
Abstract
Clinical findings and data obtained in animal models indicate that nutrient uptake and exposure to environmental agents during pregnancy may affect fetal/newborn gestational programming, thereby resulting in obesity and/or obesity-related disorders in offspring. Human amniotic mesenchymal stem cells (hA-MSCs) differentiate into adipocytes and are thus a suitable model to investigate adipocyte functions in obesity. The aim of this study was to elucidate the miRNome of hA-MSCs and its contribution to obesity in pregnancy. To this aim we used the following: (i) high-resolution small RNA sequencing to characterize the microRNA (miRNA) profiles of hA-MSCs of 13 obese (Ob-) and 7 control (Co-) pregnant women at delivery; (ii) multiple-method integrated bioinformatics to predict the metabolic pathways potentially miRNA deregulated in Ob-hA-MSCs; and (iii) microarray mRNA expression profiling to verify obese-associated mRNA alterations. In summary, 12 miRNAs were differentially expressed between Ob-hA-MSCs and Co-hA-MSCs, with a multiple-methods bioinformatic consensus on miR-138-5p and miR-222-3p, which were overexpressed in Ob-hA-MSCs versus Co-hA-MSCs. The top 20 significant pathways predicted to be deregulated through miR-138-5p and/or miR-222-3p/target interaction included fat cell differentiation and deposits, lipid/carbohydrate homeostasis, response to stress, metabolic syndrome, heart disease, and ischemia. In conclusion, our finding of miR-138-5p/miR-222-3p overexpression in Ob-hA-MSCs, together with the transcriptomic data, suggests that these miRNAs in obese pregnancy could derange metabolic pathways previously found impaired in tissues from obese adults or in obesity-associated disorders and concur to modify gestational programming as has been demonstrated in animal models. This raises the possibility of using diet-based strategies to normalize the perinatal miRNome in obesity.
Introduction
C
In particular, in pregnant animal models, high calorie diet induced miRNA-mediated variations in gene expression, and so affected the development of offspring [1,2]. In line with this finding, we previously reported a different miRNA profile in the amnion of obese women versus that of control women and predicted the deregulation of several pathways concurring to placental growth and function that could enhance the newborn's risk of obese-associated metabolic diseases [7]. Deregulated metabolic pathways have also been described in subcutaneous and visceral adipose tissues from obese adults in association with altered miRNA profiles [8,9].
The amnion is also a source of human amniotic mesenchymal stem cells (hA-MSCs). These cells have two advantages for research: (i) they have a close ontogenic relationship with embryonic stem cells but, unlike the latter, are accessible without ethical problems, as placenta is usually discarded at birth [10]; (ii) they are multipotent stem cells that have the potential to differentiate into a wide range of cells, including adipocytes. Consequently, hA-MSCs are a suitable cellular model with which to investigate the metabolic functions of adipocytes during obesity [10].
The expression of miRNAs has been investigated in MSCs isolated from such sources as adipose tissue, bone marrow, and amniotic fluid [11], mostly using a microarray or candidate gene approach [12]. To our knowledge, hA-MSCs isolated from obese women have not yet been studied using small RNA sequencing (small RNA-Seq). This technique is a powerful and a widely used tool with which to identify known and unknown miRNAs and to quantify miRNA levels at a higher resolution than that possible with the array technique [13]. Furthermore, thanks to powerful software, we can avoid false-positive results in the small RNA-Seq of miRNA profiles and predict, with high statistical significance, the putative effect of miRNAs in deregulating cell functions.
The aim of this study was to elucidate the miRNome of hA-MSCs from obese and control women and look for abnormalities associated with obesity in pregnancy.
Materials and Methods
Patients and controls
Thirteen morbidly obese (Ob) and seven lean control (Co) pregnant women aged [mean ± standard error mean (SEM)] 33.7 ± 1.0 and 30.7 ± 1.5 years, respectively, with a prepregnancy body mass index (kg/m2) >30 and <25 kg/m2, respectively, were recruited at the Dipartimento di Neuroscienze e Scienze Riproduttive ed Odontostomatologiche, University of Naples Federico II. The clinical and anamnestic data of the enrolled women were recorded upon hospitalization during a medical interview conducted by an expert. Mean (SEM) gestational age was 38.8 (0.3) and 38.7 (0.2) in obese and control women, respectively. Five of the thirteen (46%) obese women and 1/7 (14%) control women were at their first pregnancy. The exclusion criteria were neoplasia, viral infection, diabetes, and metabolic syndrome.
