
Abstract
All mammalian cells display an array of surface glycans that can modulate cellular interactions and regulate the development of an organism. In spite of their important implications in health and disease, investigations of glycans pose a great challenge given their inherent heterogeneity and the frequently observed cross-reactivities of antibodies against glycosphingolipids (GSLs) with multiple glycans, which may lead to erroneous interpretation and conclusion. We employed matrix-assisted laser desorption–ionization mass spectrometry and tandem MS/MS analyses to systematically delineate changes in GSLs during differentiation of human embryonic stem (ES) and induced pluripotent stem cells into various derivatives. In addition to the well-known human ES-specific markers, stage-specific embryonic antigen (SSEA)-3 and SSEA-4 and several globo- and lacto-series GSLs (Gb4Cer, Lc4Cer, fucosyl-Lc4Cer, Globo H, and disialyl-Gb5Cer) not reported previously were identified. A close association of the switching of core structures of GSLs from globo- and lacto- to ganglio-series during ES differentiation was revealed, consistent with the observed changes in GSL expression during murine embryonic development. Such switching was accompanied with the concerted changes in the expression of glycosyltransferases during differentiation. Finally, since aberrant glycosylation is a general feature of cancer, targeting tumor-associated surface glycans for cancer immunotherapy is gaining international attention. While GD2-targeted immunotherapy of neuroblastoma represents the first antiglycan monoclonal antibody to obtain FDA approval for standard medical care, Globo H-targeted immunotherapy of breast cancer has generated promising results in the ongoing multinational clinical trials. In this study, we also describe the studies of the scientific rationales for the use of glycans as anticancer immunotherapeutics.
Introduction
E
In addition, the ES cells established from the preimplantation embryos and those generated from slightly later embryonic epiblast stages have distinctly different culture requirements, gene expression, and epigenetic features [4]. Therefore, the terms “naïve” and “primed” were used to describe the early and late phases of epiblast ontogeny and these naïve and primed ES cells represented, separately, the different stages of developmental potency [4]. Under inductions in vivo or in vitro, these ES/iPS cells can differentiate into tissue- or organ-specific cells with specialized functions, thus presumably becoming excellent candidates for use in cell-based therapy and regenerative medicine. Despite numerous studies, our understanding of the regulation of pluripotency and differentiation of ES/iPS cells remains limited.
All mammalian cells display an array of surface carbohydrates that have the ability to modulate cellular interactions and to regulate the development and functions of an organism. More than 85% of the cell surfaces in mammalian cells are glycosylated, and aberrant glycosylation is often regarded as a general feature of cancer [5,6]. However, relatively little is known about the functional roles of these surface carbohydrates in normal and cancer cells. Therefore, we have employed glycomic approaches to interrogate the glycan profiles of carbohydrates in ES/iPS cells and their differentiated derivatives, as well as tumor cells.
Based on the expression of unique glycans in various stages of differentiation for ES/iPS cells, particular biosignatures of surface glycans are potentially useful for the use of development of cancer therapeutics. In spite of these important implications for health and disease, investigations of surface glycans pose a great challenge as they are extremely heterogeneous and stereochemically complex; furthermore, glycosylation is an event involving post-translational modifications to various extents.
Most of the previous studies of surface carbohydrates of ES cells came from experiments performed during embryonic development using biochemical extraction, thin-layer chromatography, and immunostaining. These studies found that globo-series stage-specific embryonic antigen 3 (SSEA-3, Galβ1-3GlcNAcβ1-3Galα1-3Galβ1-4Glcβ1-Cer) and SSEA-4 (NeuNAcα2-3Galβ1-3GlcNAcβ1-3Galα1-3Galβ1-4Glcβ1-Cer) were expressed at the four-cell stage of mouse embryos and then declined during further development of embryo [7]. In contrast, SSEA-1 (Lewisx; Fucα1-3(Galβ1-4)GlcNAcβ1-3Galβ1-4Glcβ1-Cer; type 2 chain) was not expressed until the morula stage of mouse embryos [7]. These studies revealed the presence of ganglio-series GM3, GD3, GT3, GM2, and GD2 in later stages upon neural crest formation [8,9], and GD3, GD1a, and GT1b were also expressed after somite formation in mice [8,10].
It should be noted, however, mouse ES cells differ from human ES cells in several aspects; most mouse ES cells arise from the inner cell mass before embryo implantation [11], whereas human ES cells are derived from epiblasts of postimplantation embryos [12,13]. Only a few studies have investigated the chemical structures of the carbohydrate antigens on the cell surface of human ES cells [14 –17].
Specific surface markers of human ES/iPS cells are valuable for monitoring the pluripotency of these stem cells and their differentiation progress. For example, SSEAs, SSEA-3 and SSEA-4, are important markers of human ES/iPS cells [18], while O4, O1 antigen [19], A2B5 antigens, and ganglioside GD3 are markers of neural lineage cells [20]. However, many markers for ES/iPS cells currently in use were adopted from phenotypic characteristics of cell types unrelated to ES cells and can only offer limited insight into the functional roles of stem cells.
