
Abstract
Cell-based gene therapy holds a great promise for the treatment of human malignancy. Among different cells, mesenchymal stem cells (MSCs) are emerging as valuable anti-cancer agents that have the potential to be used to treat a number of different cancer types. They have inherent migratory properties, which allow them to serve as vehicles for delivering effective therapy to isolated tumors and metastases. MSCs have been engineered to express anti-proliferative, pro-apoptotic, and anti-angiogenic agents that specifically target different cancers. Another field of interest is to modify MSCs with the cytokines that activate pro-tumorigenic immunity or to use them as carriers for the traditional chemical compounds that possess the properties of anti-cancer drugs. Although there is still controversy about the exact function of MSCs in the tumor settings, the encouraging results from the preclinical studies of MSC-based gene therapy for a large number of tumors support the initiation of clinical trials.
Background
T
It has been recently shown that MSCs also have a natural ability to migrate toward tumors, being attracted by the plethora of chemo-attractants facilitating cell homing to active cancer sites with posterior transdifferentiation due to the local microenvironmental cues [4]. The population of cancer-attracted MSCs actually support the tumor growth and progression in different cancer types [5,6]. However, anti-tumor properties of MSCs have also been reported, rendering them very attractive to researchers and clinicians [7,8]. To circumvent the problem with the duality of MSC influence on the tumor cells, a delivery of exogenous, engineered MSCs could present some solution for converting them into the unequivocal therapeutic tools.
The engineering strategies of MSCs equip them for targeted delivery of different factors using more focused biological approaches. MSCs can be modified to become the carriers of suicide genes, which, in turn, would produce toxic products that would inhibit tumor expansion, whereas the surrounding healthy tissues remain intact [9 –11]. MSCs may also be employed as the carriers of anti-angiogenesis factors that contribute to the inhibition of tumor growth and to prevent metastasis [12,13]. Yet another approach is the induction of cytokine gene expression in MSCs, which, in turn, will attract and modulate processes, making the tumor cells more exposed to the host immune system response [14 –16]. Besides this, anti-mitotic factors could be a rational target for the MSC-based anti-cancer engineering [17].
Ultimately, growing interest is focused on the use of exosomes as biological delivery vehicles for miRNA transfer, as exosomes do not elicit acute immune rejection and risk of tumor formation [18].
In this article, we will focus on some recent advances in cell-based cancer therapies using genetically engineered MSCs as well as on the potential side effects of MSC delivery strategies.
Heterogeneity of MSCs
In the 1970s, Friedenstein and his coworkers identified within the bone marrow a subpopulation of nonhematopoietic cells with a fibroblast-like morphology designated as colony-forming unit fibroblasts [19]. Afterward, the term “MSCs” was adopted by the Caplan group to define a population of stem cells with a three-lineage differentiation potential [20].
In 2006, the International Society for Cell Therapy (ISCT) proposed the minimal criteria for MSCs: adherence to plastic when cultured in vitro; possession of a trilineage mesodermal differentiation capacity toward chondrocytes, osteocytes, and adipocytes. Additional requirements for MSCs include the expression of the cell surface molecules such as CD73 (ecto 5′ nucleotidase), CD90 (Thy-1), and CD105 (endoglin) as well as the absence of hematopoietic markers, including CD45, CD34, CD14 or CD11b, CD79α, and the MHC II class cellular receptor HLA-DR [21].
However, these criteria have been proved to be inadequate. The expression of this broad set of markers was also found on fibroblasts and on the surface of the other cell types [22]. In fact, the isolation of MSCs according to ISCT criteria produces heterogeneous, nonclonal cultures of stromal cells containing stem cells with different multipotential properties, committed progenitors, and differentiated cells [23]. In addition, it was recently postulated that only a minor subpopulation of pluripotent stem cells among MSCs, called multilineage-differentiating stress-enduring (Muse) cells, are responsible for the broad spectrum of differentiation abilities previously attributed to the whole MSC population [24].
Bone marrow remains one of the major sources of MSCs for clinical use; however, these cells can be successfully isolated from other tissues such as adipose, umbilical cord blood, and Wharton's jelly. MSCs from various sources share some common features but exhibit many differences, including the variable potential for differentiation and functional abilities. Moreover, a study by Lv et al. demonstrated that only a small part of cells among the MSC population are stem cells, with trilineage differentiation potential, whereas others represent a mixture of different cell types with support functions. Researchers proposed novel specific markers that associate with the stemness of MSCs, including Stro-1, SSEA-4, and CD146. They revisited the antigens expressed on the surface of MSCs from different sources, aiming in their potential as MSC markers, and suggested to define the relevant panel for future investigation [25].
Some recent papers demonstrated significant differences between MSCs obtained from neonatal and adult sources, favorable neonatal tissues as less differentiated with higher proliferation rate and clonality [26]. Recently, specific markers regarding the source of MSCs have also been identified. CD271 has been described as the best marker for characterization of the bone marrow MSC population [27].
Moreover, despite the similar pattern of surface antigen expression, global expression patterns vary significantly in MSC population isolated from bone marrow, adipose tissue, and umbilical cord blood. Numerous publications indicate that MSCs have multiple developmental origins and belong to pericytes, fibroblasts, or neural crest cells but issues regarding ontogeny of MSCs are still very controversial [28,29]. Clonal assays demonstrated that in an MSC population, multiple types of cells with different developmental potential exist [30].
Due to this heterogeneous nature of MSCs, their precise characterization in the absence of known accurately defining biomarkers poses a challenge for their further use in cell therapy. However, the problems with detailed identification should not interfere with future investigation of their therapeutic properties, even if the results among studies somewhat vary.
Inherent Anti-Cancer Properties of MSCs
Several studies postulated that naïve, nonengineered MSCs may exert anti-tumor activities [31 –33]. However, it should be noted that this alluring from the therapeutic point-of-view feature of MSCs is still under debate due to contradictory data reported on MSC influence on tumor cells [34]. For instance, MSCs derived from different tissues may stimulate or suppress glioblastoma cell proliferation as reported by Akimoto et al. that adipose tissue–derived MSCs (AT-MSCs) induced and umbilical cord blood–derived MSCs (UCB-MSCs) inhibited the progression of the glioblastoma cells [35].
Even more remarkably, MSCs of the same origin, cultured in the same conditions in vitro, promote or restrain tumor progression depending on the protocol applied during the experiment, that is, human fallopian tube MSCs (hftMSCs) used in a murine breast adenocarcinoma study [36]. In this study, hftMSCs participated in tumor progression when coinjected subcutaneously with tumor cells, but they exerted antitumor effects when administrated intraperitoneally to the animals already bearing tumor cells.