All patients and controls gave their informed consent to the study that was performed according to the Helsinki II Declaration and approved by the Ethics Committee of the School of Medicine, University of Naples Federico II (authorization n. 248/08, 23/02/2009; amendment n. 248/08/ES1, 1/10/2014). A second cohort of 14 placental tissue sections (amnion) from nine obese and five control pregnant women was used for validating purposes [7].
Sample collection
Two maternal blood samples were collected in the morning after a 12-h fast from all enrolled women immediately before delivery. One sample was used for DNA extraction, whereas the other was centrifuged and the serum was stored at −80°C until required for the measurement of the main biochemical parameters and leptin/adiponectin concentrations by routine methods or immunoassay (Bio-Rad, Hemel Hempstead, Herts, United Kingdom), respectively. Term placentas were collected at delivery by cesarean section and immediately processed to isolate the hA-MSCs, according to Parolini et al. [14].
Briefly, we removed the maternal decidua and separated the chorion from amnion. Then, we washed the amnion five times in 40 mL phosphate-buffered saline (PBS) containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 250 μg/mL amphotericin B (all from Sigma-Aldrich, St. Louis, MO) and mechanically minced it into small pieces. The latter was digested overnight at 4°C in ACCUMAX® reagent (Innovative Cell Technology, San Diego, CA), a combination of DNase, protease, and collagenolytic enzymes [15] containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 250 μg/mL amphotericin B. Digestion enzymes were then inactivated with complete culture medium constituted by low-glucose Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich) supplemented with 10% of heat-inactivated bovine serum [fetal bovine serum (FBS)], 1% of nonessential amino acids, and 2% of UltraGlutamine (all from Lonza, Basel, Switzerland).
After centrifugation at 300 g for 10 min, cell pellets and digested tissue fragments were seeded in a cell culture dish (BD Falcon, New York, NY) in complete culture medium and incubated at 37°C in 5% CO2 for 7 days. We removed digested tissue fragments and isolated cells that formed distinct fibroblast colony-forming units. Colonies reaching 70% confluence were washed with PBS, detached with trypsin/ethylenediaminetetraaceticacid (EDTA; Sigma-Aldrich), and counted and reseeded in complete medium for expansion at a concentration of about 5,000/cm2 [15]. hA-MSCs were further expanded, and when 70%–80% confluent cultures reached about four population doublings [15], they were detached with trypsin/EDTA and resuspended in PBS with 10% FBS.
The fetal origin of hA-MSCs was verified by typing DNA obtained from hA-MSCs, amnion, and mother's peripheral blood leucocytes with the AmpFlSTR® Identifiler™ PCR Amplification Kit (Applied Biosystems, Foster City, CA). DNA was extracted using the Nucleon BACC2 extraction kit (Illustra DNA Extraction Kit BACC2; GE Healthcare, Calfont St. Giles, Bucks, United Kingdom) and its concentration evaluated with a NanoDrop® ND-1000 UV-Vis spectrophotometer (NanoDrop Technologies, Wilmington, DE). Polymerase chain reaction (PCR) was performed with a GeneAmp PCR System 9700 (Applied Biosystems) instrument, and PCR primers and conditions were according to the manufacturer's instructions. Analysis of PCR products by capillary electrophoresis performed with the ABI Prism 3130 Genetic Analyzer (Applied Biosystems) confirmed the fetal origin of all the isolated hA-MSCs [15].
