
Abstract
Hypertrophy is a key component of endochondral ossification, the process controlling skeletal (cartilage, bone) development by differentiation of mesenchymal stem cells. Hypertrophic events also occur on cartilage injury such as during osteoarthritis (OA) and to a certain extent in focal lesions that may lead to OA if let untreated. Strategies based on the delivery on therapeutic genes and progenitor cells (the cells mostly recruited in spontaneous and guided repair) offer potential tools to delay or even prevent such undesirable events in sites of cartilage damage. The goal of this review is to revisit the mechanisms of hypertrophy during skeletal development and diseases and to provide an overview of the most recent advances in gene and stem cell therapy in the field of cartilage repair.
Introduction
A
Articular Cartilage and Hypertrophy
Structure and functions of the articular cartilage
The articular cartilage is the tissue that supports the smooth gliding of articulating surfaces in diarthrodial joints and allows to withstand local mechanical loading forces by maintaining a balance between the production and degradation of extracellular cartilage matrix (ECM) components by the articular chondrocytes. The adult, hyaline articular cartilage is an aneural tissue devoid of vascularization and lymphatic drainage that is separated from the underlying subchondral bone by a thin calcified layer. This particular tissue has a typical zonal organization, from the superficial to the deep zone (Fig. 1A). The superficial zone at the articular surface contains premature chondrocytes and thin collagen fibrils aligned parallel both to each other and to the surface area. In the middle zone, lower cell densities are present, with a rounded and mature phenotype surrounded by randomly arranged collagen fibrils with slightly larger diameters. The deep zone contains proliferating chondrocytes in columnar arrangement vertical to the articular surface, with collagen fibrils showing maximal diameters. The underlying calcified layer acts as a crossover to the subchondral bone and is characterized by the presence of chondrocytes with a hypertrophic phenotype. The chondrocytes (the only cell population in the cartilage) are embedded in this dense ECM made of proteoglycans bound to 70%–80% water and of collagen fibrils (mostly type-II collagen but also of type-VI, -IX, -XI, and -XIV collagen) associated with noncollagenous proteins, including the cartilage oligomeric matrix protein (COMP), link protein, fibromodulin, fibronectin, decorin, and tenascin [10 –13]. A narrow (pericellular) matrix (PCM) rich in proteoglycans and collagens (type-II but especially type-VI collagen) surrounds the chondrocytes, mediating interactions with membrane receptors at the surface of the cells that also serve as sensors of biochemical and mechanical stimuli (integrins, CD44, syndecan-4, discoidin domain receptor 2 [DDR2]) to adapt the cartilage homeostasis to physiological or altered environmental conditions [14,15].
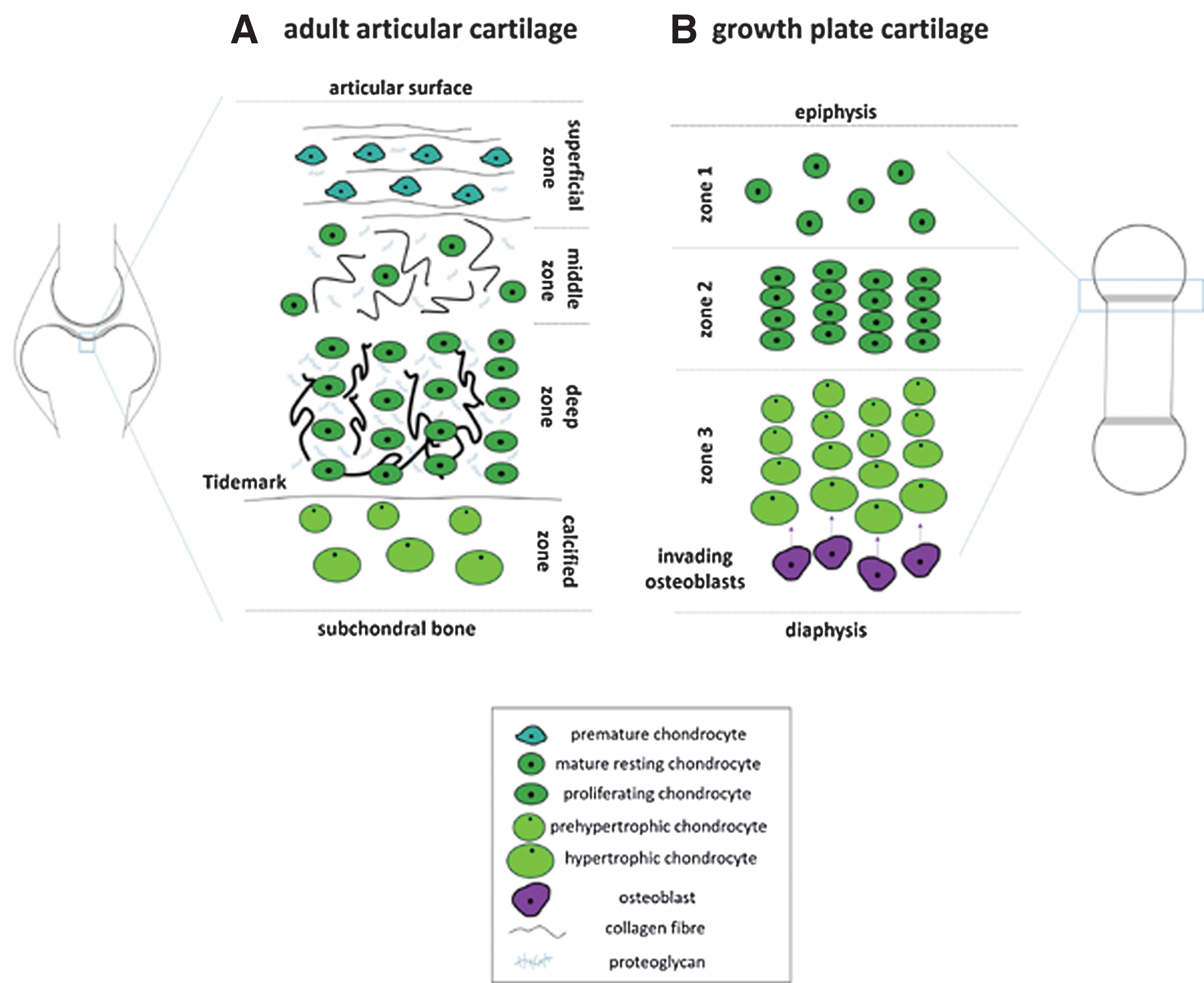
Organization of the cartilage at different developmental stages. Adult hyaline articular cartilage
Skeletal development and hypertrophy
The increasing knowledge on the processes controlling skeletal development (endochondral ossification) has allowed for a better understanding of the steps and pathways implicated in cartilage formation while providing key insights into the mechanisms of cartilage repair [3 –5,16]. Endochondral ossification is based on the complex model of the limb bud development involving mesenchymal stem cells (MSCs) and is divided in steps of MSC condensation, differentiation from chondroprogenitors to mature chondrocytes, terminal differentiation to hypertrophy, and ultimately, bone formation [2,17 –23]. The growth plate is a tripartite organization consisting of resting chondrocytes (zone 1) that undergo maturation and rapid proliferation, resulting in columns of rapidly dividing cells (zone 2), and then increase their volume, terminally differentiate, and exit the cell cycle, representing prehypertrophic and hypertrophic cells (zone 3) (Fig. 1B).