In addition, attention must be paid to senescent MSCs with therapeutic purposes, due to the contradictory data obtained, as on the one hand, that type of cells promotes cancer cell migration and proliferation, that is, by the galectin-3 secretion in the case of AT-MSCs [37], or by the secretion of IL-6 in the case of umbilical cord–derived MSCs (UC-MSCs) [38]; however, when UC-MSCs with tumor progression-fomenting properties are pretreated with IL-6, they exert anti-tumor properties [39]. On the other hand, senescent AT-MSCs were proved to inhibit tumor growth, but when these cells were primed by tumor cells, their anti-tumor effect was abolished [40].
Furthermore, senescent bone marrow–derived MSCs (BM-MSCs) triggered senescent phenotype in proliferating MSCs, confirming the notion that secreted factors from MSCs might promote senescence in neighboring cells that, in turn, might have an application in the anti-cancer cell-based approaches [41]. Thus, taking into consideration several aspects of the MSC biology, including altered molecular characteristics caused by cellular senescence [42], in spite of the anti-tumor properties, naïve MSCs ought to be used with caution [43].
Nonetheless, to limit the story to the productive side of MSCs, there are several examples of beneficial effects elicited by the unmodified MSC administration in various cancer types, suggesting that MSCs do possess intrinsic anti-tumor properties that are worthy of interest and further investigation (Fig. 1A). There are several prominent examples from anti-glioblastoma experiments [44 –47], followed by other anti-cancer MSC application such as in breast cancer [48], liver cancer [49], pancreatic cancer [50], prostate cancer [7], colon cancer [51], myeloma [52], and sarcoma [53], up to the case of the human BM-MSCs use in an anti-lymphoma [54]. Interestingly, neither positive nor negative effects were attributed to human Wharton's jelly-derived MSC secretome on lung cancer cells in vitro [55].

Complex interaction between MSCs and cancer cells.
Due to their inherent ability to migrate toward lesion sites, MSCs seem to be very attractive in future anti-tumor therapies. For instance, UC-MSCs contributed to the increased overall anti-tumor effects when they were administrated in vivo into tumor-bearing mice followed by the therapeutic irradiation exposure. In this case, human tumor cells had been implanted to the dorsal skin folds to generate bilateral xenotumors followed by the intraperitoneal MSC administration. In the next step, only one tumor-containing site was irradiated, leaving the opposite tumor site for the study of “bystander effect” [56]. Due to their positive impact on radiotherapy effects, UC-MSCs might find their application in the tumor radio-sensitization approaches [56].
After BM-MSC administration into mice bearing hepatocellular carcinoma (HCC), upregulation of p53 and caspase-3 genes was reported in liver tissue, which eventually led to the apoptosis induction [32]. In addition, tumor cells were found to be in their majority in the G0/G1 phase, with the concomitant S-phase decline.
Furthermore, MSCs could limit the growth of liver cancer cells by releasing Dickkopf-1 (Dkk-1) factor, which was reported to be an inhibitor of Wnt/β-catenin signaling pathway [57], which, in turn, is frequently elevated in various tumors and contributes to their expansion [58]. Dkk-1-secreting MSCs caused a decline in the expression of Wnt/β-catenin signaling pathway-related factors, that is, bcl-2 and survivin in human liver carcinoma cell line in vitro [59]; likewise, Dkk-1 released from UC-MSCs elicited the suppression of the Wnt/β-catenin signaling pathway in human breast adenocarcinoma cells in vitro [60].
Time-dependent anti-tumor activity was noticed in an in vivo study with MSC intravenous administration [61]. In this study, implanted BM-MSCs contributed to the elevated HCC cell apoptosis; however, changes in tumor apoptotic and anti-apoptotic genes were not conclusive, and reported effects were reduced with time. In another in vivo study with BM-MSC tail vein injection in artificial pulmonary metastatic mouse and ascitogenous hepatoma model, BM-MSC treatment brought more encouraging results in the form of reported substantial improvement of the lifetime of mice and inhibition of the development of both tumor types [62].
In vivo, UCB-MSCs triggered PTEN stimulation not only in glioblastoma cells that were in direct contact but also in those in the vicinity [44]. PTEN upregulation influenced Akt expression pattern, causing PI3K/Akt pathway disruption and leading to the inhibition of growth and migration of cancer cells [44].
Glioblastoma tumor progression needs new blood vessel formation. Ho et al. indicated that BM-MSCs could exert anti-tumor effects in this regard [45]. It was shown that glioma tumor angiogenesis might be unsettled by BM-MSC paracrine action on endothelial progenitor cell recruitment, with concomitant downregulation of proangiogenic factors [45]. Further, BM-MSC secreted factors that inhibited endothelial cell tube formation in vitro and participated in the decline of microvessel density in vivo in the subcutaneous glioma tumor mouse model.
In an in vitro study, glioblastoma multiforme stem-like cells entered senescence, but not apoptosis, with a hallmark of cyclin D1 increased level on account of exposure to BM-MSC conditioned media [47]. An increased sensitivity to the anti-cancer drugs, that is, Temozolomide and 5-Fluoro-Uracil, was also noticed [47]. Similar results were observed in the case of human breast cancer cells that were co-cultured with AT-MSCs [63].
Another in vitro study brings data on apoptosis and differentiation induction in the U251 human glioma cell line after stimulation with MSC conditioned media from different MSC origins: AT-MSCs and UC-MSCs. In this study, after the MSC conditioned media exposure, elevated levels of mRNAs for caspase-3 and -9 were detected with the simultaneous decline in mRNA levels of survivin and X-linked inhibitor of apoptosis protein (XIAP) in U251 cells. Additionally, G0/G1 growth cell arrest had been reported in these cells. Besides, MSC conditioned media called out U251 cell differentiation toward normal phenotype glial cells that were manifested with GFAP presence and cell migration decline [46]. On the other hand, there are some data claiming that MSC origin is crucial, because different tissue-derived MSCs act in completely different ways [35].
UC-MSCs exert their inhibitory effect by initiating cell cycle arrest on breast cancer cells both in vitro and in vivo [48], and BM-MSCs were proved to reduce metastasis in vivo [64]. Yoon et al. performed experiments on BM-MSC activation to arouse their anti-cancer properties [65]. BM-MSCs were stimulated with TNF-α, and DNA/RNA was released from apoptotic cancer cells.
Culture medium from osteo-induced AT-MSCs contributed to the in vitro proliferation inhibition and apoptosis induction in breast cancer cells [66]. Such a positive influence might be dependent not only on the differentiation state of AT-MSCs but also on the origin of the therapeutic MSCs [67]. These data are reinforced by observation of the immortalized MSC line that produced and secreted tissue inhibitor of metalloproteinase-1 and -2 (TIMP-1, TIMP-2). The elevated presence of TIMP-1 and TIMP-2 in the extracellular matrix, undoubtedly, interferes with the tumor cell migration processes that are crucial for tumor progression through metastasis [68].