Several previous reports have indicated that these markers for ES/iPS cells currently in use are also expressed in differentiated progeny and tissues [15,20,21]. For example, SSEA-1 is expressed in more differentiated tissues [20,21] and neural progenitors [22,23]. It is found on neutrophils, which mediate phagocytosis and chemotaxis [24], and is also expressed in patients with Hodgkin's disease [25], some B cell chronic lymphocytic leukemias [26], acute lymphoblastic leukemias [27], and most acute nonlymphocytic leukemias [28]. The markers, SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81, currently used to characterize human ES cells have similar limitations because of their delayed downregulation during differentiation [11,15].
Furthermore, many studies have reported cross-reactivities of monoclonal antibodies (mAbs) with different glycoforms or glycoconjugates, containing the same glycosyl epitopes [29,30]; thus, positive immunostaining with these mAbs alone might not necessarily reflect a particular entity of glycosphingolipids (GSLs) on ES/iPS cells. Therefore, it is critically important to (1) systematically identify new markers for stem cells that define ES/iPS cells and (2) to perform detailed mass spectrometric (MS) analysis to decipher the precise profile of GSLs in these cells.
GSLs are amphiphilic membrane lipids consisting of a polar oligosaccharide chain attached to a hydrophobic ceramide moiety with various fatty acid chains. GSLs are ubiquitous components of cell membranes among all eukaryotic species, including humans. GSLs in animal tissues can be divided into three major groups: (i) the ganglio- and isoganglio-, (ii) the lacto- and neolacto-, and (iii) the globo- and isoglobo-series [7,31]. GSLs serve as mediators of cell adhesion and signal transduction [5,7], as well as cell type-specific differentiation markers [8,32]. Cells often possess specific well-defined GSL antigens and can be detected by antibodies.
For example, Lewisx (CD15, also called SSEA-1) represents a marker for murine pluripotent stem cells, in which it plays an important role in adhesion and migration of cells in the preimplantation embryo. Human natural killer cell antigen-1 (HNK-1) (sulfo-3GlcAβ13Galβ14GlcNAcCer), expressed at the cell surface, is uniquely enriched in neural cells and natural killer cells and is thought to play important roles in cell–cell interactions [33]. Sonnino and Prinetti proposed that GSLs form clustered microdomains at the cell surface and interact with membrane proteins such as caveolin-1, integrins, growth factor receptors, and tetraspanins, thus modulating adhesion, growth, and motility of cells [34,35]. Furthermore, GSLs may also be involved in the epithelial–mesenchymal transition [36,37], an important process during embryonic development and tumor metastasis.
Furthermore, the ceramide formation of GSLs may relate to the expression of ceramide synthase found in endoplasmic reticulum, while the synthesis of glycan moiety of GSLs may incur glycosylation in the Golgi apparatus [38]. In this respect, it should be noted that sphingolipidoses may be attributed to specific enzyme disorders in lysosome [39,40]. The iPS cells derived from patients with Niemann–Pick disease can be efficiently differentiated into disease-relevant cell types, which might be useful in mechanistic and therapeutic studies [41]. In addition, iPS cells derived from Fabry disease, globoid cell leukodystrophy, and mucopolysaccharidosis VII showed defects in disease-specific enzyme activities and significant accumulation of substrates for these enzymes [42].
Aberrant glycosylation is considered a general feature of cancer [5,6]. These abnormal glycans contribute to tumorigenesis and metastasis. Overexpression of these glycoconjugates in tumors with limited expression in normal tissues makes them targets for immunotherapy of cancers. Indeed, since 1990s, many tumor-associated glycans have been exploited as targets for cancer immunotherapy in clinical trials, but the majority terminated in early phase studies. So far, GD2 is the only tumor-associated glycan shown to be an effective target antigen for cancer immunotherapy with the mAb, Dinutuximab [43]. Another hotly pursued glycan target is Globo H based on our recent findings supporting its important role in cancer as a marker for cancer stem cells (CSCs) [44], immune checkpoint molecule [45], and angiogenic factor [46]. Promising results from a phase II/III clinical trial [47] provide further impetus for the development of a global clinical trial.
Changes of GSL Profiling During Differentiation of Human ES Cells to Embryoid Body
Current studies for human ES/iPS cells often rely on the use of mAbs for immunostaining or flow cytometric analysis. SSEA-3 (Galβ1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ1-1′Cer) and SSEA-4 (sialylated SSEA-3) are two well-known markers for ES/iPS cells. MC-631 and MC-813-70 mAbs recognize, separately, glycan epitopes for SSEA-3 and SSEA-4. However, many reports have observed cross-reactivities of these mAbs to various glycoforms or glycoconjugates [29,30]. Therefore, it is difficult to use these mAbs alone to determine if particular entities of GSLs are present in ES/iPS cells. For example, the epitopes of MC631 and MC813-70 mAbs for SSEA-3 and SSEA-4 have been delineated, respectively, as GalNAcβ1-3Galα1-4Gal and NeuAcα2-3Galβ1-3GalNAc [29,30]. It was shown that MC-631 mAb for SSEA-3 is able to cross-react with Forssman, Gb4Cer, Globo H [29,30], and fucosyl Lc4Cer [16]. MC-813-70 mAb for SSEA-4 cross-reacts with GM1b, GD1a, and a common structure of the core 1 O-glycan glycoprotein, carrying the NeuAcα2-3Galβ1-3GalNAc epitope [29,30].