To immunophenotype hA-MSCs, we analyzed 38 hematopoietic, mesenchymal, endothelial, epithelial, and no-lineage membrane antigens (CD14, CD15, CD16, CD19, CD28, CD33, CD34, CD45, and CD117; CD9, CD10, CD13, CD26, CD29, CD44, CD47, CD49d, CD54, CD56, CD58, CD71, CD81, CD90, CD99, CD105, CD151, CD166, CD200, HLA-ABC, PECAM-1/CD31; thrombospondin receptor/CD36, Bp50/CD40, Prominin-1/CD133, MDR-1/CD243, NGFR/CD271, ABCG-2/CD338, and HLA-DR), using an unmodified Becton Dickinson FACSCanto II flow cytometer and FACS-DiVa software (Becton Dickinson, San Jose, CA) as previously reported [15]. Monoclonal antibodies were from Becton Dickinson, except anti-CD338-APC (R&D, Minneapolis, MN), anti-CD-133-PE, and anti-CD271-APC (Miltenyi Biotec, Bergisch Gladbach, Germany). The immunophenotype characterization confirmed the mesenchymal origin of the isolated hA-MSCs (Supplementary Table S1; Supplementary Data are available online at
Small RNA deep sequencing and analysis
Total RNA was purified from hA-MSCs using the mirVana™ miRNA isolation kit (Ambion, Austin, TX) and quantified with the Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). RNA quality was evaluated with the Agilent 2100 BioAnalyzer (Agilent, Santa Clara, CA). Small RNA library preparation and sequencing were carried out according to the manufacturer's instructions (Illumina, San Diego, CA). Briefly, 1 μg of total RNA was ligated to specific adapters, required for library amplification and sequencing, then retrotranscribed and amplified to obtain double-stranded cDNAs. These cDNAs were loaded on a 6% polyacrylamide gel electrophoresis for size purification of small RNAs. The gel band corresponding to fragments ranging from 140 to 160 bp was cut, ethanol-precipitated, and eluted in ultrapure water.
The libraries were then evaluated for quality on the Agilent 2100 BioAnalyzer (Agilent) and sequenced using the MiSeq instrument (Illumina).
Bioinformatics
The quality of raw sequencing data was evaluated with FastQC (
Unique sequence reads of 15 nucleotides or more in length, with a minimum read count of three, were aligned, without any mismatch, with precursor human miRNA sequences from miRBase version 20 [23]. Differentially expressed miRNAs were identified using three statistical methods: Fisher's exact test, DESeq [24], and edgeR [25]. Read counts were normalized to minimize systematic technical bias. Quantile normalization and Fisher's exact test were performed with the iMir pipeline. The two Bioconductor packages (DESeq and edgeR) were chosen as a negative binomial distribution model with which to test the differential expression between Ob- and Co-hA-MSCs. In all analyses, a P-value <0.05 was considered significant. Hierarchical biclustering of samples, based on annotated miRNAs and their normalized read counts, was performed using MATLAB's Clustergram algorithm [26,27]. Venn diagrams were obtained using the Jvenn viewer [28].
Target prediction and validation
We used the miRanda [29,30] and TargetScan [31] tools to predict putative targets of the miRNAs that differed significantly between Ob- and Co-hA-MSCs. TargetScan computationally predicts miRNA gene targets by searching for 8-mer and 7-mer sites matching the seed region of each miRNA, whereas miRanda target prediction incorporates current knowledge on target rules and uses a compendium of mammalian miRNAs. The targets predicted by miRanda were filtered according to two main criteria: the score was set to ≥130 and the free energy ≤−16 kCal/mol. The targets predicted by TargetScan were filtered according to a total context score ≤−0.30.
We used MirTarBase [32] to search for experimentally validated interactions. Functional annotation of targets was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [33]. This Functional Classification Tool is a rapid means with which to organize large lists of genes into functionally related groups and thus to help unravel the biological content captured by high-throughput technologies. The following annotation categories were selected: Gene Ontology, Genetic Association DB Disease, OMIM disease, BIOCARTA, KEGG pathway, and PANTHER pathway. Statistics related to over-representation of functional categories was based upon a False Discovery Rate statistic methodology. A P-value <0.05 was considered significant.
Validation of miRNA expression by TaqMan miRNA assay
Real-time (RT) PCR was performed on the 7900 HT Real-Time PCR System (Applied Biosystems) by using the TaqMan miRNA Assay Protocol to assess the relative expression of miR-138-5p and miR-222-3p. For RT reactions, 10 ng of total RNA was used in each reaction (15 μL) and mixed with the corresponding TaqMan miRNA assays RT primer (3 μL). The RT reaction conditions were as follows: 16°C for 30 min; 42°C for 30 min; 85°C for 30 min; and then hold at 4°C. After the RT reaction, 1.33 μL of cDNA was used for the PCR together with TaqMan primer (2 μL). The PCR was carried out at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. The miRNA expression values were normalized to RNU48, and then the relative expression values were obtained versus Co-hA-MSCs using the ΔΔCT method (Relative Quantification, RQ = 2−ΔΔCT) with Sequence Detection System v2.3 and RQ Manager 1.2 software (Applied Biosystems).
