2, and ACVR2B (Fig. 2D). Brachyury expression was enriched in LIF-FGF2 cESCs, but did not reach the threshold of statistical significance. The primed PSC markers SOX17 and LEFTY2 were not detected by RT-qPCR in either LIF-FGF2 or 2iL cESCs (data not shown). Conversely, an induction of naïve pluripotency markers REX1, TBX3, GBX2, PECAM1, and STAT3 (Fig. 2E) was observed after 7 days of culture in 2iL. Expression of transcripts for KLF4 and LIN28 did not differ between LIF-FGF2 and 2iL cESCs.
Reduction of naïve pluripotency-associated transcripts following 2i removal is consistent with destabilization of a naïve-like pluripotency program. These results suggest that 2iL promotes a pluripotent phenotype with a unique array of transcriptional circuitry that is dependent on partial repression of GSK3β and MEK.
Altered signaling and divergent cell fate decisions in the presence of BMP4
To examine signaling pathways associated with maintenance of primed and naïve pluripotency in LIF-FGF2 and 2iL cESCs, we assessed the phosphorylation status of FGF-ERK, Activin-SMAD2/3, and LIF-STAT3 signal transduction proteins. The proportion of phosphorylated ERK1/2 (Thr202/204) was moderately reduced in cESCs propagated in 2iL, but did not reach statistical significance (Fig. 4A). However, there was a significant increase in the ratio of phospho-STAT3 (Tyr 705) to total STAT3 in 2iL cESC lysates compared with LIF-FGF2 cESCs (Fig. 4A). These correlative results may reflect the signaling cascades stimulated or repressed to induce and maintain 2iL cESCs.
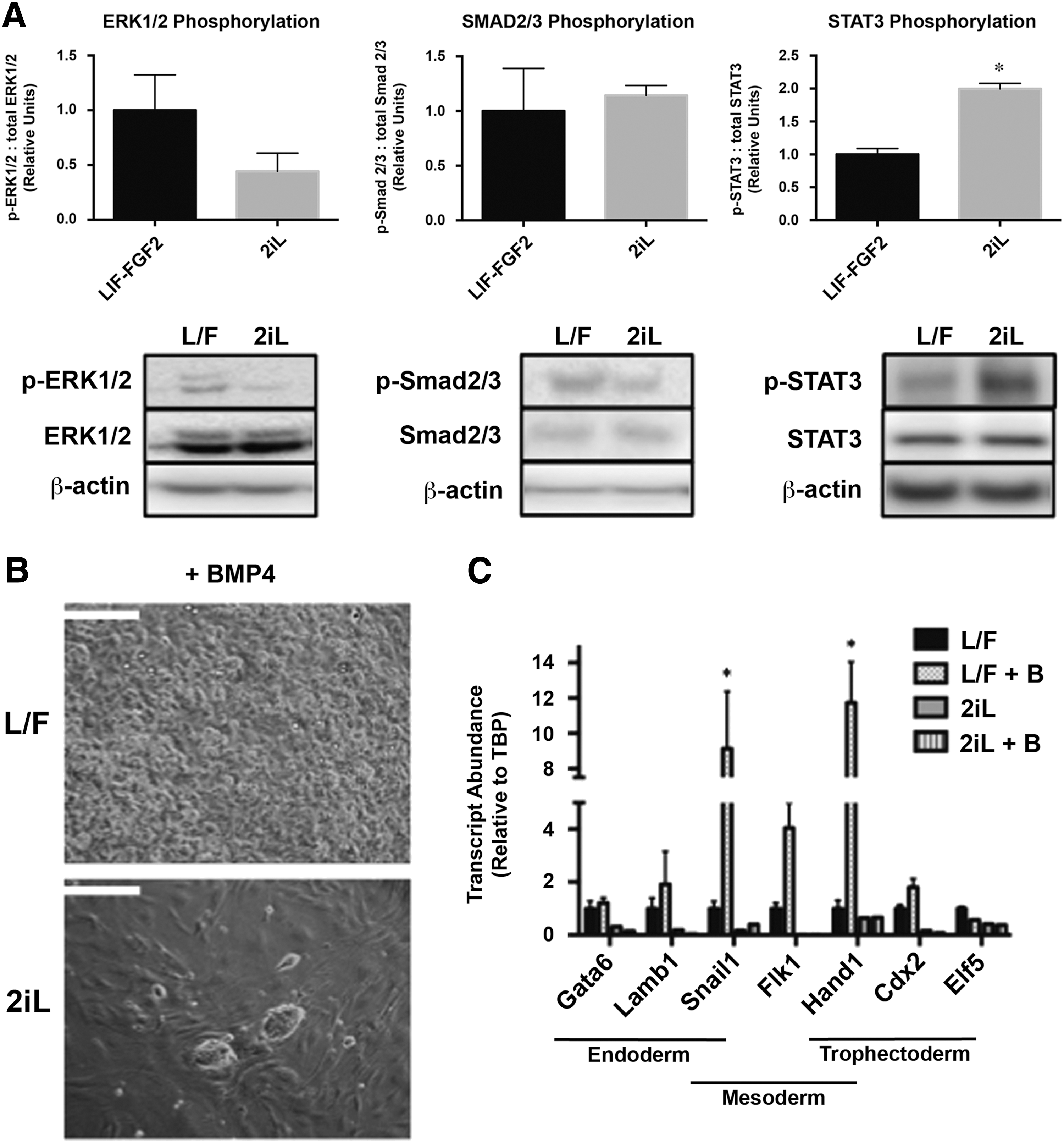
Altered signal transduction in pluripotency-associated pathways and response to BMP4 exposure.