Cell condensation
MSCs first undergo cell proliferation and clustering (Fig. 2), resulting in an increase in cell volume, based on cell–matrix and cell–cell interactions via cell adhesion molecules, including N-cadherin (Ca2+ dependent) and neural cell adhesion molecule (N-CAM). At this stage, the cells produce ECM molecules such as fibronectin and type-I collagen. MSC condensation is initiated by the transforming growth factor beta (TGF-β).
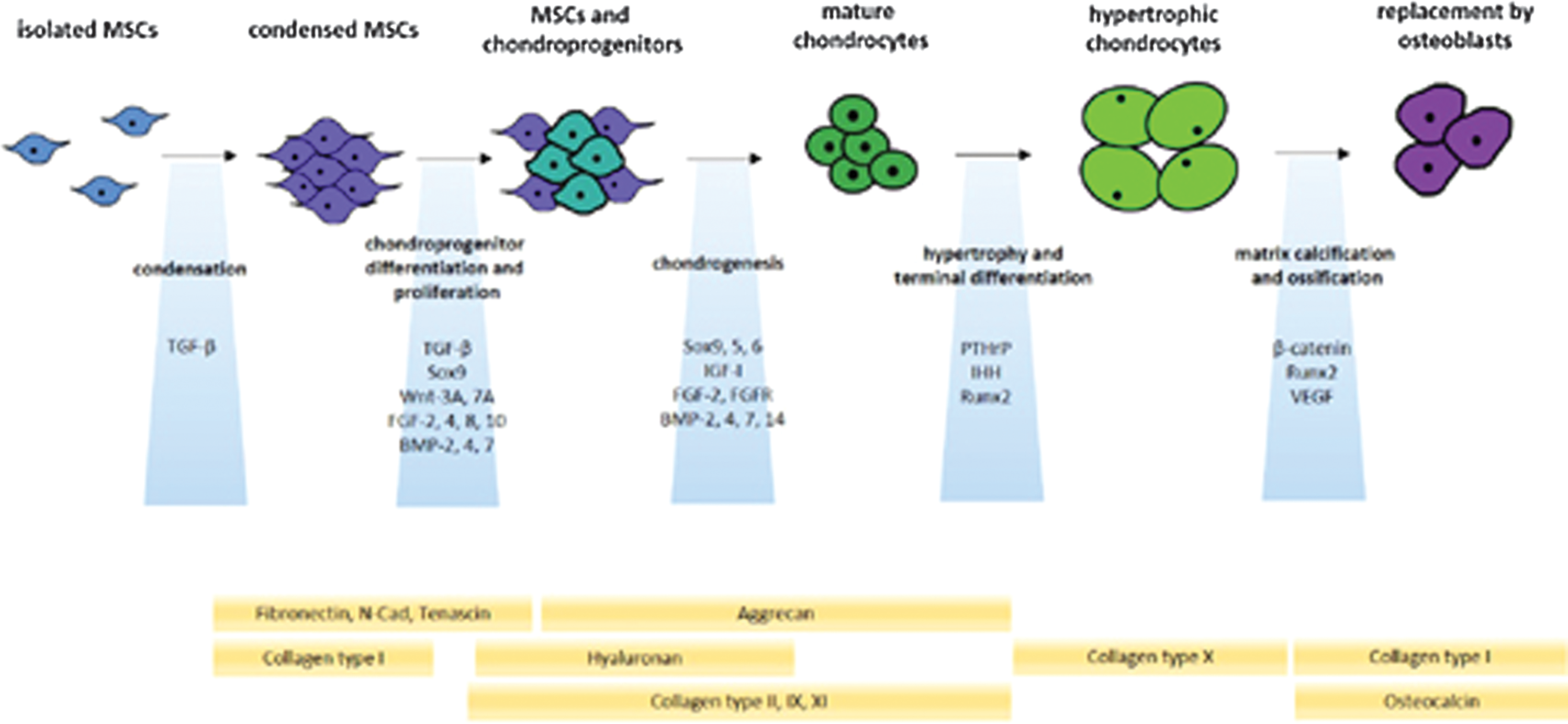
Endochondral ossification during embryonic development. Condensed MSCs undergo differentiation as chondroprogenitors with subsequent cell proliferation. Mature chondrocytes next develop by chondrogenic differentiation with increases in cell volume, leading to hypertrophic chondrocytes that ultimately undergo apoptosis and subsequent replacement by osteoblasts (including by differentiation of hypertrophic chondrocytes). These complex processes involve several participating factors (blue boxes) and matrix molecules (yellow boxes). BMP, bone morphogenetic protein; FGF, fibroblast growth factor; FGFR, FGF receptor; IGF, insulin-like growth factor; IHH, Indian hedgehog; MSCs, mesenchymal stem cells; N-Cad, N-cadherin; PTHrP, parathyroid hormone-related protein; Runx2, runt-related transcription factor 2; SOX9, sex-determining region Y-type high-mobility group box 9; TGF-β, transforming growth factor beta; VEGF, vascular endothelial growth factor; Wnt, Drosophila segment polarity gene wingless and vertebrate homolog integrated int-1 (c, canonical; nc, noncanonical).