Furthermore, the identities and roles of many surface glycan antigens on ES/iPS cells have not yet been elucidated. It is unclear whether these known glycan markers such as SSEA-3 or SSEA-4 on ES/iPS cells appear simply as GSLs by conjugation with lipids or also as glycans conjugated with proteins. Therefore, positive immunostaining with mAbs alone would not necessarily validate the presence of particular entities of glycoconjugates on ES/iPS cells. Instead, detailed MS analyses coupled with immunostaining will be essential to decipher a precise profile of GSLs on ES/iPS cells. In Table 1, we provide a list of chemical structures of various GSLs with IUPAC-style abbreviations that are commonly found in undifferentiated human ES/iPS and differentiated derivatives.
The color symbols and nomenclatures for structure representation of glycans follow the instructions of Consortium for Functional Glycomics [137].
ES, embryonic stem; SSEA, stage-specific embryonic antigen.
We used matrix-assisted laser desorption–ionization mass spectrometry (MALDI-MS) and tandem mass spectrometry (MS/MS) analyses to systematically delineate changes in GSL profiles during differentiation from undifferentiated human ES cells to the 16-day differentiated embryoid body (EB) outgrowths [14]. The total crude GSL extracts after Folch partitioning were permethylated and subjected to MALDI-MS and MS/MS analyses without further purification. This approach has the advantage of neutralizing all carboxylic groups on sialic acids by methyl esterification, thus allowing a simultaneous semiquantitative profiling of both neutral and negatively charged sialylated species.
As shown in Fig. 1, the profiles of GSLs on MALDI-MS analysis obtained from human ES and EB cells differed remarkably from each other, with each being dominated by several major peaks that occurred in signal clusters due to the expected heterogeneity of the ceramide moieties. After the annotations of GSLs were assigned based on m/z values of each of the major sodiated molecular ion signals, the values of m/z for the respective GSLs were fitted to the expected core structures of the three major GSL series (globo-, lacto-, and ganglio-series) along with the most common permutation of sphingosine and fatty acyl chains (Fig. 1) [14]. Such assignments were further confirmed by additional MS/MS analyses of each of the major peaks [14].
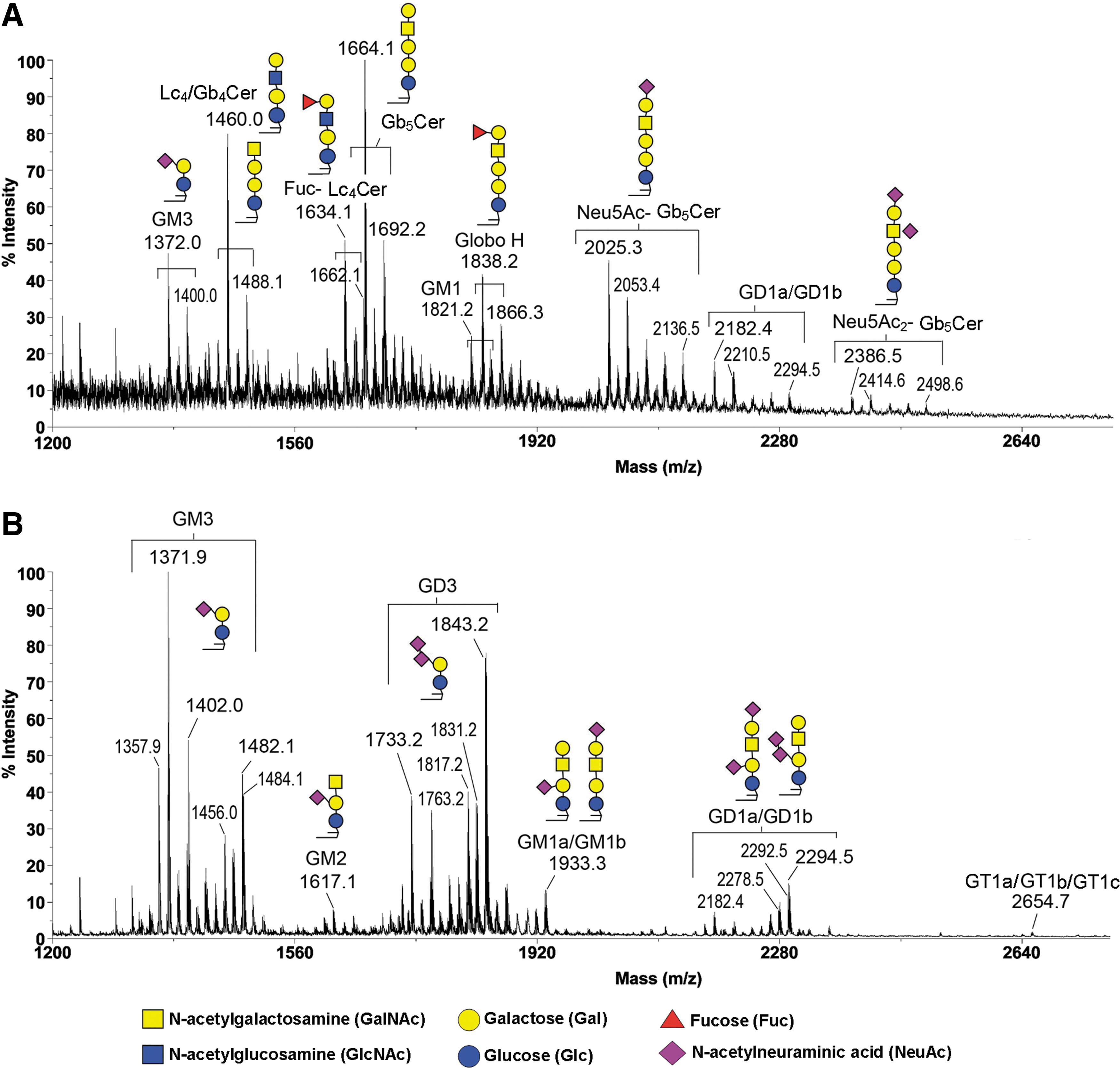
MALDI-MS profiles of permethylated GSLs from undifferentiated human ES cells and differentiated EB outgrowth. MALDI-MS profiling analyses of permethylated GSLs were carried out by using the crude extract of Folch upper phase obtained from undifferentiated human ES cells
The MALDI-MS and MS/MS analyses of GSLs in undifferentiated human ES cells revealed that GSLs comprise Gb5Cer and its sialylated version, corresponding to SSEA-3 and SSEA-4 epitopes, respectively. The structure of SSEA-3 is Gb5Cer (Table 1) (Fig. 1), which was derived from its precursors, Gb4Cer and Gb3Cer (Table 1). In turn, SSEA-4 was the sialyl-Gb5Cer (Table 1) (Fig. 1), whereas disialyl Gb5Cer was the further sialylated SSEA-4. Globo H, a hexasaccharide, which was overexpressed in epithelial cell tumors such as breast, colon, and lung cancers, was the fucosylated SSEA-3 (Fig. 1). Overall, the globo series of GSLs in the undifferentiated human ES cells all contained the common core structure as Galα1-4Galβ1-4Glcβ1-1′Cer (Table 1).