The investigated parameters were expressed as mean (SEM). Student's t test was used to compare group means, and a P-level <0.05 was considered statistically significant. Statistical analyses were carried out with the PASW package for Windows (Ver.18; SPSS, Inc., Headquarters, Chicago, IL).
Microarray analysis
Total RNA from Co- and Ob-hA-MSCs was hybridized on a Human HT-12V4.0 Gene Expression Bead Chip (Illumina) containing 47,732 probes, and procedures were according to the manufacturer. The binary intensity data files were read with Illumina's GenomeStudio software and converted into intensity matrices. Probes without a significant detection P-value <0.05 in at least one sample were removed. Differential expression analysis (obese subjects vs. controls) was performed using the limma package [34]. Genes were considered to be differentially expressed at a significant level in case of Benjamini–Hochberg multiple testing correction adjusted P-value <0.05 and with a log2FC >0.5 or <−0.5. Gene lists were analyzed using the DAVID [33].
Results
The clinical and biochemical characteristics of obese and control pregnant women are reported in Table 1. The prepregnancy body mass index was higher (mean [SEM] 41.5 [2.2] vs. 22.4 [1.0] kg/m2), P < 0.0001, and the weight gain in pregnancy lower (mean [SEM] 10.0 [1.1] vs. 14.3 [1.8] kg) in obese than in control pregnant women, as recommended by guidelines [35]. Clinical and biochemical characteristics were similar in the two groups of pregnant women, whereas serum leptin (P < 0.0001) and the leptin to adiponectin ratio were significantly higher (P < 0.05), and serum adiponectin lower, although not significantly, in obese women than in control women.
Data are expressed as mean (SEM). Biochemical parameters were evaluated in serum.
Statistically significant difference at Student t test: a P < 0.0001, b P < 0.05.
AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALP, alkaline phosphatase; GGT, γ-glutamyl transferase; Ob, obese; Co, control; BMI, body mass index; SEM, standard error of mean.
miRNA sequences from small RNA libraries
The miRAnalyzer detected from 147 to 411 known miRNAs, whereas miRDeep* identified from 136 to 320 miRNAs, the range of the reads number being 78,093–3,588,828 (Table 2). In all samples, the miRAnalyzer detected more miRNAs than miRDeep* did, therefore leading to an increased risk of false-positive results, which we subsequently reduced considering only the miRNAs identified as being differently expressed with both tools. At Spearman correlation analysis, there were no significant differences between Co- and Ob-hA-MSC miRNAs (Supplementary Fig. S1). We carried out a hierarchical clustering analysis using the miRNA expression data of each individual sample, excluding those miRNAs whose read count was equal to 0 in more than 15 samples (Fig. 1A, B, miRNAs identified by MiRAnalyzer and by miRDeep, respectively).