Suicide Gene Approaches
The herpes simplex virus-thymidine kinase (HSV-TK) became one of the most used enzymes employed in suicide gene anti-cancer approaches [69]. In the case of MSCs, the advantage of this approach is that HSV-TK modified MSCs could be effectively delivered to the area of interest and ganciclovir, a substrate for HSV-TK, could be safely administrated systemically. The toxic product is exclusively produced from ganciclovir by the HSV-TK-bearing MSCs, which, in practice, would target the region occupied by tumor cells (Fig. 2A). As a consequence, only tumor cells will be affected, whereas surrounding tissues would remain intact [70].
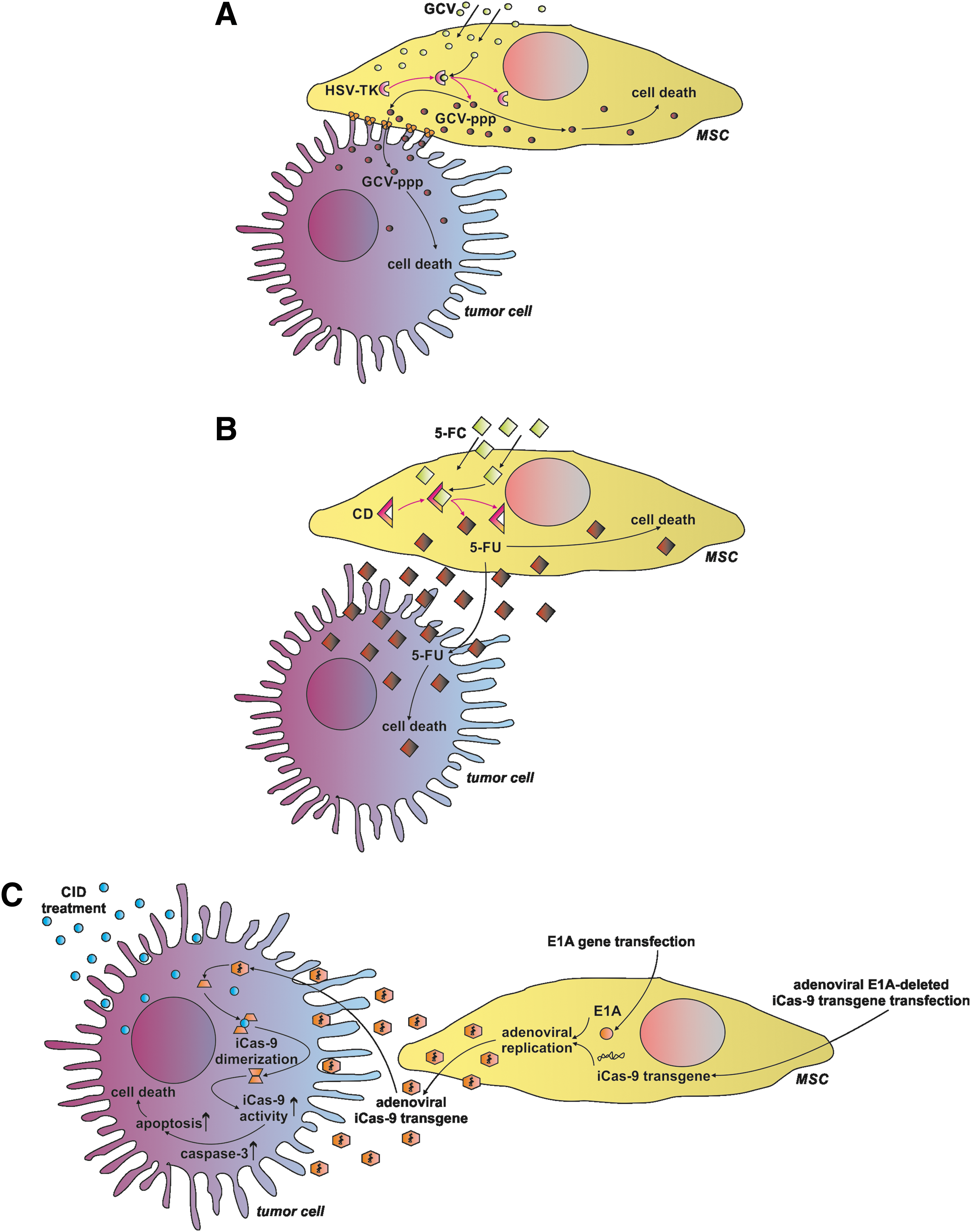
Suicide gene delivery system mediated by MSCs.
HSV-TK engineered MSCs act mainly through the “bystander effect,” which consists of an action of the toxic compound not only in the cells where it had been produced but also after its delivery to the surrounding target cells [71,72]. Additionally, this mechanism is reported to be very effective for anti-tumor acting BM-MSCs [73]. In the case of HSV-TK-based anti-cancer cell therapy, the gap-junctional intercellular communication system between therapeutic and target cells is crucial due to its involvement in the toxic compound transmission [74]; thus in practice, this phenomenon may lead to the insufficient toxic compound penetration throughout the tumor site, leading to incomplete treatment. For this reason, advanced monitoring methods are desired to exert control over ongoing processes in vivo [75,76].
On the other hand, the HSV-TK bystander effect might be enhanced by the simultaneous connexin 43 overexpression in targeted glioma cells, leading to the increase in the number of gap-junctional connections between those cells and therapeutic BM-MSCs [77], or through chemical stimulation such as by histone deacetylase inhibitor 4-phenylbutyrate (4-PB) [78], or valproic acid [79].
In spite of the reported drawbacks, this kind of MSC modification poses an attractive solution for anti-cancer treatment, since no adverse effects were reported for tumor neighboring normal cells [80] and no considerable negative side effects were found for HSV-TK engineered BM-MSCs themselves [81]. The HSV-TK engineered MSCs were applied in a variety of studies on different tumor types such as pancreatic carcinoma [82,83], HCC [84], prostate tumor [9], pulmonary melanoma metastasis [85], and a cluster of experiments with the aim of battling gliomas [86 –91]. Besides, HSV-TK BM-MSCs have been already employed in I/II clinical trials on gastrointestinal tumors [92].
In many cases, HSV-TK engineering might be enforced by additional modifications aiming at the increase of therapeutic effects of modified cells, that is, HSV-TK overexpression was accompanied with cytosine deaminase (CD) expression with an example of the lung metastases treated with engineered AT-MSCs [93] and ovarian carcinoma studies utilizing UCB-MSCs [94]. Furthermore, the HSV-TK engineering of BM-MSCs could be improved by the co-transfection of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene [95,96]. To improve the efficiency of gene therapy, some investigators have combined HSV-TK suicide gene therapy with classical radiotherapy or Temozolomide, both of which are commonly used to treat glioblastoma patients. It was shown that this combination was more efficient than HSV-TK alone [97].