There was no discernable difference in the ratio of phospho-SMAD2 (Ser465/467) to total SMAD2/3 in both LIF-FGF2 and 2iL cESC cultures (Fig. 4A). Fluorescent immunocytochemistry revealed no obvious difference in phospho-SMAD2 staining intensity, but unexpectedly revealed a clear pattern of nuclear exclusion under both conditions (Supplementary Fig. S2A).
Because Activin A has a well-characterized role in long-term maintenance of primed PSCs [55], we further assessed dependence of primed-like LIF-FGF2 cESCs on SMAD2/3 signaling. We examined the abundance of phospho-SMAD2 and co-SMAD member SMAD4 in the presence of exogenous Activin A or ALK inhibitor SB431542. LIF-FGF2-cultured cESCs were responsive to culture supplementation with Activin A because relative SMAD2 phosphorylation was enhanced after 96 h of treatment (Supplementary Fig. S2B). However, LIF-FGF2 cESCs also tolerated ALK inhibitor SB431542 at 10 μM with no overt differentiation over the same time course. Furthermore, SB431542 treatment did not significantly alter the ratio of phospho-SMAD2 to total SMAD2/3 protein levels in LIF-FGF2 cultures (Supplementary Fig. S2B). In summary, LIF- and FGF2-dependent cESCs have intact Activin-SMAD2/3 signal transduction machinery, but exhibit low basal levels of SMAD2/3 phosphorylation.
BMP4 has been reported to support the self-renewal of naïve PSCs [56] and to promote nonectodermal differentiation of epiblast-like primed PSCs [57 –59]. Based on our findings suggesting that 2iL conditions maintained cESCs in a more naïve pluripotent state, we predicted that exogenous BMP4 would support the self-renewal of 2iL cESCs, whereas BMP4 would promote the differentiation of LIF-FGF2 cESCs. LIF-FGF2 cESCs supplemented with 20 ng/mL BMP4 lost PSC-like morphology and form highly proliferative derivatives (data not shown) with marked cytoplasmic vacuolation after 96 h (Fig. 4B). In contrast, cESCs propagated in 2iL exhibited no observable morphological differentiation after 96 h of BMP4 treatment (Fig. 4B).