In addition to globo-series, lacto- series GSLs, including Lc4Cer (type 1) and fucosyl Lc4Cer (H type 1), not previously known to be expressed in the undifferentiated ES cells, were also identified (Fig. 1) [14]. For example, Lc4Cer had been determined as Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-Cer (Table 1). It was further observed by flow cytometric analysis that mAbs against H type 1 epitope (ie, Fucα1–2Galβ1–3GlcNAc-) stained the undifferentiated human ES cells, but not the differentiated EB cells, confirming the presence of lacto-series GSLs in human ES cells carrying a terminal H type 1 antigen.
Later, Tang et al. [48] also reported the findings of the H type 1 chain GSL as an embryonic antigen (fucosyl Lc4Cer in Table 1) designated as SSEA-5. Furthermore, the anti-SSEA-5 mAb, which binds the H type 1 glycan in undifferentiated ES cells, enabled the partial removal of teratoma-initiating cells [48]. More recently, Matsumoto et al. reported another mAb R-17F that recognized the same H type 1 antigen (LNFP I) and that displayed a cytotoxic effect on human ES and iPS cells [49]. To comprehensively examine GSL expression in ES/iPS cells, Barone et al. studied GSL profiles based on the use of a large amount of iPS cells (1 × 109 cells) [16,17]. Taking into consideration the nature of negatively charged sialyl- and sulfo-GSLs, they separated the total lipid extract into nonacidic and acidic GSL fractions and demonstrated the presence of several nonacidic GSLs (eg, GluCer, GalCer, LacCer, Ga2Cer, Gb3Cer, and type 2 chain neolacto-series) [16] and acidic GSLs (eg, sulfatide, sialyl Lc4Cer) [17], which were not previously found in human ES/iPS cells.
It was also demonstrated that sialyl Lc4Cer (sialyl lactotetra) was detectable in human ES/iPS lines, but not in ES-derived hepatocytes or cardiomyocytes, and differentiated derivatives of iPS cells [17], confirming the presence of type 1 chain structures in lacto-series GSLs for undifferentiated ES/iPS cells. The fucosyl Lc4 (also as SSEA-5/LNFP 1) and sialyl Lc4 antigens are lacto-series GSLs masked, respectively, with α1-2 fucose and α2-3 sialic acid in the nonreducing terminal; both are derived from the common type 1 chain (Galβ1-3GlcNAc) core structure.
On the other hand, after differentiation of human ES cells, the differentiated EB cells were dominated by GSLs of the ganglio-series, especially GM3 and GD3 (Fig. 1) [14]. GD3 was highly expressed in EB cells and not in human ES cells; GM3, along with GD1a/GD1b, was commonly expressed in both. Other less abundant ganglio species, including GM2, GM1, and GT1a/GT1b/GT1c, were also more obvious in EB outgrowths (Fig. 1). Additional MS/MS analyses revealed that isomers of GM1a/GM1b and GD1a/GD1b were present in EB-differentiated cells, which had the core structure as Galβ1-3GalNAcβ1-4Galβ1-4Glcβ1-Cer, but with additional sialic acid residues. The complexities of gangliosides in the differentiated EB outgrowth could be attributed to the complex mixture of various amounts of the three different germ cells in the samples.