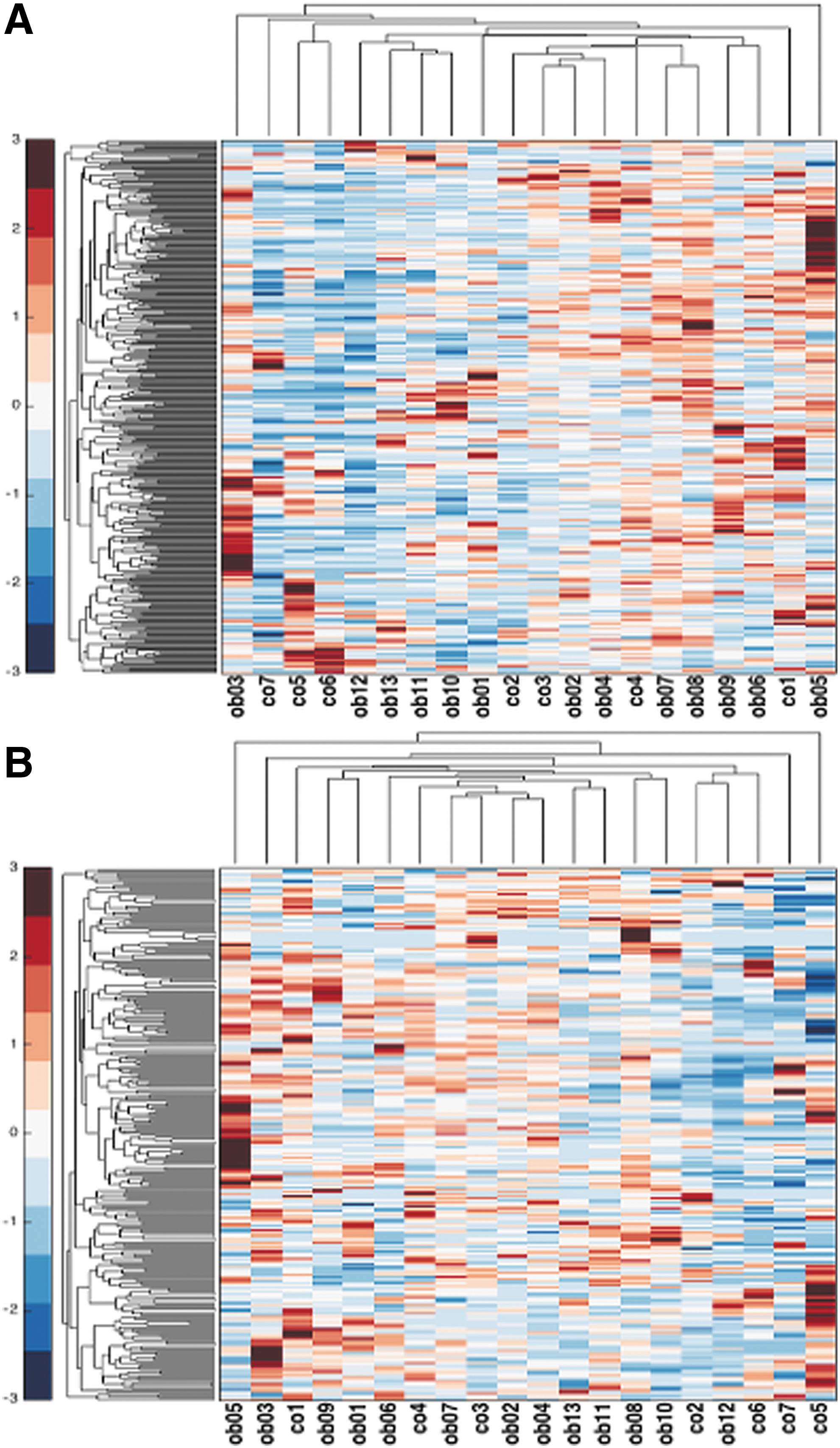
Hierarchical clustering of Ob- and Co-hA-MSCs based on miRNA expression data of each individual sample, identified using miRAnalyzer
Ob, obese; Co, control; miRNA, microRNA.
There was a high variability among Ob- and Co-hA-MSC miRNAs, and no miRNA cluster was detected in any sample. We grouped the identified miRNAs in families, according to the MiRBase database, and calculated the read frequencies by dividing specific miRNA read counts by the sum of all miRNA read counts for each sample. We conducted hierarchical clustering for all the miRNA families that represented at least 5% of the total miRNA reads in at least one sample. miRNA families did not differ significantly between Ob- and Co-hA-MSCs.
miRNA expression profiling analysis
The number of differentially expressed (P < 0.05) miRNAs was calculated by three methods: Fisher's exact test, DESeq, and edgeR. miRAnalyzer identified 224, 13, and 41 differentially expressed (P < 0.05) miRNAs, using one-by-one Fisher's exact test, and the DESeq and edgeR methods (Fig. 2A). Only six miRNAs were identified by all the three methods. Thirty-four miRNAs were identified by Fisher's exact test and the edgeR method and four miRNAs by Fisher's exact test and the DESeq method (Fig. 2A, B). The miRdeep* tool identified 210, 13, and 21 differentially expressed (P < 0.05) miRNAs using Fisher's exact test, the DESeq method, and the edgeR method, respectively. Eight miRNAs were identified by all the three procedures, two miRNAs were identified by Fisher's exact test and the DESeq method, and 12 miRNAs were identified by Fisher's exact test and the edgeR procedure (Fig. 2C, D).

Venn diagrams of miRNAs identified by miRAnalyzer
Intersection of the miRNAs identified with both miRAnalyzer and miRDeep* and, differently expressed (P < 0.05) by all the three statistical methods, resulted in consensus for two miRNAs: miR-138-5p and miR-222-3p, both overexpressed in Ob- versus Co-hA-MSCs. It should be noted that the mature fragment miR-138-5p, as reported in miRBase, could arise from two different stem loop precursors, miR-138-1 and miR-138-2. While miRAnalyzer correctly identified miR-138-5p (Fig. 2B), miRDeep* assigned the reads to the precursors (Fig. 2D).