The expression of mesenchymal markers SNAIL1 and HAND1 was significantly upregulated (P < 0.05) following BMP4 addition to LIF-FGF2 cESCs (Fig. 4C), but not for 2iL cESCs. Transcripts for pan-endodermal markers (SOX17, FOXA2) and visceral endoderm marker HNF4A were not detected in BMP4-treated cESCs by RT-qPCR (data not shown). Similarly, 2iL cESCs exposed to BMP4 were negative for the trophoblast and posterior mesoderm marker CDX2 (Supplementary Fig. S3). Whereas BMP4-treated LIF-FGF2 cESCs formed small CDX2-positive subpopulations (∼1.66%) (Supplementary Fig. S3). Collectively, these findings suggest that application of 2iL to LIF-FGF2 cESCs can augment STAT3 signaling and promote self-renewal on BMP4 stimulation.
2iL culture enhances colony-forming efficiency of cESCs
Population doubling time and colony-forming efficiency were quantified to determine if the growth kinetics and clonality of 2iL cESCs were practical for scaling and clonal isolation procedures. Culture with 2iL prolonged the calculated linear-phase population doubling interval from 33.52 h in LIF-FGF2 cESCs to 51.77 h (P < 0.05) (Fig. 5A). Moreover, the population doubling times of LIF-FGF2 and 2iL cESCs were significantly greater (P < 0.01) than that of mouse and hESC lines. Individual 2iL cESCs have marginally improved viability compared with LIF-FGF2 cESCs following enzymatic disaggregation (Fig. 5B).

Modulated growth kinetics and clonogenicity of 2iL cESCs.
SSEA4 labels primed- and naïve-like cESC colonies in live culture (Fig. 5C) enabling rapid identification of pluripotent cells. LIF-FGF2 cESCs were highly sensitive to single-cell dissociation (0.03% cloning efficiency), and clonogenic ability was not significantly different from control media without LIF and FGF2. Naïve-like cESCs seeded as single cells into 2i media without LIF formed SSEA4+ colonies at a significantly (P < 0.0001) lower efficiency (0.25%) compared with 2iL cESCs (3.18%) (Fig. 5D). The low but appreciable cloning efficiency observed in 2i (without LIF) culture at clonal density was significantly (P < 0.05) more than LIF-containing or base media alone, which did not yield any pluripotent colonies. Our findings implicate reduced cESC proliferation in 2iL, but a substantial increase (∼100-fold) in their ability to form clonal populations.
The time interval between each cell passage of 2iL cESCs (∼7 days) was prolonged compared with mouse PSCs (∼2–3 days), indicating a disturbance in proliferation. Indeed, 2iL cESCs have a limited replication capacity in the undifferentiated state and could not be maintained for more than 20 passages before in vitro growth stalled.
To assess the possibility that karyotypically abnormal cells were accumulating in extended cESC culture, metaphase spreads were prepared for Giemsa staining. Although extended culture (>13 passages) of cESCs resulted in appreciable levels of polyploid cells alongside karyotypically normal (2n = 78 XY) cESCs, propagation in 2iL did not alter the percentage of euploid cESCs (Supplementary Fig. S4A). Therefore, modified growth kinetics in 2iL does not enrich for gross karyotypic abnormalities, despite detectable levels of polyploidy in expanded cESC culture.
Reduced genome methylation and repressive H3K27me3 marks in 2iL cESCs
Generally, PSCs in the naïve pluripotent state demonstrate a lower frequency of repressive histone modifications and CpG dinucleotide methylation, as well as a high degree of histone acetylation correlated with gene activity [16,60,61]. Culture of cESCs in 2iL induced a modest, but statistically significant (P < 0.05) decrease in the percentage of genomic 5-mC compared with LIF-FGF2 cESC cultures (Fig. 6A). In addition, the ratio of histone H3 lysine 27 tri-methylation (H3K27Me3) to total histone H3 was increased in LIF-FGF2 cESCs compared with 2iL cESCs (Fig. 6D). Pan histone H3 acetylation (H3Ac) did not significantly differ between LIF-FGF2 cESCs and 2iL cESCs (Fig. 6C). There was also no apparent difference in the overall intensity of H3K27Me3 immunostaining between LIF-FGF2 cESCs and 2iL cESCs (Supplementary Fig. S3B).