CD was proposed to be a good candidate for the pro-drug anti-cancer strategies, even claimed to be the most effective pro-drug system compared with others [98], and considered harmless for basal biologic properties of CD-producing AT-MSCs [99] and BM-MSCs [100] (Fig. 2B). CD converts a low-toxic substrate 5-fluorocytosine into a 5-fluorouracil, which is known as a potent anti-cancer agent, but the systemic administration is associated with the adverse side effects such as severe myelin degeneration [101]; hence, the MSC cell-based therapy is an interesting proposal to address this question. However, CD-producing cells induce immune responses, leading to the rapid clearance of the engineered cells and causing limited duration of their anti-cancer action [102].
Nonetheless, CD overexpressing MSCs were successfully used in glioma [103 –106], glioblastoma [10], osteosarcoma [107], gastric cancer [108], medullary thyroid carcinoma [109], and melanoma experiments [110], showing the evident anti-cancer activity of the therapeutic cells with good prognosis for future clinical trials and with an example of the ongoing clinical trial NCT01156584 on malignant glioma. Concordantly, a significant inhibition of tumor growth was observed in the colon cancer and prostate cancer mice models after the CD-engineered AT-MSC administration [111,112].
Finally, the inducible Caspase-9 (iCas-9) suicide gene system might find its application in the MSC-based anti-tumor therapies (Fig. 2C). The iCas-9 system includes activation of the Caspase-9 by the small-molecule chemical inducer of dimerization (CID), which is designed to interact with the iCasp9 [113]. For instance, iCas-9-producing BM-MSCs treated with CID initiated apoptotic pathways, causing their relatively fast clearance in vivo [114]. This feature was utilized in the anti-lung cancer BM-MSC-based experiment. In this case, the Bortezomid stimulation of the iCas-9-producing BM-MSCs led to the increase of pro-apoptotic activity via the Caspase-3 stabilization, confirming feasibility of the iCas-9 system usage in the field of anti-tumor activities [115].
Anti-Angiogenesis Approaches
The tumor progression is not possible without the participation of new blood vessels. For this reason, the induction of angiogenesis inhibition is one of the worth considering issues in the field of anti-cancer treatment (Fig. 3A). The vascular endothelial growth factor (VEGF), a core element involved in the angiogenesis [116], was found plausible as a potential target in the anti-angiogenic cancer therapy [117].

Diversity of anti-tumor MSC-based approaches.
Apart from this, the application of a soluble VEGF receptor-1 (sFlt-1) in the treatment of solid human ovarian carcinoma in the mice model was found to be beneficial, contributing to the reduction of tumor growth and vascularity [118]. In the case of BM-MSC-based studies, the sFlt-1 modified BM-MSCs were administrated to the metastasis mice model of Lewis lung cancer and colon cancer carcinoma. sFlt-1 was secreted by the engineered cells in the tumor loci, participating in the anti-angiogenic and pro-apoptotic processes within tumor tissue [119].
The physiological function of endostatin is the inhibition of endothelial cell proliferation, angiogenesis, and tumor growth [120]. Besides, endostatin was described as an inducer of endothelial cell apoptosis [121]. The anti-tumor effects of endostatin were investigated in the colorectal peritoneal carcinomatosis mouse model, where the endostatin overexpressing placenta-derived MSCs (plMSCs) were intraperitoneally administered. In in vivo studies, a significant reduction in the tumor nodules and the prolongation of survival after the transgenic cell administration were observed [122].
It is believed that Kringle 1–5 protein (K1-5) is a stronger inhibitor of angiogenesis than endostatin and angiostatin [13]. Human plMSCs expressing K1-5 showed effective inhibition of angiogenesis both in vitro and in vivo. Furthermore, this result suggested that the fiber-modified viral vector transfection method could be effectively adapted in an anti-angiogenic strategy in cancer therapy.
Based on the data that the high-mobility group box1 proteins (HMGB1) are involved in the tumor angiogenesis as strong modulators [123,124], these proteins were selected as yet another target for the anti-angiogenic therapies. It was established that another DNA-binding domain of HMGB1, the A box, competed with the HMGB1 for the same binding sites. For this reason, the A box protein was utilized in the anti-HMGB1 trials [125].
The A box encoding gene was chosen for the BM-MSC transfection to produce the A box protein secreting cells, which were found to efficiently migrate toward colon cancer and to block the activity of HMGB1, cooperating with significant tumor growth [17].
Another important factor is thrombospondin-1 (TSP-1), a matricellular protein that is widely considered to be involved in the inhibition of angiogenesis and tumorigenesis [126,127]. A recent study demonstrated that BM-MSCs expressing a novel variant of TSP-1 significantly inhibited tumor progression and had an anti-angiogenic effect on glioblastoma cells [128].
The pigment epithelium-derived factor (PEDF) is a potent antagonist of VEGF in the regulation of angiogenesis [129], thus another anti-angiogenic strategy was proposed [130]. BM-MSCs were engineered to overexpress the PEDF and used in studies on the HCC in nude mice. The exogenous cells properly migrated to the sites of tumor growth with the posterior inhibition of tumor progression and participated in the decline of pulmonary metastases development.
In a different study, the PEDF overexpressing AT-MSCs were reported to reduce the tumor growth and endothelial cell tube formation in a prostate cancer model [131].
In both cases, the engineered MSCs had strong anti-angiogenic activity [130,131]. Based on a recent report, BM-MSCs overexpressing PEDF could inhibit tumor angiogenesis and increase apoptosis of gliomas [132]. The study conducted in mice reported that intravenously administrated PEDF-modified MSCs migrated and delivered PEDF to target glioma cells, prolonging survival of glioma-bearing mice. It is believed that this strategy might be a novel and promising therapeutic approach for refractory brain tumors.
Recent reports confirmed the effectiveness of IL-12 as a powerful strategy in MSC-mediated tumor therapy [133,134]. IL-12 is considered the most efficient cytokine; it exhibits anti-tumor effects, has antiangiogenic effects, and activates natural killers and T lymphocytes [133]. The anti-tumor effect of IL-12 was shown in the mouse orthotopic glioblastoma model, where IL-12 overexpressing BM-MSCs were intracranially administrated. In this study, the significant retardation of tumor growth and an increase of mouse survival were observed [133]. In another report, the anti-tumor effect of MSCs that produced IL-12 was also confirmed in kidney cancer, cervical cancer, and Ewing sarcoma models [135 –137].
Another protein with potential future application in MSC-based therapy trials is the interferon-inducible protein-10 (IP-10), which was previously found to be a strong anti-angiogenic chemokine [138]. AT-MSCs were successfully transfected with the IP-10 encoding gene and initially characterized in terms of changes in the expression levels of other genes that are potentially important in anti-cancer therapies such as VEGF, CXCR4, and SDF-1 [139].