Chondrocyte differentiation, maturation, and proliferation
MSCs next undergo important changes in their patterns of ECM production (hyaluronan production, shift from type-I collagen to aggrecan, type-II, type-IX, and type-XI collagen) while proliferating and adopting the rounded shape characterisic of differentiated chondrocytes (Fig. 2). These processes are mostly controlled by TGF-β and transcription factors of the cartilage-specific sex-determining region Y-type high-mobility group box family (SOX9, SOX5, SOX6- SOX trio) and also probably by other transcription factors such as Nk3 homeobox 2 (Nkx3.2). Beyond this stage, cells either form the hyaline cartilage or undergo further steps of differentiation that may lead to matrix calcification and ossification.
Hypertrophy and mineralization
Cells may next undergo hypertrophy, with conversion from prehypertrophic to hypertrophic cells by terminal differentiation and size increase (up to 20-fold), with replacement of type-II by type-X collagen on expression of matrix metalloproteinase 13 (MMP13), expression of alkaline phosphatase (ALP), and calcium-dependent hydroxyapatite deposition (mineralization) (Fig. 2). These processes are mostly regulated by the parathyroid hormone-related protein (PTHrP)/Indian hedgehog (IHH) pathway and by the bone-specific runt-related transcription factor 2 (Runx2), and also probably by other transcription factors, including members of the myosin enhancer factor 2 family (MEF2C), forkhead box A 2/3 (FOXA2/3), and CCAAT/enhancer-binding protein beta (C/EBPbeta). This stage is a prerequisite to matrix calcification and ossification.
Matrix calcification and ossification
The final steps of endochondral ossification consist of (1) further ECM remodeling [expression of type-I collagen, osteocalcin (OCN), osteopontin (OP)], (2) apoptosis of hypertrophic chondrocytes, (3) invasion of hypertrophic zones by newly synthesized blood vessels on expression of the angiogenic vascular endothelial growth factor (VEGF), and (4) replacement with osteoblasts, including by differentiation of hypertrophic chondrocytes with further expression of MMPs (Fig. 2). These processes are mostly determined by the Wnt (Drosophila segment polarity gene wingless and vertebrate homolog integrated int-1)/β-catenin pathway and Runx2.
Articular cartilage injuries and hypertrophy
Injuries of the articular cartilage are defined as disruptions of its structural and functional integrity and can appear either as a generalized disease such as in OA or as localized (focal) defects [1,24 –26].
OA is a progressive, multifactorial joint disease characterized by the gradual degradation of the articular cartilage surface and also associated with pathological changes in all other joint tissues (subchondral bone, synovial membrane, meniscus, tendons and ligaments, muscles). OA is thus also associated with sclerosis and osteophyte formation and hypertrophy, by activation of inflammatory, biomechanical, and catabolic cascades and with crosstalks between tissues, showing also a genetic background [27 –32]. OA cartilage shares hypertrophic features similar to those involved in the process of endochondral ossification (chondrocyte proliferation and differentiation with loss of SOX9, type-II collagen, and aggrecan expression, ECM remodeling and mineralization with increased expression of type-X collagen, MMP13, and Runx2, chondrocyte apoptosis, invasion of blood vessels, and replacement with osteoblasts). This may be seen as a switch from the articular to a growth plate chondrocyte phenotype except for hypertrophic cell size increase [3,5,33 –38].
Focal lesions instead, such as resulting from trauma, are generally restrained to defined areas, involving the cartilage alone (chondral defects) or reaching through the subchondral bone (osteochondral defects), leading to OA if left untreated. While hypertrophic features similar to those occurring during endochondral ossification or as observed in OA have not been clearly defined in such defects, several lines of evidence indicate hypertrophic-like changes in traumatic defects. They include the advancement of the subchondral bone plate within the cartilage surface and the formation of intralesional osteophytes and subchondral bone cysts with possible type-X collagen deposition [6,7,39 –43], even on cartilage repair procedures [6,44].
Control Mechanisms of Hypertrophy
Hypertrophy is a complex process that is regulated at different levels by various signaling pathways, with an influence of the microenvironment and of epigenetic factors (Fig. 3).