Loss of DNA methylation and H3K27me3 modifications in 2iL cESCs.
However, there were subtle differences in the pattern of nuclear H3K27Me3 localization between cell populations. LIF-FGF2 cESCs showed punctate H3K27Me3 immunostaining (Fig. 6E), while H3K27Me3 immunofluorescence appeared to be more uniform in 2iL cESCs (Supplementary Fig. S3B) and H3K27me3 clouds could represent heterochromatic foci (Fig. 6F). Collectively, our results suggest that cESCs maintained in 2iL culture conditions undergo epigenetic reprogramming associated with removal of generalized repressive modifications on DNA and histones, including a transition from diffuse H3K27Me3 localization to nuclear condensations of H3K27Me3.
Survival and differentiation competence in vivo
We [39] and others [37,62 –64] have struggled to generate mature teratomas from canine PSCs following injection into immune-deficient mice. We posited that naïve-like conversion by 2iL culture conditions may permit in vivo differentiation competence of cESCs. Control mice injected with hESCs (n = 2/2) formed teratomas ∼1.5 cm in diameter by 6 weeks (Supplementary Fig. S4B), composed of mature derivatives of mesoderm (hyaline cartilage), endoderm (intestine, ciliated respiratory epithelium), and ectoderm (melanocytes). However, no overt tumor growth was observed 10 weeks following intramuscular injection of LIF-FGF2 (n = 0/3) and 2iL (n = 0/3) cESCs into NOD/SCID/IL2Rγ−/− mice.
Discussion and Conclusion
cESCs with divergent morphological and molecular properties have been derived from early embryos [38,39], but these cell lines were established from different genetic backgrounds. In addition, previous reports have focused on the presence of general pluripotency factors, rather than discriminatory markers of naïve versus primed pluripotency. In an attempt to establish cESCs with naïve-like characteristics, we applied the well-studied combination of LIF, PD, and CHIR (2iL) to basal cESC culture media. Our data indicate that culture in 2iL reverts LIF- and FGF2-dependent cESCs to a pluripotent state resembling PSC lines designated as naïve. This naïve-like phenotype was associated with distinct epigenomic organization, change in signaling pathway transduction, and resetting of transcriptional circuits compared to LIF-FGF2 cESCs. These findings provide the first evidence that partial repression of MEK and GSK3β may support an earlier developmental state in cESCs.
Stabilization of naïve pluripotency without constitutively expressed transfactors in nonrodent species, such as humans, typically requires a more comprehensive cocktail of inhibitors and growth factors. However, in hESCs, the 2i component is indispensable regardless of basal media composition and other supplements [24,65 –70]. Persistence and growth of a cESC subset in 2iL media may be bestowed by pre-existing STAT3 signaling in our LIF-FGF2 cESCs. Indeed, primed-like cESCs are also LIF-STAT3 dependent and may exhibit a mixture of yet unrevealed features representative of the pre- and postimplantation epiblast. Positive regulation of STAT3 signaling by 2i may be due to other rate-limiting components of the LIF-signaling pathway such as the coreceptor GP130 [71]. However, we cannot formally exclude the role of extrinsic regulatory factors secreted by feeder cells in stabilizing a naïve-like phenotype because of the obligatory relationship between 2iL cESC induction and the presence of MEFs.
The possibility that pluripotency-associated signaling pathways may have divergent evolutionary functions in the dog must also be considered. Fundamental experiments in canine iPSC lines derived by Luo et al. [63] stipulated that LIF and FGF2 have antiapoptotic and proproliferation roles, respectively. However, we provide the first characterization of the phosphorylation status of key pluripotency signal transduction proteins in canine PSCs. These experiments suggest that distinct signaling cascades are active and possibly responsible for self-renewal of LIF-FGF2 and 2iL cESCs in vitro. Interestingly, LIF-FGF2-dependent cESCs appear to have adopted a self-renewal program largely unaided by Activin-SMAD2/3 signaling. Low Activin-SMAD2/3 signaling in LIF-FGF2 cESCs starkly contrasts with canonic primed PSC lines and could represent a canine-specific feature. Alternatively, selective trophic pressures throughout the derivation and expansion of cESC lines may have instilled these unique culture requirements.