MSCs Modified to Express Anti-Tumor Cytokines
Another field of interest in the anti-cancer attempts employing MSCs as therapeutic cells deals with the MSCs overexpressing the various cytokines (Fig. 3B). It is well known that inflammation is one of the consistent features of the tumor microenvironment. Immune cell infiltration of tumors can have a dual role, leading to either an anti-tumor response or inhibition of an immune reaction, promoting growth and spread of malignant disease. Thus, in noninflammatory “silent” conditions, therapeutic strategy to activate pro-tumorigenic immunity, based on the local production of cytokines by MSCs transduced to express specific genes of interest, is intensively explored [140]. It was shown that MSCs overexpressing different cytokines have suppressed tumor growth by the selective induction of apoptosis of cancer cells or activation of immune response [141].
According to the type of cancer, a series of anti-cancer genes, including interferons (IFNs), tumor necrosis factor (TNF) super family proteins, or interleukins (ILs) genes, have been transfected into MSCs. Interferons released by engineered MSCs demonstrated anti-proliferative activities on tumor cells [142].
IFN-β is the most therapeutically attractive member of the IFN family. It was proved that IFN-β gene modified BM-MSCs (BM-MSCs/IFN-β) effectively inhibited the proliferation of HCC or prostate cancer (PC-3) cell lines in vitro [143]. Co-culture of BM-MSCs/IFN-β with tumor cells accompanied with high levels of IFN-β secretion decreased the percentage of hepatoma cells with incorporated BrdU [143]. Systemic transplantation of BM-MSCs/IFN-β caused a significant reduction of tumor burden, suppression of metastasis, and prolonged animal survival in HCC xenograft-bearing mice [143].
A co-injection of MSCs and cancer cells was also described. The authors used transfected human MSCs with IFN-β to treat melanoma xenografts in mice [144]. An injection of MSCs/IFN-β into the peripheral circulation resulted in reduced tumor growth and an increased animal lifespan. These results are in concordance with other findings, in which MSCs/IFN-β potentially inhibited breast carcinoma [145], glioma [146], melanoma [147], pancreatic cancer [50] and gastrointestinal carcinoma [148]. In each of these tumor experimental models, the treatment showed efficacy in the inhibition of local tumor growth.
The mechanism by which IFN-β caused cancer cell growth attenuation still remains an open question. Dedoni et al. have shown that IFN-β treatment downregulates the PI3K/Akt pathway in neuroblastoma cells [149]. These data provide a line of evidence for the regulatory effect of MSCs/IFN-β on Akt signaling cascade inhibition in HCC cells [143].
The extent of the CD8+ T-cell infiltrate positively correlates with good prognosis in many cancers. Tumor-specific IFN-γ-producing CD8+ T-cell immune response is important to the outcomes of cancer-bearing animals [150]. It supports the notion that to inhibit tumor growth, delivery of immune-stimulatory genes by MSCs is valuable [151]. ILs that regulate immune response are known to have an anti-tumor effect via positive modulation of the endogenous immune system [152]. The delivery of ILs via MSCs has been advantageous to improve the anti-cancer immune surveillance by activating cytotoxic lymphocytes and natural killer cells [152]. The value of IL-12, IL-15, IL-18, and IL-21 engineered MSCs for immunotherapy of different cancers has been confirmed [15,136,137,153,154].
It is well known that IL-12 is a key cytokine inducing a pro-inflammatory pattern of immune response [155]. BM-MSCs engineered to express IL-12 have been shown to slow tumor progression in mice bearing renal cell carcinoma [136] or head-and-neck tumors [137]. In vivo administration of MSCs/IL-12 prevented metastases into the lymph nodes and the other organs, as well as increased tumor cell apoptosis in different tumor models of melanoma or hepatoma tumors [156]. Similarly, murine BM-MSCs retrovirally engineered to secrete IL-12 significantly reduced breast cancer growth when delivered subcutaneously in syngeneic and allogeneic hosts [157]. This treatment resulted in high density of intratumoral T-cell infiltration, suggesting that the therapeutic effect was dependent on a T-cell immune response.
MSCs have also been engineered with other immune stimulatory ILs, such as IL-15, which is associated with long-lasting T-cell anti-tumor immunity and cancer cell apoptosis in pancreatic tumor-bearing mice [153]. It was observed that in addition to NK and CD8+ T cells, γδ lymphocytes were also expanded in the tumor site after MSCs/IL-15 treatment. Furthermore, tumor-specific memory T cells were induced by IL-15 gene modified MSCs, giving the possibility to reject subsequent re-challenges with identical tumor cells by mice cured of previous tumors in such a setting [153].
In another study, human MSCs engineered to express IL-18 were tested in mice bearing noninvasive and invasive gliomas [154]. In these settings, transplantation of IL-18 secreting MSCs was associated with enhanced T-cell infiltration and long-term anti-tumor immunity in the hosts.
More recent studies have revealed the anti-tumor effects of MSCs expressing IL-21. Previously, IL-21 has been shown to activate NK and T cells and to induce a strong cell-mediated immune response in the tumor vaccine approaches [158]. In experimental settings, human UC-MSCs (hUC-MSCs) transduced by the IL-21 gene induced tumoricidal activities in ovarian tumor-bearing mice [15]. This was accompanied with enhanced NK cytotoxicity and elevated serum levels of IFN-γ and TNF-α, which were probably related to the secretion of IL-21 from transplanted hUC-MSCs/IL21. Both cytokines have an important function in anti-tumor immunity by increasing cytotoxic potential of NK cells.
Moreover, hUC-MSCs/IL-21 could inhibit tumor growth through the negative regulation of the Wnt signaling pathway being involved in metabolism, proliferation, and cell cycle. Hence, transplantation of hUC-MSCs/IL-21 downregulated the expression of β-catenin and cyclin D1 in ovarian cancers [15]. This alteration may refer to the inhibition of tumor growth confirmed histopathologically.
TNF-α is another major mediator of tumor growth. This cytokine was originally named owing to its anti-tumor properties; however, TNF-α has been shown to have a paradoxical effect on cancers by both inhibiting and promoting tumor growth depending on the tumor type and the level of TNF-α protein [159]. At the cellular point, the positive anti-cancer role of TNF-α has been shown to induce apoptosis and necrosis in the malignant cells in vitro.
In vivo, TNF-α exerts a stronger anti-tumor activity, because it acts toward other cells in the tumor microenvironment, especially toward endothelial cells, inducing their apoptosis and leading to vessel destruction [160]. Moreover, TNF-α can enhance anti-tumor immunity via stimulating T-cell infiltration into the tumor tissue [161]. TNF-α is also capable of regulating other cytokines, showing synergism with their cytotoxic effects [162].