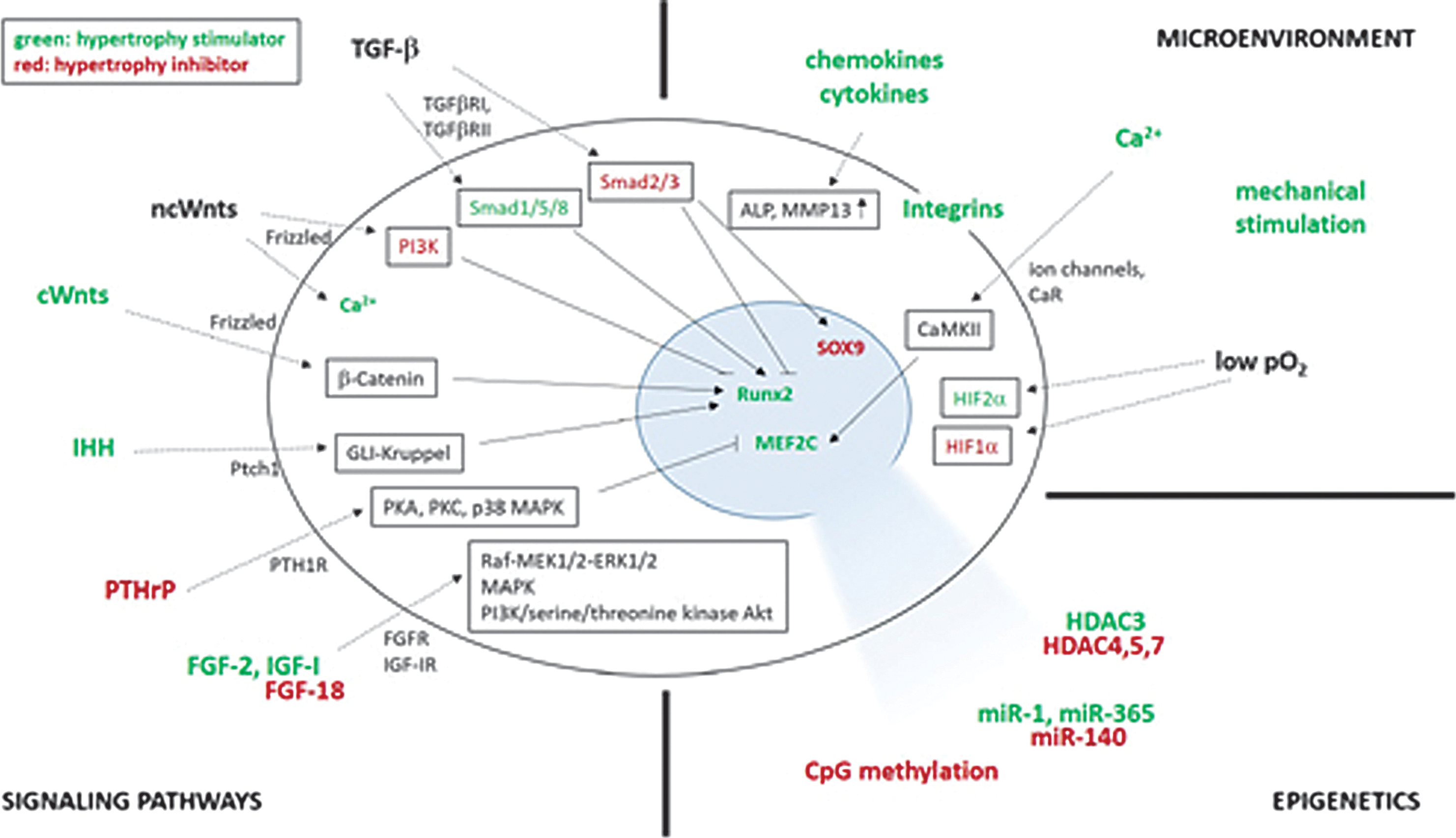
Control mechanisms of hypertrophy. Hypertrophy is regulated by signaling pathways, the microenvironment, and by epigenetic factors. Hypertrophy regulators are represented outside of the cell, uptake mechanisms at the cell membrane, and downstream signaling in frames inside of the cell, including the effects on target transcription factors in the nucleus. Factors that stimulate hypertrophy are illustrated in green, inhibiting factors in red. CaMKII, calcium/calmodulin-dependent protein kinase; CaR, calcium-sensing receptor; Gli, glioblastoma-isolated protein; HDAC, histone deacetylase; HIF, hypoxia-inducible transcription factor; IGF-IR, IGF-I receptor; MAPK, mitogen-activated protein kinase; MEF2C, myosin enhancer factor 2 family; miR, microRNA; MMP, matrix metalloproteinase; PI3K, phosphatidylinositol-bisphosphate 3-kinase; PKA/PKC, protein kinase A/C; Ptch, patched receptor; PTH1R, PTHrP receptor; Raf-MEK1/2-ERK1/2, Raf-mitogen-activated protein kinase kinase 1/2-extracellular signal-regulated kinase 1/2; Smad, Sma- and Mad-related protein; TGFβR, TGF-β receptor.
Signaling pathways
Various pathways have been identified for their involvement in the process of hypertrophy, including signaling from PTHrP/IHH, Wnt, TGF-β/bone morphogenetic protein (BMP), fibroblast growth factor (FGF), and insulin-like growth factor I (IGF-I) (Table 1).
BMP, bone morphogenetic protein; CaR, calcium-sensing receptor; DDR, discoidin domain receptor; ECM, extracellular cartilage matrix; ERG, ETS-related transcription factor; ETS, erythroblast transformation-specific transcription factor; FGF, fibroblast growth factor; GDF-5, growth differentiation factor 5; Gli, glioblastoma-isolated protein; HDAC, histone deacetylase; HIF, hypoxia-inducible transcription factor; IGF, insulin-like growth factor; IHH, Indian hedgehog; IL-1, interleukin 1; MEF2C, myosin enhancer factor 2 family; miR, microRNA; NF-κB, nuclear factor kappa-light chain enhancer of activated B cells; Nkx3.2, Nk3 homeobox 2; PI3K, phosphatidylinositol-bisphosphate 3-kinase; PTHrP, parathyroid hormone-related protein; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; Runx2, runt-related transcription factor 2; Smad, Sma- and Mad-related protein; SOX9, sex-determining region Y-type high-mobility group box 9; TGF-β, transforming growth factor beta; TNF; tumor necrosis factor; Wnt, Drosophila segment polarity gene wingless and vertebrate homolog integrated int-1.
PTHrP/Indian hedgehog
PTHrP and IHH are key factors controlling the balance between chondrocyte differentiation and hypertrophy. PTHrP is a critical antihypertrophic factor acting via its receptor (PTH/PTHrP receptor expressed in prehypertrophic and hypertrophic chondrocytes) to counteract the effects of prohypertrophic IHH by a negative feedback loop [45 –47]. The chondrocytes stay in a mature state as long as PTHrP expression is higher than that of IHH, but they undergo hypertrophy when IHH expression increases, that is, when chondrocytes become fully hypertrophic. PTHrP signaling acts via the PTH1 receptor (PTH1R) and protein kinases A and C (PKA, PKC) and mitogen-activated protein kinase p38 (p38 MAPK) that reduce the phosphorylation of MEF2C [48 –50]. PTHrP signaling also blocks hypertrophy by stimulating the expression of Nkx3.2 [51] and preventing Runx2 expression [52]. IHH signaling acts via the Patched 1 receptor (Ptch1) that inhibits the Smoothened (Smo) receptor, leading to the activation of transcription factors of the GLI-Krüppel family [glioblastoma-isolated protein (Gli) to induce Runx2 expression] [53].