We extracted the fasta sequences of the reads matching miR-138-1 and those matching miR-138-2, based on the miRDeep* results. We found that all the reads aligned to miR-138-2 by BLAST local alignment mapped to the mature miRNA miR-138-5p, and the reads aligned to miR-138-1 mapped mostly to miR-138-5p, and to a lesser extent to miR-138-1-3p. Thus, we confirmed that the mature miR-138-5p was identified by both miRAnalyzer and miRDeep. Regarding miR-222-3p, miRDeep* also detected the stem loop precursor miR-222, and we checked that most of the reads, according to BLAST, mapped to the mature fragment miR-222-3p, in accordance with the data obtained by miRAnalyzer.
Validation of miR-138-5p and miR-222-3p expression by quantitative RT-PCR
We measured the expression levels of miR-138-5p and miR-222-3p also using TaqMan miRNA assay and found that the mean RQ was higher in Ob-hA-MSCs than in Co-hA-MSCs [RQ = 2−ΔΔCT (SEM): 5.77 (1.08) and 4724.93 (1252.22), for miR-138-5p and for miR-222-3p, respectively]. This finding confirms that both miRNAs are overexpressed in Ob-hA-MSCs in agreement with the data obtained using the small RNA-Seq approach (Supplementary Fig. S2).
We also evaluated the expression of miR-138-5p and miR-222-3p in a second cohort of 14 placental tissue sections (amnion) from nine obese and five control pregnant women using TaqMan miRNA assay. We obtained in Ob- respect to Co-amnion the following mean RQ (SEM) values: miR-138-5p: 5.3 (2.0) and miR-222-3p: 1.1 (0.1). Consequently, the expression of miR-138-5p confirmed the trend observed in Ob-hA-MSCs, whereas the expression of miR-222-3p was similar in Ob- and Co-samples.
Identification of miR-138-5p and miR-222-3p validated gene targets
Validated gene targets of miR-138-5p and miR-222-3p were identified using the mirTarBase database. We investigated in greater depth the meaning of the top 20 significant biological processes most pertinent to hA-MSC epigenetic regulation (enriched terms) (Tables 3 and 4, respectively). The enrichment analysis of the validated targets of miR-138-5p revealed terms related to fat cell differentiation, fat deposits, lipid and carbohydrate homeostasis, metabolic syndrome, heart disease, ischemia, as well as terms involved in response to stress, cytoskeleton, and the p53 signaling pathway. The enrichment terms of miR-222-3p referred to several cell functions: cell differentiation, transport, DNA replication, DNA damage response, signal transduction by p53, and stress response.
Gene expression profiling analysis
We used microarray-based expression profiling to identify the genes differentially expressed between Ob- and Co-hA-MSCs. We found that 241 genes were upregulated and 33 downregulated probes (Benjamini–Hochberg adjusted P-value <0.05, log2FC >0.5 or <−0.5) in obese versus controls, some of which were targeted by miR-138-5p and/or miR-222-3p as predicted by miRanda, TargetScan, and miRTarbase (Supplementary Table S2). The differently expressed genes were functionally annotated with DAVID (Tables 5 and 6), and only those with a significant P-value (<0.05) were considered. The 20 significant biological processes involving upregulated genes included cytoskeleton organization, blood vessel development, and immune response, while those involving downregulated genes included steroid and lipid metabolism (Tables 5 and 6).
Discussion
Experimental evidence obtained in animal models suggested that the placental epigenome is involved in gestational programming and that gestational programming of obesity results in offspring obesity and/or in obesity-related disorders [2,3]. Given the ban on experimentation with human embryo tissue, studies to evaluate whether intrauterine maternal obesity in humans is related to obesity in childhood or in adult life would entail long-term follow-up monitoring. Thus, given their capability to differentiate in adipocytes, hA-MSCs could represent a surrogate cellular model with which to investigate the impact of maternal obesity on the programming of adipose tissue dysfunctions. In the attempt to shed light on the role of miRNAs in human pregnancy during obesity, we used high-resolution small RNA-Seq to unravel the entire miRNA expression profiles of Ob- and Co-hA-MSCs.