AT-MSCs transduced to express TNF-α gene induced apoptosis of tumor cell lines of different origins in vitro, and when injected subcutaneously with melanoma cells into nude mice resulted in tumor growth inhibition [16]. This was in concordance with the previous studies of Al-Zoubi et al., in which delivery of TNF-α by engineered MSCs completely prevented breast cancer tumor formation in mice [163]. Significant inhibition of tumor mass described in vivo may be probably also a result of MSCs/TNF-α stimulation to secrete other inhibitory factors and cytokines by the host [163].
In addition to TNF-α presence, the upregulation of gene transcripts associated with inflammation and senescence, that is, of IL-1β, IL-6, IL-8, and MCP-1, were observed in vivo [163]. We can even speculate that an insufficient level of TNF-α could be a factor facilitating tumorigenesis in people.
Among the other factors could be TRAIL, which is a molecule that selectively kills transformed and cancer cells but not the normal cells, making it an attractive target for anti-cancer therapy [164,165]. A number of studies have shown the therapeutic efficacy of MSCs engineered to express TRAIL in either cell lines or different experimental models of cancers both in vitro and in vivo.
Loebinger et al. have shown that human BM-MSCs transduced with the TRAIL gene caused lung, breast, squamous, and cervical cancer cell apoptosis and death in co-culture experiments [140]. Subcutaneously xenotransplanted TRAIL-expressing MSCs contributed to the tumor mass reduction and inhibited metastases of breast cancer cells in NOD/SCID mice. Similarly, human MSCs genetically engineered to express TRAIL induced apoptosis in glioma cell lines, had profound anti-tumor effects in orthotopic glioma tumors in mice, and resulted in a significant increase in animal survival [166 –168].
Human pancreas–derived MSCs (pan-MSCs), modified to express a soluble form of recombinant TRAIL, have also been explored for anti-tumor effects in gastrointestinal carcinomas [169]. TRAIL has been shown to induce cytotoxic effects on pancreatic cancer cells [169]. Similarly, in a human pancreatic carcinoma mouse xenograft model, the engineered MSCs expressing TRAIL slowed tumor growth and reduced the incidence of metastases [170]. Similarly, MSCs/TRAIL induced apoptosis of human colorectal cancer cell lines, including DLD1 in vitro; however, they failed to affect colorectal adenocarcinoma xenograft growth secondary to pulmonary engraftment in mice after systemic MSCs/TRAIL transplantation [171]. This observation highlights not only the strong potential of TRAIL-mediated anti-tumor effect but also some limitations of therapies using genetically engineered MSCs.
LIGHT, another member of the TNF super-family [172], was used in the anti-tumor studies, exposing its role as a strong inducer of anti-tumor immunity. Systemic administration of UCB-MSCs transfected by the LIGHT gene into gastric cancer-bearing nude mice suppressed tumor growth [173]. The average volume of tumors in UCB-MSCs/LIGHT was smaller than in the control group with a larger tumor necrosis area. LIGHT, homologous to lymphotoxin, can enhance the extravasation and homing of naïve T cells [174].
Further data demonstrated that forced expression of LIGHT in the tumor induces a massive infiltration of T lymphocytes into tumor tissue, leading to its rejection [175]. It was shown that LIGHT sustains effector T-cell functions at the site of various tumors without defining tumor antigens [176]. Delivering LIGHT protein in genetically engineered MSCs into breast cancer-bearing mice resulted in tumor growth repression [177]. The stable proliferation rate of cancer cells indicated that tumor growth arrest was not involved in the therapeutic efficacy of MSCs/LIGHT. However, the tumors showed CD4+ and CD8+ T-cell infiltration, which may contribute to maintenance of tumor control.
Moreover, MSCs-/LIGHT-treated mice induce pro-inflammatory cytokines, that is, IFN-γ, IL-6 in local tumor tissue [177]. In addition, IFN-γ-producing T-cell level also increased in lymphoid tissues of tumor-bearing hosts after MSCs-/LIGHT-mediated tumor control caused by the primed antitumor immunity. This phenomenon was not due to the direct contact inhibition of tumor cells with LIGHT-producing MSCs. Yu et al. have shown that LIGHT gene delivery inside fibrosarcoma cells inoculated in mice recruited naive T lymphocytes into the tumor microenvironment [174].
High expression of LIGHT inside the tumor promoted cytokine changes that may contribute to the activation, expansion, and conversion of CD8+ T cells into fully functional effectors. Moreover, targeting primary tumors with the LIGHT gene elicited tumor-specific cytotoxic T lymphocytes that migrate into the distal tumors and eradicated them as observed in mammary carcinoma-bearing mice [175]. It seems that LIGHT is capable of generating comprehensive immune responses against established tumors and spontaneous metastases [176].
Another example of the apoptosis induced in tumor cells is the case of the tumor-suppressor phosphatase with tensin homology (PTEN) overexpressing pan-MSCs [178]. PTEN acts as an inhibitor of the anti-apoptotic pathway, so its overexpression drives the induction of the apoptotic signaling.
Janus Face of MSC-Derived miRNA in Oncology
There is constantly growing evidence of miRNA involvement in the process of oncogenesis [179]. In that context, it has been shown that tumor-recruited MSCs enhance tumorigenic properties of breast cancers through an increase of miR-199a [180]. The miR-221 paracrine secretion from gastric cancer-derived MSCs supports growth and migration of the tumor HGC-27 cell line [181]. In turn, let-7 family miRNA has been downregulated in prostate cancer–derived MSCs [182]. In both cases mentioned earlier, the genetic intervention to normalize miRNA expression/secretion abolished the tumor-promoting properties of cancer-derived MSCs. Moreover, the exogenous miRNA can have anti-tumor activity [183,184]. The exosomes derived from naïve MSCs carry the miR-16, which downregulates VEGF, and through that mechanism inhibits angiogenesis and limits the growth of tumors [185] (Fig. 3C).
Furthermore, the MSCs can be engineered to express specific and desired anti-tumor miRNA, and such prepared MSCs through active recruitment by a cancer tissue could also serve as a Trojan horse. The one strategy for tumor targeting is finding miRNAs downregulated in cancer cells and supplying them with missing miRNA. Particularly, the low expression of miR-145 and miR-124 was found in glioma cells [186]. The genetically engineered MSCs were capable of efficient delivery of these miRNAs through gap junctions and exosomes to the glioma cells, which resulted in the migratory inhibition properties of the latter cells [187]. Instead of genetic engineering, the MSCs can be equipped with synthetic miRNA, which then will be enveloped in exosomes and released. It was shown that effective supplementation of miR-143, downregulated in osteosarcoma cells, suppressed their migratory properties [188].