These results thus showed that there was an almost complete makeover of the GSLs when the original ES cells differentiated to the more developed EB outgrowths. In other words, the globo- and lacto-series of GSLs became rare and replaced by ganglio-series upon differentiation of human ES cells. This finding of a switch in the core structures of GSLs from globo- to ganglio-series during human ES differentiation (Fig. 2) is consistent with the observed changes in expression patterns of GSLs during embryonic development of mice [7]. In addition, roles of gangliosides during the stages of mouse embryonic development, including ovulation, spermatogenesis, and embryogenesis, had been studied in detail [50]. Furthermore, differentiation of neural stem cell could also be regulated by lipids in the lipid rafts or other subcellular sites, acting on receptors, protein kinases/phosphatases, and other cell signaling proteins [51]. Finally, the results of MALDI-MS indicated that human ES lines, HES5 and H9, both exhibited similar expression patterns of GSLs either before or after induction of differentiation.
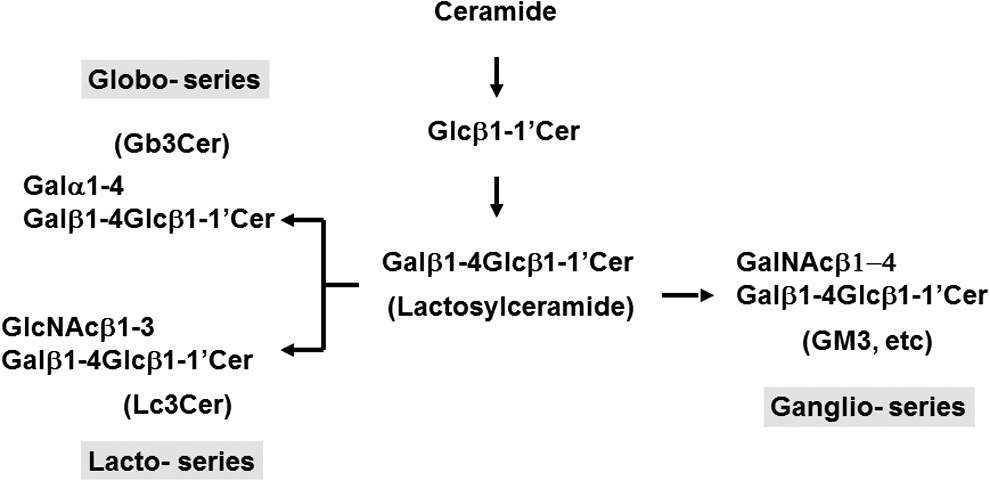
Switching of the core structures of GSLs from globo- and lacto- to ganglio-series upon human ES differentiation. Stepwise biosynthesis of GSL occurs first with glucose added to ceramide, and then subsequently galactose was transferred by a key glycosyltransferase to form lactosylceramide. Further biosynthesis of GSLs provides an example of how competing biosynthesis pathways can lead to alterations of the core structures of GSLs from globo- and lacto- (Gb3Cer and Lc3Cer) to ganglio- (GM3, etc.) series upon human ES differentiation. For example, when the overall Gb3Cer synthesis from lactosylceramide decreased in favor of the switch toward ganglio-series upon differentiation, it should follow that the expression of both SSEA-3 and SSEA-4 is reduced. On the other hand, the observed serial changes in GTs involved in GSL biosynthetic pathways underlie the switch in the core structures of GSLs in favor of ganglio-series upon human ES differentiation. SSEA, stage-specific embryonic antigen.
We also examined the expression of these unique surface GSLs during reprogramming of human fibroblasts into iPS cells and subsequent induction for differentiation [3]. As shown (Fig. 3) [52], the fibroblasts expressed no Globo H, Lc4Cer, and H type 1 antigens, but fully reprogrammed iPS cells that had reactivated endogenous pluripotent genes (eg, Oct4) displayed expression of GSL markers comparable with those in ES cells. On the other hand, after differentiation of iPS cells, the expression level of these markers declined to those typical of fibroblasts before reprogramming (Fig. 3). Therefore, it was concluded that similar to ES differentiation, there was also a striking switch in the core structures of GSLs from globo- and lacto- to ganglio-series during differentiation of iPS cells (Fig. 3). Collectively, these data point to a strong association of surface GSL markers with the pluripotent state in both ES and iPS cells.