It is not known whether there are unique cells within a nonuniform LIF-FGF2 cESC population that are intrinsically poised for pluripotent state resetting. This population heterogeneity may be compounded by multiple possible routes by which LIF-FGF2 cESCs obtain a unique pluripotent state in the presence of 2iL. Although most cESC lines have not been taken beyond a few passages after 2iL-mediated reversion, the OVC.EX.BE5 line was subcultured up to 20 times at a 1:6 passage ratio, which represents at least 50 doublings. Finite proliferation of 2iL cESCs is similar to what has been described for naïve PSC lines independent of ectopic transfactors [48] and may reflect the inability of the culture conditions to sustain the naïve transcriptional circuitry long term. Senescence in 2iL conditions may be related to genomic instability and notably the GSK3β inhibitor CHIR has been associated with karyotypic changes in other mammalian cells [72]. We are currently investigating these possibilities to shed light on cell cycle regulation and genome stability of cESCs.
Orthologous transcription factors that define naïve pluripotency in mice and humans are enriched in 2iL cESCs, including REX1, TBX3, and GBX2. Although the epistatic relationship between these genes is unknown in canine preimplantation development, we suspect that naïve pluripotency-associated genes are directly regulated by 2iL signaling in our model system. We anticipate that naïve pluripotency transcriptional networks will be constructed and/or operate differently in canines compared to other mammals. Future experiments should dissect the naïve pluripotency transcriptional programs in canine preimplantation embryos and PSCs derived from embryos or reprogramming technologies.
Comparative transcriptome analyses in mESCs have linked TCFCP2L1 [73] and estrogen-related receptor β (ESRRB) [74] to naïve pluripotency GRNs. Interestingly, novel ground state hPSCs have a limited ability to activate endogenous ESRRB using small molecules compared to their rodent PSC counterparts [24]. Knowledge of the naïve pluripotency-associated factors that are poorly induced by 2iL supplementation could be useful to tailor the composition of chemical induction protocols for canine PSCs. In the current study, transcriptional profiling experiments were performed on bulk cultures of cESCs and therefore represent a population average of these cellular characteristics. Although intercellular heterogeneity was not investigated, we anticipate that appreciable transcriptional variation exists within our cESC populations. Future work should focus on analysis of individual sorted cESCs to decipher the subpopulations that arise during steady-state culture.
BMP4 is critical for both maintenance of the undifferentiated state and lineage restriction, depending on the recipient embryonic compartment [58,75]. The differentiation status of BMP4-treated primed PSCs is a point of contention with phenotypic evidence of trophectoderm [58], definitive and extraembryonic mesoderm [57], or primitive endoderm [59]. The direction of lineage commitment likely depends on the context of FGF or Wnt signaling and the culture matrix utilized [76,77]. Hence, in the absence of small-molecule repression of lineage specifying factors, a mixed developmental population would arise [78].
The lack of pan-endodermal transcripts and negative antibody staining for SOX17 and GATA4 in BMP4-treated cESCs suggests that primed-like LIF-FGF2 cESCs were not directed toward an endodermal cell fate. Instead, the gene expression changes are characteristic of precursors undergoing epithelial-to-mesenchymal transition, which is consistent with the role of BMP4 in murine gastrulation [79]. The presence of a CDX2+ subpopulation may indicate the presence of primitive streak mesoderm cells, which express caudal genes during posterior axis elongation [80,81]. Unlike previous studies in canine [63] and porcine [82] iPSCs, trophoblast markers were not upregulated on bulk differentiation and no trophoblast giant cell-like morphology was observed.