It has been previously shown that miR-122 can also determine resistance to chemotherapy of HCC cells [189]. In fact, the MSC-derived exosomes bearing miR-122 revealed chemosensitizing properties [189]. Such a strategy of supplementation of tumor cells with lacking miRNA for therapeutic purposes could be considered a personalized medicine approach. The contribution of EGFR amplification to the glioma invasiveness is a well-known phenomenon [190], and the delivery of EGFR-mRNA-silencing miR-146b through MSC-derived exosomes reduced its invasion, migration, and viability, thus abolishing a negative consequence of EGFR amplification [191]. The production of miR-21 by MSCs also tunes the supportive/inhibitory influence of MSC-derived exosomes on tumor growth [192].
Although the miRNA-centered approaches mentioned earlier were focused on the supplementation of tumor cells with missing miRNA to facilitate their homeostasis and reduce malignancy, the continuous search in using miRNA as therapeutics revealed that miRNA is capable of having a direct cytotoxic effect on the glioblastoma cells, whereas not on normal astrocytes [193]. It is likely that this miRNA could be delivered to the tumor through MSC-derived exosomes. Although the suppression of tumor cell invasion is highly desirable, it has also a dark side—it can produce a dormant state of tumor cells in metastatic niches, not approachable to the current chemotherapeutic regimes.
In particular, the exosomal transfer of miR-23b from BM-MSCs promoted the long-lasting local presence of breast cancer cells, which might be responsible for delayed recurrence on the unknown as yet signaling mechanism [194]. Therefore, the miRNA may also determine the chronicity of cancer disease and may require therapeutic targeting. The power of miRNA can be best presented in the case of miR-145. Its loss in glioma cells upregulates a palette of cell proliferation and migration-related proteins: cyclinD1, c-myc, N-myc, N-cadherin, and E-cadherin, which emphasizes the central role of miRNA in the regulation of cellular fate through simultaneous action on many genes [195].
Although the multigene regulatory characteristic of miRNA predisposes it as an anti-cancer weapon, there are also positive effects with the use of more specific small RNA molecules such as siRNA, and myeloma-stimulating naïve MSCs have been converted into anti-cancer acting cells by IL-6 siRNA transfection to silence myeloma arousal effects of IL-6 production [196].
MSC-Assisted Nanodrug Delivery to Tumor Sites
Additionally, MSCs can be used as carriers for the traditional chemical compounds that possess the properties of anti-cancer drugs, with the example of the paclitaxel (PTX)-loaded nanoparticles delivered to MSCs (Fig. 3D). MSCs loaded with nanoparticles containing paclitaxel, a cytotoxic drug, showed active tumor-tropism so that the release of paclitaxel took place locally [197]. This strategy characterized by high efficiency and low systematic toxicity has the potential to be developed as a cancer targeting tumor therapy in humans.
Recently, Pessina et al. demonstrated that paclitaxel-releasing MSCs had a potent therapeutic effect and suppressed the growth of a murine B16 melanoma in vitro. They also confirmed that paclitaxel-loaded MSC treatment inhibited lung metastasis [198]. Another study showed that PTX-loaded MSCs, in a mouse orthotopic glioblastoma model, migrated to the tumor microenvironment and exerted an anti-tumor effect [199]. A significant number of recent approaches demonstrated new methods of injecting nanoparticles containing classical anti-cancer drugs to MSCs [200,201]. However, up to now, no effective therapeutic treatment, anchoring drug-loaded nanoparticles to MSCs, has been discovered.
Similarly, the study of Li et al. has proved that the silica nanorattle system may be a promising vehicle for drug delivery to MSCs. Silica nanorattle-doxorubicin (SN-DOX) anchored MSCs are capable of accumulating in the vicinity of tumor tissues to slowly release the stored drug. The study also reported that increased and prolonged doxorubicin levels secreted by MSCs significantly enhanced apoptosis in human glioma cell xenografts compared with the effect elicited by the drug alone [202]. An SN-DOX drug delivery system was efficiently conjugated to MSCs by specific monoclonal antibodies that bind to the membrane proteins CD73 and CD90 [202]. Uploading with up to 1,500 nanoparticles did not affect the viability of MSCs [202]. The intracellular retention time of the silica nanorattle was sufficient, as cell-directed tumor tropism amounted to no less than 48 h [202].
In vivo experiments proved that the loaded MSCs could trace the glioma tumor cells and deliver doxorubicin with wider distribution and longer retention lifetime in tumor tissues, compared with free DOX and silica nanorattle encapsulated DOX [202]. The increased and prolonged DOX distribution into the tumor site further contributed to the enhanced tumor cell apoptosis. This strategy has potential to be developed as a high, efficient, and meaningful method for tumor targeting therapy in humans.
Malignant gliomas are aggressive and difficult-to-treat tumors. Therefore, the effective drug delivery methods that are also characterized with the capability of infiltration of the tumor site are highly desirable. One of the biggest limitations is low targeting efficiency for nanoparticle drug delivery system-based cancer therapy. Thus, in brain tumor therapy, alternative strategies are applied to enhance the delivery and homing activity. Recently, researchers have demonstrated that MSCs in an organometallic complex Ferrociphenol lipid nanocapsule delivery system can be used as promising treatment. In that study, a subpopulation of human BM-MSCs isolated adult multilineage inducible cells (MIAMI), combined with nanoparticles, migrated to the tumor microenvironment and exerted a cytotoxic effect on glioma cells [203]. In addition, this strategy could be effectively combined with classical radiotherapy.
Another alternative anti-cancer therapy is to use MSCs as the targeting vehicles to uptake and release another important drug, gemcitabine (GCB). GCB-loaded MSCs integrated into the tumor mass, therefore, were capable of delivering much higher amounts of the drug in situ than could be achieved by intravenous injection [204].
Inherent Potential of MSCs for Oncogenic Transformation
The achievement of therapeutic effect in clinical use of MSCs requires transplantation of large amounts of cells. However, the limiting factor in terms of single-dose injection is the number of isolated MSCs. Therefore, in vitro propagation is often required to reach a sufficient number of cells. During this process, MSCs may undergo genetic alterations that, subsequently, increase the probability of spontaneous malignant transformation. For that reason, there is a presumable risk for patients receiving in vitro expanded MSCs.
Issues regarding MSC transformation are very controversial, and both spontaneous and induced transformation have been reported [205,206]. On the one hand, there are some reports demonstrating that MSC expansion in long-term in vitro culture is safe and deprived of acquired chromosomal aberrations [207 –210].
One of the first results confirming stability of human MSCs in long-term in vitro culture was published by Bernardo et al.; researchers studied genetic changes in human MSCs at different stages with the use of karyotyping and comparative genomic hybridization methods [207]. MSCs obtained from bone marrow of 10 healthy donors were propagated in vitro until reaching senescence or till passage 25 and did not exhibit any sings of malignant transformation. Alternatively, other studies that investigated both osteosarcoma patient and healthy donor showed no evidence of neoplastic changes in human MSCs during long-term in vitro culture [210].