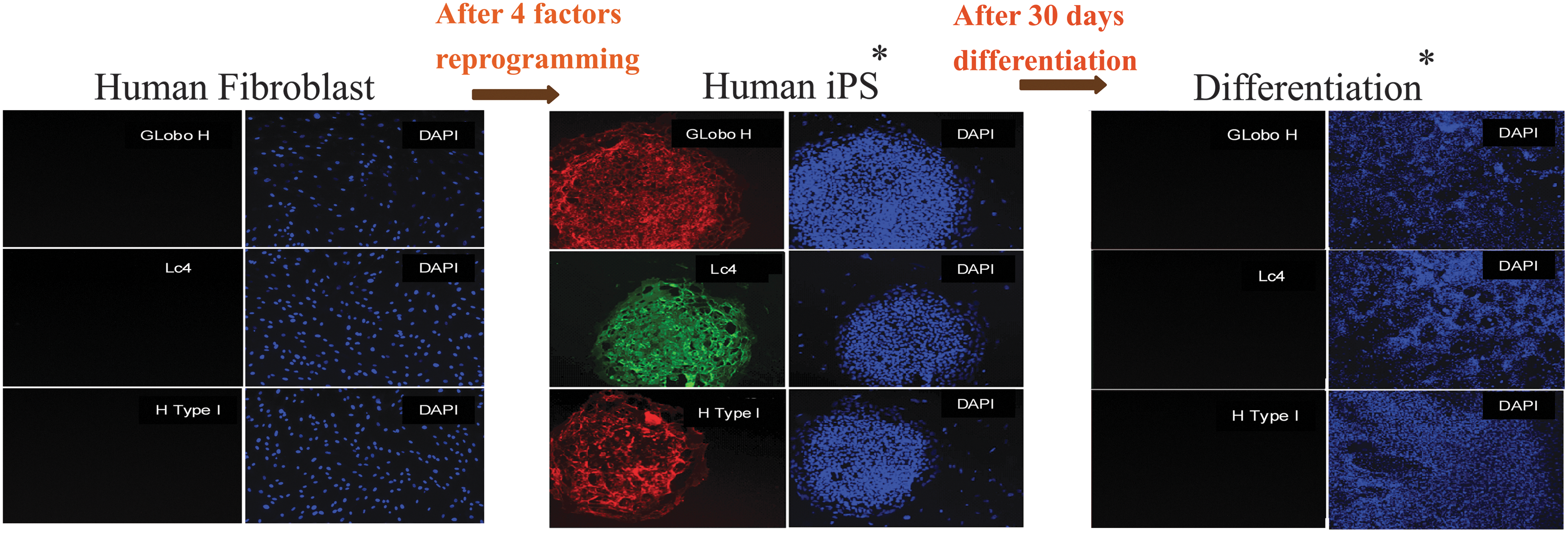
Alterations of GSL expression in human iPS cells and differentiated derivatives. The samples of fibroblasts, fibroblast-derived iPS cells, and their differentiated derivatives were stained, separately, with antibodies against Globo H, Lc4Cer, and H type 1 antigen. Changes of GSL profiles were observed during reprogramming of cells and exhibited a strong association with the pluripotency of iPS cells. Reproduced from Fig. 17-3 of Ref. [52] with permission. iPS, induced pluripotent stem.
Alterations of GSL Expression Patterns After Lineage-Specific Differentiation
Similarly, we further investigated the alterations of GSL expression when human ES cells underwent lineage-specific directed differentiation. ES cells were differentiated specifically toward neural progenitor cells (Nestin+, 99% Pax6+, Sox1+ cells) and definitive endoderm cells (SOX17+, FOXA2+) separately [15]. The mass spectra obtained from permethylated GSLs from neural progenitor cells and definitive endodermal cells differed drastically from each other [15]. As described above, the MS/MS analysis confirmed the presence of SSEA-3, SSEA-4, Globo H, Lc4Cer, and fucosyl Lc4Cer in undifferentiated human ES cells.
In contrast, when human ES cells differentiated into neural progenitor cells, the GSL profiles became dominated by ganglio-series GD3 and GM3, but other gangliosides, such as GM1 and GD1, were also detected [14]. However, GD3 was not detectable in human ES cells and definitive endoderm cells, but was highly expressed in neural progenitor cells, thus qualifying it as a distinctive marker [15], whereas the expression of other ganglio series GSLs such as the GM3, GM1, and GD1 was more universal. MALDI-MS analysis confirmed the absence of Lc4Cer and fucosyl Lc4Cer in neural progenitor cells [15]. Moreover, in flow cytometric analysis, anti-SSEA-3 MC631 was reactive with essentially all undifferentiated ES cells (93.9%) and became mostly undetectable in neural progenitor cells [15]. These results were confirmed by the absence of MALDI-MS signal at m/z 1664 assigned to Gb5Cer (SSEA-3) [15].
In contrast, when human ES differentiated into definitive endodermal cells, patterns of GSLs completely differed from those observed in EB outgrowth and neural progenitors [15]. Flow cytometric analysis indicated that lactose-series of GSLs, such as Lc4Cer and fucosyl Lc4Cer, were found both in human ES (99.9% and 95.2%, respectively) and definitive endoderm cells (79.7% and 80.4%, respectively) and these expression levels disappeared in neural ectoderm cells [15]. However, epitopes for mAbs against Lc4Cer and fucosyl Lc4Cer had been shown to be Lec and H type 1, which represent two members of blood group antigens and are also known to be present on both glycolipids and glycoproteins [53] as well.
In MALDI-MS analyses, however, the most prominent GSL signal for endodermal lineage was found at m/z 1460, presumably corresponding to a mixture of Lc4Cer and Gb4Cer. Since many complex GSLs have various degrees of glycan isomers, MALDI-MS analysis alone for isomers such as Lc4Cer and Gb4Cer with identical primary structure would not exhibit significant differences in the acquired spectrum. Therefore, further MS/MS analysis revealed the relative abundance of Lc4Cer versus Gb4Cer that share the same molecular weight and hence give common molecular ions at m/z 1460 in ES versus definitive endoderm [15]. The results showed that the Gb4Cer structure was more abundant in definitive endoderm, whereas the Lc4 was more abundant in human ES cells [14]. Therefore, the signal attributed to Gb4/Lc4Cer at m/z 1460 in human ES cells was contributed more by Lc4Cer; but on the other hand, in definitive endoderm, Gb4Cer was the more abundant component instead [15]. For endodermal lineage, minor signals, which could be assigned as GM3, GM1, GD1, Globo H, and fucosyl Lc4/nLc4Cer, were also detected [15].