Wnt
The canonical Wnt/β-catenin prohypertrophic pathway acts by intracellular transport of canonical Wnts (Wnt4, Wnt8, Wnt9) via the Frizzled receptor, preventing proteosomal degradation of β-catenin that subsequently translocates in the nucleus to induce Runx2 expression [54]. Noncanonical Wnts (Wnt5a, Wnt11) have dual functions, being prohypertrophic early on by induction of calcium release by G-protein-coupled receptors (GPCR; Wnt/Ca2+ pathway) and antihypertrophic later on by inhibiting Runx2 expression through activation of phosphatidylinositol-bisphosphate 3-kinase (PI3K) and of the nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) [55].
TGF-β/bone morphogenetic protein
Even though TGF-β is a critical inducer of chondrogenesis, it can display prohypertrophic effects on interaction with receptors (TGFβRI, TGFβRII). Subsequent signaling via specific members of the Sma- and Mad-related family (Smad1/5/8) regulate Runx2 expression [56] possibly by epigenetic changes, while signaling via Smad2/3 instead has antihypertrophic effects by SOX9 stabilization and Runx2 inhibition by epigenetic regulation [57]. A tight balance between Smad2/3 and Smad1/5/8 is thus critical to the processes of chondrocyte differentiation and hypertrophy, and a shift from Smad2/3 to Smad1/5/8 signaling has been reported to lead to hypertrophic phenotype in OA [58]. BMPs (BMP-2, -4, -5, -6, -7, -9) belong to the TGF-β superfamily of factors that act through Smad1/5/8 and growth arrest and DNA damage-inducible protein 45beta (GADD45beta) signaling on binding to the receptors [BMP receptor type I (BMPR-I), BMPR-II], leading to Runx2 expression [59]. The growth differentiation factor 5 (GDF-5), another member of the BMP family of factors, also displays hypertrophic activities by activation of the ETS (erythroblast transformation-specific transcription factor)-related transcription factor (ERG) [60,61].
Fibroblast growth factor
Signaling via members of the fibroblast growth factor (FGF) family and their receptors (FGFR) is another important component of the control of hypertrophy [62]. FGF-2 interacts with FGFR1, activating Raf-mitogen-activated protein kinase kinase 1/2 (MEK1/2)-extracellular signal-regulated kinase 1/2 (ERK1/2) signaling that positively regulates Runx2 expression [63,64]. FGF18 acts via FGFR3 and MAPK signaling to downregulate proliferation and maturation, with inhibition of IHH expression [65,66] and with FGFR1 to regulate vascular invasion by inducing the expression of VEGF [67].
Insulin-like growth factor
The IGF-I acting via its receptor (IGF-IR) also enhances hypertrophy via the PI3K/serine/threonine kinase Akt (also known as protein kinase B) pathway, inducing the expression of type-X collagen, ALP, and Runx2 [68 –70].
Microenvironment
The microenvironment is also a critical source of factors that regulate hypertrophy (Table 1).
Soluble mediators
A number of cytokines and chemokines are capable of inducing hypertrophy. They include interleukin 1 (IL-1), tumor necrosis factor (TNF), receptor for advanced glycation end products (RAGE), reactive oxygen species, leptin, chemokine-ligands (CXCL1, CXCL8, ie, IL-8), possibly acting via p38 MAPK to enhance the expression of type-X collagen, ALP, and MMP13 [71 –76].
ECM-related compounds
Cell–matrix and cell–cell interactions are particularly important during skeletal development and maturation. Various ECM receptors play key roles in cell arrangement and signaling during cartilage formation. They include the integrins (major collagen receptors that act via integrin-linked kinase and the Rho GTPase Rac1/CdC42 to promote hypertrophy) [77] and the DDRs (tyrosine kinases activated by type-II and type-X collagen that modulate the expression of MMP13) [78,79]. ECM fragmentation products play also crucial roles in hypertrophy such as type-II collagen breakdown products that are capable of upregulating type-X collagen expression [80].
Mechanical stimulation
Hypertrophic processes are also regulated by biomechanical stimuli [81 –83]. Cyclic mechanical stress and weight loading activate the Wnt/β-catenin pathway and enhance the expression of IHH, Runx2, MMP13, type-X collagen, OP for ossification, and VEGF for vascularization [84 –89]. Fluid shear forces are also capable of activating the Wnt/β-catenin pathway, leading to an upregulation of Runx2 expression [90 –93]. Pathways of mechanotransduction regulating chondrogenesis and hypertrophy may include TGF-β/Smad, IHH, and integrin signaling [94].
Oxygen tension
Adult articular cartilage has a natural hypoxic (2%–8% O2) environment. Oxygen tension plays critical roles in hypertrophy as hypoxia induces MSC chondrogenesis by activating p38 MAPK and the hypoxia-inducible transcription factor 1 (HIF-1α), promoting the expression of sox9, type-II collagen, and aggrecan while inhibiting that of Runx2 and preventing hypertrophy [5,21,95,96]. HIF-2α instead has prohypertrophic activities, enhancing the expression of Runx2, type-X collagen, MMP13, and VEGF [5,21,97 –99].
Calcium
Calcium also plays important roles in hypertrophy, especially via cytoplasmic accumulation. Extracellular Ca2+ can penetrate either by direct transfer through cell membrane ion channels [100] or by activating GPCR such as the calcium-sensing receptor (CaR) expressed in hypertrophic chondrocytes. Such processes lead to Ca2+ release from the endoplasmic reticulum [101], to the subsequent secretion of ALP-containing vesicles required for mineralization, and to the activation of the calcium/calmodulin-dependent protein kinase (CaMKII) that promotes the upregulation of type-X collagen expression [102].
Epigenetic factors
Several epigenetic events have been also reported for their impact on hypertrophy (Table 1). Histone deacetylases (HDACs) prevent the access of DNA to transcription factors through maintenance of the DNA/histone complex in a highly packed form. HDAC4, HDAC5, and HDAC7 inhibit the expression of Runx2, MEF2C, type-X collagen, and MMP13 via the TGF-β and PTHrP pathways [50,103 –109]. HDAC3 instead promotes hypertrophy by activating the PI3K/Akt pathway that impacts Runx2, type-X collagen, and VEGF expression [110]. microRNAs (miRNAs) also play important roles in the regulation of hypertrophy, such as miR-140 that inactivates p38 MAPK and inhibits MEF2C expression [111], while miR-1 and miR-365 inhibit HDAC4, consequently enhancing Runx2, IHH, and type-X collagen expression [112,113]. DNA methylation also participates in the control of hypertrophy by silencing of the type-X collagen gene at CpG sites [114].