Low clonogenicity related to slow growth kinetics and sensitivity to enzyme-induced apoptosis is generally understood as a property of primed PSCs [48]. Improvements in cloning efficiency with 2iL cESCs are consistent to what other groups have reported in naïve porcine cell lines (∼12%) [83], but not identical to studies in human and mouse systems (>40%) [65,84]. However, utilization of prosurvival compounds such as Rho-associated kinase inhibitors, timing of treatment, seeding density, and other factors influencing clonality are not consistent between studies [85]. The single-cell cloning efficiency results in our model indicate that 2i cannot sustain naïve-like cESC self-renewal alone and LIF signaling is essential for maintenance of clonal growth. The low efficiency of colony formation observed with 2i alone may be related to the soluble or adsorbed LIF secreted from the MEF feeder layer used in this experiment [22].
Expansion of cESCs in 2iL did not confer teratoma-forming ability in immunodeficient mouse models. Canine PSCs have had low success and poor reproducibility with these traditional in vivo assays of self-renewal and differentiation into mature lineages. Fundamental questions regarding the physicochemical requirements for proliferation should be resolved with further investigation into the metabolic properties and anabolism of canine PSCs.
Naïve-like cESC colonies emanate following 2iL culture over a prolonged time course, which is consistent with a selection or epigenetic reprogramming event rather than microenvironmental adaptation. The ∼35% reduction in 5-mC on expansion in 2iL is similar to the degree of global demethylation observed following mESC culture in 2i [15]. Lower genomic DNA methylation is a hallmark of naïve PSCs [16] and correlates with generalized developmental plasticity [12].
In a porcine model of naïve- and primed-like iPSCs, hypomethylation of a panel of differentially methylated promoters in pluripotent cells was associated with the ability to form chimeric embryos [86]. The ability of 2iL cESCs to contribute to embryonic development is yet to be assessed by embryo microinjection or aggregation experiments to generate intraspecies chimaeras. Formation of similar indices may identify canine PSCs capable of forming chimeric embryos. Repressive histone modifications, including H3K27Me3 and H3K9 methylation, demarcate transcriptionally silent domains and regulate pluripotency genes and early developmental regulators [87,88]. These repressive domains are posited to expand and/or increase in frequency throughout the genome during differentiation and the transition of ICM-like naïve PSCs to epiblast-like primed PSCs [67,89]. Consistent with the possibility that 2iL cESCs adopt an earlier developmental identity, we observed a decline in repressive H3K27Me3 modifications.
Histone H3 acetylation was maintained in 2iL culture compared to LIF-FGF2 cESCs, reflecting high levels of global transcription characteristic of the pluripotent state. Further experimentation in female lines remain to be completed to confirm the X-chromosome activation status of primed- and naïve-like cESCs. Interestingly, Whitworth et al. detected two active X-chromosomes in female canine iPSC lines and multipotent stromal cells, which may indicate species-specific regulation of X-chromosome activation status [90].
In summary, the described work represents the first evidence for population-level metastability in canine PSCs and reveals an opportunity to alter pluripotent characteristics of cESCs using small-molecule kinase inhibitors. Distinctions remain between our primed- and naïve-like cESCs and conventional human and mESCs, and so, the applicability of knowledge from human and murine systems to pluripotent canine cells is unclear. This study lends additional support to a conserved role for 2iL in promoting a naïve-like pluripotent state in mammals. However, the 2iL culture medium appears to be unable to fully support the self-renewal of reset cESCs long term. Given the recent success of empirically consolidated kinase inhibitor combinations for hPSCs [65,67], we suspect that subtle modifications to the 2iL culture system, alone or in combination with genetic modification, may support a stable canine PSC phenotype. Defining the optimal conditions to obtain and propagate naïve cESCs is essential for their application in biotherapeutic screening platforms and transplantation-based medicines.
Footnotes
Acknowledgments
The authors thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support. Ian Tobias is supported by an NSERC Post-Graduate Scholarship - Masters (PGS-M) and an Ontario Graduate Scholarship (OGS). They recognize the technical expertise of Karen Nygard for integrated microscopy services at the Biotron facility. Reagents and the guidance provided from Drs. Lina Dagnino and John DiGuglielmo are gratefully acknowledged. The assistance of Gillian Bell and Madelyn Harvey with animal care and surgeries is also deeply appreciated.
Author Disclosure Statement
The authors are not aware of any competing financial interests or membership that would affect the credibility of this research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.