As mentioned, many antibodies against GSLs cross-react to a different extent with multiple glycans. Such cross-reactivity is caused by antibodies cross-reacting with the same glycosyl epitope included in different glycoconjugates. In flow cytometric analysis, MC631, mAb against SSEA-3, stained the undifferentiated ES cells. In contrast, the apparent SSEA-3+ cells detected with mAb decreased for definitive endoderm cells, but were still present in as many as 62.9% of cells with MC631 [15]. On the other hand, MALDI-MS analysis of this lineage of cells indicated little, if any, amount of m/z 1664.2 in the spectrum for Gb5Cer (SSEA-3+) [15]. Therefore, the MC631-stained SSEA-3+ cells in definitive endoderm could be attributed to cross-reactivities with glycoconjugates, which carried the MC631 epitope. In other words, the MS analysis of definitive endoderm cells confirmed the presence of signal corresponding to Gb4Cer GSL at m/z 1460, but not at m/z 1664.2 for Gb5Cer (SSEA-3) [15].
Another example of the problems involved with cross-reactivity is mAb MC813-70. In undifferentiated ES cells, SSEA-4 (Neu5Ac-Gb5Cer) was highly expressed by immunostaining with mAb MC813-70, but the immunopositive cells decreased to 86% of cells after differentiation into neural progenitor cells. In addition, ∼96% of definitive endoderm cells also showed positive immunostaining with this mAb. However, the mass spectrometry analysis failed to detect the presence of SSEA-4 (Neu5Ac-Gb5Cer) in either neural progenitor or endodermal cells [15]. Instead, GD1a and GM1b cross-reacted with SSEA-4 mAb in both neural progenitor and definitive endoderm cells. In other words, mass spectrometry analysis did not detect SSEA-4 in cells committed toward ectoderm or endoderm lineages, while specific mAb seemed to reveal the presence of SSEA-4+ cells after lineage differentiation in flow cytometric analysis. This discrepancy, however, can be reconciled in view of cross-reactivity of mAbs used with various glycoconjugates in the cells containing the specific glycan epitopes.
While it is possible that the MS profile may be biased against the SSEA-4 epitope, the lack of immunostaining with mAb MC631 (against SSEA-3) in the neural progenitor cells, which was known to recognize SSEA-4 equally well [29,30], attested to the absence of SSEA-4 in neural ectoderm cells. Therefore, the SSEA-4+ cells in immunostaining of neural progenitor cells could be attributed to cross-reactivity of mAb MC813-70 with GD1a, GM1b, or even the extended core 1 O-glycan glycoproteins [54]. In other words, cautious interpretations of the presence of particular GSL markers especially SSEA-3 or -4 should also be guided with detailed MS analysis.
Concerted Changes in the Expression of Glycosyltransferases and GSLs Accompanied with ES Differentiation
To elucidate the mechanism underlying GSL changes during ES differentiation, we analyzed the expression of glycosyltransferases involved in GSL biosynthesis during differentiation of ES cells. Knockdown of the Ugcg gene, which encodes a key glycosyltransferase to convert ceramide to glucosyl Cer in the initial step of GSL biosynthesis, resulted in many embryonic defects in mice, suggesting a crucial role of GSLs in normal development [55]. Jung et al. reported that suppression of Ugcg in mouse ES cells led to significant abnormalities in neural differentiation, especially related to GFAP and MAP-2 expression [56].
We found that several glycosyltransferases for the globo-series GSLs on cell surface (B3GALT5, FUT1, FUT2) were downregulated during human ES differentiation; in contrast, enzymes responsible for the synthesis of GM2, GD3, and GT1b (ie, B4GALNT1, ST8SIA1, and ST3GAL1, respectively) were upregulated. As the overall Gb3Cer synthesis from lactosyl Cer decreased in favor of the switch toward ganglio-series upon differentiation, it should follow that the expression of both SSEA-3 and SSEA-4 was also reduced (Fig. 2). Since lacto-series GSLs were expressed in undifferentiated human ES cells, glycosyltransferases associated with such expression, Lc3 synthase (B3GNT5) and Lc4 synthase (B3GALT5), were found in undifferentiated cells, but downregulated after differentiation (Fig. 2). Therefore, the altered expression of these key glycosyltransferases may account for the switch in core structures of GSLs in favor of ganglio-series GSL biosynthesis during ES differentiation (Fig. 2).
When human ES cells were induced toward lineage-specific differentiation, there were also changes in the expression of key glycosyltransferases. In neural progenitor cells, the expression of glycosyltransferases such as FUT1, FUT2, and B3GALT5 for biosynthesis of globo-series was found to be downregulated. In contrast, the expression of glycosyltransferases for ganglio-series GSLs, such as ST3GAL1, ST3GAL5, and ST8SIA1, increased [15]. In definitive endoderm cells, the expression of glycosyltransferases was downregulated, especially Gb5 synthase, leading to the accumulation of Gb4Cer [15]. These results indicated that ES differentiation, which was accompanied with a drastic switch of the profiling of GSLs on cell surface, might occur as a result of concerted changes in the expression levels of key glycosyltransferases involved in the manufacture of GSLs [14].