Integrated bioinformatics based on a consensus method previously showed that the expression of miR-138-5p and miR-222-3p was significantly higher in Ob-hA-MSCs than in Co-hA-MSCs [20 –22]. Using quantitative RT-PCR, we confirmed the higher expression of these two miRNAs in Ob-hA-MSCs and that of miR-138-5p also in amnion from obese pregnant women, thus enlarging the panel of miRNAs previously found deregulated in this multicellular placental tissue [7]. Interestingly, the most significant pathways predicted to be deregulated by miR-138-5p and/or miR-222-3p included such obesity-associated functions and disorders as fat cell differentiation, fat deposits, lipid and carbohydrate homeostasis, stress response, metabolic syndrome, heart disease, and ischemia.
Transcriptome analysis of hA-MSCs highlighted a panel of differently expressed mRNAs in Ob- versus Co-hA-MSCs and some of them were targeted by miR-138-5p and/or miR-222-3p. The most significant mRNAs were engaged in cytoskeleton organization, vessel development, immune response, and steroid and lipid metabolism, all of which are involved in adipose tissue remodeling in obesity [36]. Thus, our hA-MSC miRNome data indicate that miRNAs could play a role in the adverse metabolic consequences associated with obesity.
Support for this hypothesis comes from our previous observation of miR-138-5p overexpression in subcutaneous adipose tissue and of miR-222-3p overexpression in both subcutaneous and visceral adipose tissues from obese adults [8,9]. Interestingly, both miR-138-5p and miR-222-3p are involved in the regulation of adipogenesis, obesity, and inflammation [37,38]. In particular, miR-222-3p was reported to be overexpressed in inflammation-induced adipocytes and macrophages, and in subcutaneous adipose tissue from obese adults [38]. Notably, activation of the inflammatory and immune systems, together with oxidative stress, is associated with abdominal obesity and has been implicated in the pathogenesis of obesity-related metabolic disorders [39,40]. miR-222-3p was also associated with adipose tissue oxidative stress, apoptosis, and inflammation, and endothelial dysfunction and adhesion, with infiltration of inflammatory cells into the endothelial space [38,41].
In addition, overexpression of miR-138 during obesity, induced by secreted proinflammatory cytokines, causes endothelial cell dysfunction, which is the most significant predisposing risk factor for cardiovascular disease [42]. In this context, it is noteworthy that our enrichment analysis of miR-138-5p targets resulted in the terms “stress response,” “heart disease,” and “ischemia.”
The prediction effect of miR-222-3p and miR-138-5p on deregulating gene expression in hA-MSCs during obesity is in agreement with the altered protein profile that we recently reported in the same cohort of hA-MSCs [43], which suggests that the response to stress and to inflammation is impaired in obesity. In particular, we found several heat shock proteins downexpressed, including HPSA8, which is a target of miR-222-3p [43].
Accordingly, in animal models, intrauterine maternal obesity has been shown to represent a stress state for embryo that leads to fetal programming of obesity and/or of obese-associated diseases [44]. In detail, maternal obesity was associated with increased oxidative stress and reduced antioxidants in placental and fetal tissues, thereby impairing mitochondrial biogenesis and glucose tolerance [44,45]. Furthermore, obesity-related alterations of oxidative stress markers have been demonstrated, both prenatally and postnatally, in offspring muscle, liver, pancreas, and adipose tissues [46].
In conclusion, our finding of miR-138-5p/miR-222-3p overexpression in Ob-hA-MSCs, together with the transcriptomic data, suggests that these miRNAs in obese pregnancy could derange metabolic pathways found to be impaired in tissues from obese adults and in obesity-associated disorders [37,38,41,42] and concur to modify gestational programming as has been demonstrated in animal models. Taken together, these findings raise the possibility of using diet-based strategies to normalize the perinatal miRNome in obesity.
Footnotes
Acknowledgments
The authors thank Jean Ann Gilder (Scientific Communication srl, Naples) for editing the text and Vittorio Lucignano, CEINGE–Biotecnologie Avanzate for technical assistance related to graphics. This work was supported by grant POR CAMPANIA FSER 2007–2013 Project DIAINTECH and grant CAMPUS-Bioframe, from Regione Campania, Italy; grant PON02_00677 (BIOGENE) Pot. lab.8 A/B-2012 from the Italian Ministry of University and Research. Centro di Ricerca per lo studio di malattie genetiche dell'uomo e loro modelli cellulari e animali–Progetto: Terapia genica delle malattie metaboliche.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.