Current Options for Cartilage Repair and Advances in Stem Cell- and Gene-Based Approaches: A Focus on Hypertrophy
Spontaneous cartilage repair and current options
Due to its avascular nature, the cartilage has only a limited capacity for self-regeneration as it has no access to reparative progenitor cells that normally migrate to sites of injury to reconstruct a damaged tissue [115 –118]. Still, spontaneous cartilage repair occurs during OA and in focal defects, depending on the type of injury but with relatively poor outcomes. OA lesions of up to 3 cm2 are filled with a blood clot that forms once the bone marrow spaces are affected by the degradation processes, while larger ones are irreparable, progressing to even more severe phenotypes with increases in the incidence of hypertrophy, angiogenesis, mineralization, and pathological remodeling of the entire osteochondral unit [115,116]. Chondral defects are invaded by cells from the synovial membrane, while osteochondral defects are filled by chondrogenically and osteogenically competent MSCs present in a blood clot originating from the bone marrow [115 –117]. However, the repair tissues generated on spontaneous repair in any kind of lesions generally do not match the structural quality and biomechanical functionality of the original, hyaline cartilage. Only a poorly organized fibrocartilaginous repair tissue is formed (type-I collagen instead of type-II collagen and proteoglycans), unable to withstand mechanical stress in the joint, and rapidly degenerating or progressing to OA [115 –117].
Various options are available in the clinics to enhance the repair of OA cartilage and of focal defects. For OA, both conservative methods (weight reduction, physical therapy), pharmacological regimens (anti-inflammatory drugs, opioid analgesics), and surgical options (osteotomy to transfer the weight load) are used [115,116,119]. Focal defects are treated depending on their nature [chondral defects: subchondral drilling, microfracturing, or abrasion arthroplasty to promote the migration of bone marrow-derived MSCs in the defect area, autologous chondrocyte implantation (ACI); osteochondral defects: ACI combined with subchondral bone grafts, implantation of osteochondral cylinders] [115 –117,120,121]. Yet again, none of these procedures fully reproduces the hyaline cartilage in the treated lesions and instead, the fibrocartilaginous repair tissue does not integrate with the adjacent, unaffected cartilage, showing mechanical resistance, leading to degenerative processes that may cause generalized OA [115 –117].
Stem cell- and gene-based approaches for cartilage repair
Stem cells for cartilage repair
The use of stem cells to treat such disorders may provide workable, clinically adapted regimens to address such issues as these are mostly the cells recruited to improve spontaneous and guided articular cartilage repair. Stem cells have an ability for proliferation, self-renewal, and multilineage differentiation potential in a specific environment from their niche [122 –124], exhibiting homing, trophic, and immunomodulatory properties [125,126].
While embryonic stem (ES) cells and induced pluripotent stem cells reprogrammed from the patient's own somatic, differentiated cells have been reported as a potentially universal and unlimited source of regenerative cells to form cartilage [127
–130], their use is still controversial due to ethical and safety reasons (rejection, teratoma formation, insertional mutagenesis) [131,132]. Apart from progenitor cells present in the amniotic fluid [133], umbilical cord blood [134], and placenta [135], adult MSCs remain the most used source of chondroreparative cells as they can be readily isolated from a wide range of tissues, including the bone marrow itself, the adipose tissue, synovium, perichondrium/periosteum, trabecular bone, skeletal muscle, and even peripheral blood [136
–140]. MSCs have been defined by the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy as follows: 1. being plastic-adherence upon maintenance in standard culture conditions, 2. expressing the surface markers CD105, CD73, and CD90 while lacking expression of CD45, CD34, CD14, CD79α, and HLA-DR, and 3. having the ability to differentiate into mesodermal lineage cells (chondrocytes, osteoblasts, adipocytes) under standard conditions [141].
MSCs have been already applied to treat patients with cartilage disorders, including for the management of OA [142 –145] and to heal focal defects [146 –151] by intraarticular injection or via surgical arthrotomy. Still, full restoration of the hyaline cartilage has not been afforded thus far with any of these techniques, showing the necessity of improving the current options of cartilage repair.
Principles of gene therapy for cartilage repair
To date, more than 2,200 phase two to three gene therapy trials have been reported to successfully treat various human diseases while the European Medicines Agency's Committee for Medicinal Products for Human Use recently approved the marketing of Glybera, a recombinant adeno-associated virus (rAAV) vector overexpressing lipoprotein lipase (LPL) for the clinical treatment of LPL deficiency [152]. Gene therapy is thus a potential tool to enhance the processes of cartilage repair by delivering chondroreparative genes in sites of irreversible, progressive cartilage injury to achieve prolonged expression of candidate factors that otherwise display very short pharmacological half-lives (some minutes) by rapid clearance from the host [117,153,154].
Different gene transfer vectors have been applied in the field of cartilage research in light of the specific advantages of each type of delivery system, including nonviral systems (electroporation, liposomes, nanoparticles) [155 –173] and vectors derived from viruses based on natural entry pathways in cell targets, including adenoviruses [162,174 –194], retro-/lentiviruses [157,195 –210], and AAV [211 –238].
While considered safe as they avoid the risk of acquiring replication competence, nonviral vectors usually result in low and short-term gene transfer efficiencies of transgene expression, making them more suited for indirect, ex vivo approaches based on the implantation of genetically modified cells [155 –173].