One of the greatest advantages of the potential MSC therapy is that these cells are safe and maintain chromosomal stability after transplantation; moreover, there are no results demonstrating tumor formation after their transplantation in humans [211]. However, there are some reports demonstrating malignant transformation of administrated MSCs in rodent models [212]. These studies confirm that genetically unmodified mice MSCs can undergo chromosomal abnormalities even at early passages in vitro and form malignant tumors when transplanted in vivo [213] (Fig. 1C).
Most surveys on the transformation process have been conducted in mice, but there have been few reports on spontaneous human MSC transformation that occurred during in vitro culture. Results published by Rubio et al. showed the primary molecular mechanism of transformation of human AT-MSCs after senescence caused by regulating c-myc and repressing p16 [214].
Many other papers confirmed deregulation of p53, Retinoblastoma, PI3K-AKT, and MAPK pathways involved in tumor transformation of MSCs [215,216]. Recently, researchers described the epigenetic mechanism implicated in spontaneous transformation of MSCs in long-term in vitro culture [217]. Particularly, the epigenetic modification of the p16 gene, including histone H3 lysine methylation (H3K27/9me) and DNA methylation, was investigated in in vitro cultured adult rat BM-MSCs at different stages during the transformation process [218]. These findings are in agreement with observations made by a number of other groups. These results suggest that MSCs used for transplantation should be carefully monitored in terms of chromosomal status during their expansion in vitro [219].
To confirm the real identity of transformed cells among the MSC population, other tests such as fingerprinting and short tandem repeat analysis should be employed [220,221]. According to the recent data, the main mechanism for protecting human MSCs against spontaneous transformation includes telomere shortening and lacks both telomerase reverse transcriptase expression and telomerase activity [222]. Therefore, the reports that support the existence of the human MSC transformation process are based on telomerase-immortalized human MSCs that give rise to a tumor formation. Thus, it appears that using telomerase to help production of large numbers of cells is effective, but on the other hand, it has an impact on neoplastic transformation [223].
Tumor Propagation Mediated by MSCs
Some features of MSCs such as immunomodulation or paracrine effects, which may be beneficial in regenerative medicine approaches, could be also destructive in cell therapy against cancer. Some studies have shown MSC contribution in tumor growth, metastasis, and development of anti-cancer drug resistance.
Results from co-culture of MSCs, breast cancer cells, and peripheral blood mononuclear cells (PBMCs) showed that MSCs negatively affected proliferation and migration of PBMCs and favored the shift from Th1 toward Th2 response [224]. An increase in the number of regulatory T cells (Tregs) was also observed, which could be at least partially due to MSC-derived anti-inflammatory cytokine, TGF-β [224] (Fig. 1B). In vivo studies on mammary carcinoma-bearing mice showed that intravenously administered human peripheral blood–derived MSCs (human PB-MSCs) might support tumor growth through stimulating the Tregs and secretion of immunosuppressive factors, that is, TGF-β, IL-4, and IL-10 with a simultaneous decrease in the cytotoxic capacity of CD8 T lymphocytes and NK cells [225].
Furthermore, MSCs infiltrate tumors and support their progression [6]. It was observed that the majority of the mouse glioma cell line (GL261) consisted of cells with mesenchymal features (Lin-Sca-1+CD9+CD44+CD166+ phenotype; trilineage differentiation capacity) [6]. However, most of the MSC-like cells in a tumor mass were of host origin, which indicated that endogenous MSCs were recruited to the tumor site. Moreover, brain tumor–derived MSCs co-injected with GL261 cells significantly reduced mice survival rate, compared with GL261 cell injections alone [6]. Similarly, other studies on different tumors such as head and neck cancer [226], colorectal cancer [227,228], or breast cancer [229] also confirmed the positive effect of MSCs on tumor growth.
Additionally, MSCs could contribute to promotion of metastasis formation [230]. Only few microscopic metastases were found in lungs of mice grafted with breast cancer cells (BCC) alone, whereas injections of a mixture of BCC and BM-MSCs increased the number of metastases in distant organs [230].
There are also a number of studies reporting MSC participation in the development of anti-cancer drug resistance. Mammary adenocarcinoma and Lewis lung carcinoma cells increased their resistance to paclitaxel or doxorubicin when exposed to BM-MSC conditioned medium [231]. Greater viability was due to decreased caspase-3 activity and Annexin-V expression.
Similarly, doxorubicin resistance was observed among triple-negative breast cancer cells after AT-MSC conditioned medium treatment [232]. Further studies revealed that the medium containing IL-8 caused an increase in BCRP protein expression (breast cancer resistance protein, one of the ABC transporters that are ATP-binding cassette transmembrane proteins engaged in the transport of a wide range of molecules across cell membranes, frequently against their concentration gradient, leading to the multidrug resistance in case of cancer cells [233]).
Other results showed that MSC-derived exosomes could restrain apoptosis of gastric cancer cells and enhanced the expression of multi-drug resistance-associated proteins after exposure to 5-fluorouracil [234]. In turn, Vianello et al. reported that co-culturing chronic myeloid leukemia cells with BM-MSCs prevented them from imatinib-induced apoptosis via the SDF-1/CXCR4 axis. Inhibition of CXCR4 abolished this resistance [235].
Because of the heterogeneity of the MSC population, Waterman et al. proposed a distinction between those cells into two phenotypes [236]. According to this hypothesis, MSCs can be polarized, depending on which Toll-like receptor (TLR) was previously activated on the cell surface, into pro-inflammatory MSC1 (TLR-4-primed) or immunosuppressive MSC2 (TLR-3-primed). Both in vitro and in vivo studies showed that MSC1 hampered tumor cell growth and invasion, whereas MSC2 supported these processes [237].
Conclusions
Genetically modified MSCs seem to be a promising strategy to improve cell-based therapy, enabling delivery of a plethora of factors that effectively repress tumor growth. A review of currently published studies has shown that the effects of MSCs engineered to express different genes or to serve as a vehicle for therapeutic agents for cancer therapy are multiple and may depend on the state of tumor and interactions with other cell types in the tumor microenvironment. These results are sometimes contradictory, and the factors released from modified MSCs have been implicated in both pro- and anti-tumor growth and/or metastases. Hence, the selective control of therapeutic gene expression by MSCs within the defined tumor settings provides new options to use modified MSCs for cancer therapy in patients.
Footnotes
Acknowledgments
This study was supported by grants from the Polish National Centre for Research and Development STRATEGMED1/235773/19/NCBR/2016 “EXPLORE ME” and NIH R01 NS091100-01A1.
Author Disclosure Statement
All authors declare that no competing financial interests exist.