Viral vectors have specific features and advantages, but also particular limitations for cartilage therapy. Even though the cartilage is considered as a tissue relatively protected from immunity, adenoviral vectors may retain the capacity of inducing host immune responses. Furthermore, while very effective early on, allowing for direct in vivo approaches, adenoviral vectors have a relatively limited duration of action by dilution of viral episomal forms in the targets [162,174 –194]. Expression from retro-/lentiviral vectors may be achieved for much longer periods of time by integration in the host genome, but they are less efficient (therefore rather applied in indirect, ex vivo strategies) [157,195 –210] and exhibit a risk for insertional mutagenesis, an issue in translational cartilage research as most of the disorders affecting this tissue are not life-threatening. Highly effective, small rAAV vectors (∼20 nm) [211 –238] can be also used in direct in vivo settings through the dense extracellular matrix present in most of tissues. They are much less immunogenic than adenoviral vectors due to the complete removal of AAV sequences in the recombinant genome and are mostly kept as stable episomes that allow for sustained transgene expression. The limited packaging capacity of rAAV (∼4.6 kb) and the rate-limiting step of rAAV genome conversion from single- to double-stranded DNA have been, respectively, addressed, at least, in part, by generating splicing vectors [239,240] and using self-complementary AAV (scAAV) that bypass the requirement for DNA synthesis in the target cell [241]. Active work is ongoing to circumvent natural humoral immune responses raised in the host against AAV capsid proteins [242 –244] by providing the vectors via controlled delivery systems that may mask the viral epitopes and allow for a temporal and spatial presentation of the vectors [214,228, 245 –250]. Yet, although first considered safer than the integrative retro-/lentiviral vectors, rAAV have recently raised concerns regarding their possible potential for oncogenic insertional mutagenesis leading to hepatocellular carcinoma [251,252]. While still controversial as many studies did not document such genotoxicity in vivo [253], the design of new, engineered rAAV vectors could improve the safety of human gene therapy protocols [254].
Gene- and stem cell-based therapy for cartilage repair: a focus on hypertrophy
A number of studies have provided evidence of the benefits of gene- and stem cell-based approaches to delay or prevent hypertrophy for the goal of articular cartilage repair (Fig. 4). The families of factors overexpressed to achieve these goals include adhesion molecules, ECM components, growth and transcription factors, signaling molecules, and epigenetic factors as single or combined treatments (Table 2). The strategies developed were mostly tested in vitro using various sources of stem cells (MSCs from the bone marrow, synovium, and adipose tissue), but also in in vivo, relevant translational models based on a wide range of gene transfer systems, including nonviral and viral (adenoviral, retro-/lentiviral, rAAV) vectors.
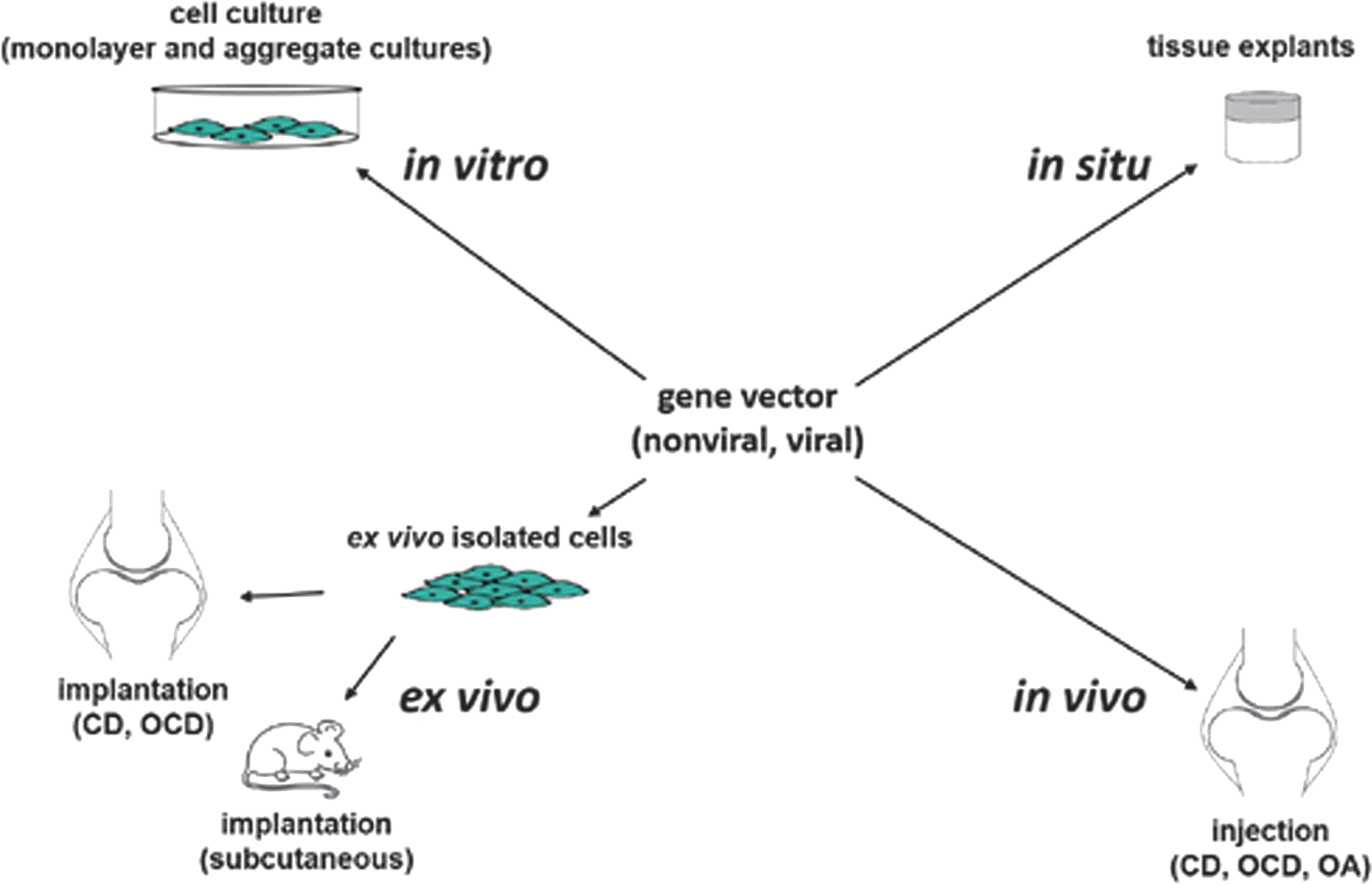
Therapeutic gene- and stem cell-based options to treat articular cartilage disorders. Approaches focusing on hypertrophy in experimental cartilage research have been performed both in cell culture in vitro, in explant tissue cultures in situ, and in vivo by either indirect transplantation of cells genetically modified ex vivo on isolation (and possible expansion) or direct injection of the gene vector. CD, chondral defect; OA, osteoarthritis; OCD, osteochondral defect.
AD, adipose-derived; AdV, adenoviral vector; BM, bone marrow; CD, chondral defect; Chm1, chondromodulin 1; COMP, cartilage oligomeric matrix protein; DLX5, distal-less homeobox 5; FGF-2, basic fibroblast growth factor; IGF-I, insulin-like growth factor I; LRP5, lipoprotein-related protein 5; LV, lentiviral vector; MSCs, mesenchymal stem cells; N-Cad, N-cadherin; NV, nonviral vector; OA, osteoarthritis; OCD, osteochondral defect; OCRL1, oculocerebrorenal syndrome of Lowe protein 1; rAAV, recombinant adeno-associated viral vector; RV, retroviral vector; scAAV, self-complementary AAV; SD, synovium-derived; shRNA, short-hairpin RNA; siRNA, small interfering RNA; SIRT6, sirtuin 6; SOX trio, SOX5/SOX6/SOX9; YAP-1, yes-associated protein 1; ZNF145, zinc-finger protein 145; Znt7, zinc transporter 7.
Inhibition of hypertrophy and osteogenic differentiation by reduction of the expression of typical markers (type-X collagen, MMP13, Runx2, ALP, OCN, OP) has been documented in vitro in various types of MSCs, including those derived from the bone marrow as isolated cells or whole cell aspirates, synovium, and adipose tissue. Isolated bone marrow-derived MSCs (human, rat, mouse) have been modified to delay or prevent such processes [201,223,225, 229,255
–269] by delivering the following: 2. FGF-2 [229], 3. SOX9 [223,225], distal-less homeobox 5 (DLX5) [268], Twist [270], yes-associated protein 1 (YAP1) [262], HIF-1α [258], 4. Wnt3a, lipoprotein-related protein 5 (LRP5) [269], zinc transporter 7 (Znt7) [263], 5. small interfering RNA (siRNA) against the zinc-finger protein 145 (ZNF145) [201], short-hairpin RNA (shRNA) against sirtuin 6 (SIRT6) [260], various miRNAs (miR-23a, -30, -31, -34a, -138, -150-3p, -320a) targeting Runx2, β-catenin, Jagged one (JAG1, a ligand for Notch 1, an osteogenic inducer in progenitor cells), the BMP-inducible gene homeobox a10 (HOXA10, a critical regulator of osteogenesis), LRP5, and focal adhesion kinase (FAK, a kinase playing a central role in promoting osteoblast differentiation) [255
–257,261,264,266,267], and 6. combinations of TGF-β/SOX9 [212], SOX trio (SOX5/SOX6/SOX9) [165], SOX9/siRNA against Runx2 [169].
Such approaches have been performed with nonviral [165,169,255 –257,259,261,263,264,266,267,269], adenoviral [258], retro-/lentiviral [201,260,262,265,268,270], and rAAV vectors [212,223,225,229]. Whole bone marrow aspirates (human, pig) have been also manipulated for the same purposes [213,217,238] by gene transfer of TGF-β [238] and SOX9 [213,217] via rAAV vectors [213,217,238]. Genetic manipulation of MSCs from the synovium (pig) [177,183] has been also successfully attempted to modulate hypertrophy and osteogenesis to overexpress HDAC4 [183] and a combination of TGF-β/siRNA against Col1 [177] via lentiviral [177] and adenoviral vectors [177,183]. Also, such processes have been reduced in MSCs from the adipose tissue (human, sheep) [175,271] on application of sequences coding for miR-100 targeting the BMPR2 [271] and by combined FGF-2/IGF-I delivery [175] via nonviral [271] and adenoviral vectors [175].
In vivo, delayed hypertrophic and osteogenic differentiation has also been reported in clinically relevant models of cartilage lesions, including experimental OA [272] and focal (chondral and osteochondral) cartilage lesions [221,223, 230]. Inhibition of hypertrophy in an experimental model of OA induced by anterior cruciate ligament transaction (ACLT) in mice has been specifically reported by delivery of the oculocerebrorenal syndrome of Lowe protein 1 (OCRL1, a Rac GTPase-activating protein) via lentiviral vector-mediated gene transfer [272], leading to a reduction in the expression of type-X collagen and MMP13 in the treated knee joints versus control knees. Delayed hypertrophic differentiation has been also observed in experimental focal lesions created in rabbits (osteochondral defects) [221,223] and pigs (chondral defects) [230] on administration of genes for IGF-I [221], chondromodulin 1 (Chm1, an inhibitor of angiogenesis) [230], and SOX9 [223] via classical rAAV vectors [221,223] or using scAAV constructs [230], mediating decreases in type-X collagen, MMP13, type-I collagen, Runx2, and β-catenin.
Conclusions
In this review, we summarized the current knowledge on hypertrophic alterations in cartilage injuries and provided evidence of the complexity and variety of mechanisms governing such events. Even though a number of studies reported the benefits of gene- and stem cell-based approaches to delay hypertrophy for the goal of articular cartilage repair, full inhibition of this process remains to be clearly demonstrated in light of the many, interconnecting pathways implicated. Indeed, many crosstalks between each independent pathway have been described [38], including FGF-IHH/PTHrP-BMP [63], PTHrP-IGF-β-catenin [70], and FGF-TGF-β-Wnt interactions [273] that likely need to be further investigated as a possible way to adequately restrain cartilage hypertrophy. Thus, generating more detailed information on and evaluations of the developmental processes and pathomechanisms involved in the development and progression of cartilage disorders (OA, focal defects) are undoubtedly critical to define new targets for improved therapies of articular cartilage lesions.
Footnotes
Acknowledgment
This work was funded by a grant from the German Osteoarthritis Foundation (Deutsche Arthrose-Hilfe e.V.).
Author Disclosure Statement
The authors have no conflicts of interest to declare.