Development of Glycan-Targeting Anticancer Immunotherapeutics
Altered glycosylation is a well-known feature of cancer cells [57]. These abnormal glycans contribute to tumorigenesis and metastasis. A variety of tumor-associated glycan structures have been identified, including those GSLs expressed on human ES cells or their differentiated derivatives, such as Globo H, SSEA-3 [44], GD3, GD2 [58], GM2, and Neu5GC-GM3 [59], and those attached to proteins such as Tn, sialyl Tn, Lewisy, sialyl Lewisx, sialyl Lewisa, Lewisx, Thomsen-Friedenreich, and polysialic acids [60]. Overexpression of these glycoconjugates in tumors with limited expression in normal tissues makes them targets for immunotherapy of cancers. Indeed, since the 1990s, many of the above-mentioned tumor-associated glycans have been exploited as targets for cancer immunotherapy in clinical trials, but the majority terminated in early phase studies.
To date, five glycan-targeting immunotherapeutics have reached randomized phase III development, as summarized in Table 2. These include passive immunotherapy with GD2-directed mAb in neuroblastoma [43], as well as active immunotherapy with sialyl-Tn-KLH (keyhole limpet hymocyanin) vaccine in breast cancer [61,62], GM2 vaccine in melanoma [63,64], Globo H vaccine in breast cancer [47], and anti-Neu5GC-GM3 idiotypic vaccine Racotumomab in lung cancer [65]. However, only Dinutuximab, a chimeric anti-GD2 mAb, has received regulatory approval for commercialization in the United States and Europe. Treatment of high-risk neuroblastoma with Dinutuximab in a phase III study (Children's Oncology Group ANBL0032) chaired by A.L.Y. has demonstrated a significant improvement in the event-free survival (EFS) and overall survival (OS) in patients with high-risk neuroblastoma, marking Dinutuximab as the first new therapeutics approved for neuroblastoma [43]. Among more than 50 mAbs available for cancer therapy, GD2 is the first tumor-associated glycan shown to be an effective target antigen for cancer immunotherapy.
GM2-KLH/QS-21 versus observation.
GM2-KLH/QS-21 versus IFNα2b.
GloboH-KLH/OPT821 versus control.
Racotumomab-Alum vaccine versus placebo.
EFS, event-free survival; GM-CSF, granulocyte–macrophage colony-stimulating factor; HR, hazard ratio; IL-2, interleukin-2; KLH, keyhole limpet hemocyanin; NSCLC, nonsmall cell lung cancer; OS, overall survival; PFS, progression-free survival; RFS, relapse-free survival; TTP, time to progression; 95% CI, 95% confidence interval.
On the other hand, phase III trials of sialyl-Tn-KLH vaccine (Theratope) and GM2-KLH vaccine failed to demonstrate any benefits, although post hoc analysis of Theratope study showed survival benefit for breast cancer patients receiving endocrine therapy [62]. A preliminary randomized trial of Racotumomab (anti-Neu5GC-GM3 idiotypic vaccine) in advanced nonsmall cell lung cancer (NCT01460472) showed a slight but significant prolongation of progression-free survival and OS [65], which led to an ongoing, large-scale, randomized phase III trial. A randomized phase II/III trial of Globo H-KLH vaccine demonstrated significant benefits in both progression-free survival and OS for those breast cancer patients who developed IgG anti-Globo H responses [47]. The encouraging data have provided the impetus for us to conduct a global phase III trial, which is under development. In summary, GD2 is the first glycan that has been verified as an effective target for cancer immunotherapy, and the approval of Dinutuximab will certainly widen the net of potential pharmaceutical targets.
Rationales for Targeting GD2 in Cancer Immunotherapy
GD2, a b-series ganglioside consisting of GalNAcβ1-4(NeuAcα2-8NeuAcα2-3)Galβ1-4Glcβ1-1′Cer (Table 1), is expressed weakly in human and mouse brain, neural progenitor cells [22], peripheral pain fibers, and skin melanocytes [66,67]. In contrast, GD2 is abundantly expressed on various cancers, including neuroblastoma (>98%), melanoma, glioma, small cell lung cancer, and sarcomas [58,68]. Notably, GD2 is also found in normal stem cells, including embryonic neural stem cells [69] and mesenchymal stem cells (MSCs) [70,71], as well as breast CSCs [72,73] and malignant phyllodes tumor of the breast [74]. In mouse, GD2+ bone marrow-derived MSCs (BM-MSCs) have been characterized as a subpopulation of BM-MSCs possessing potent stemness ability. These GD2+ BM-MSCs displayed ES cell markers, SSEA-1 and Nanog, without CD45 and CD11b. They exhibit greater clonogenic and proliferative capabilities as well as better differentiation potential compared with unsorted BM-MSCs [75].
In line with these data, we showed that malignant phyllodes tumor of the breast contains a subpopulation of GD2+ALDH+ cells exhibiting the characteristics of CSCs, as reflected by their greater mammosphere-forming ability, higher tumor-initiating frequency, and capability of differentiation into neural cells of various lineages [74]. Similar findings have been reported for GD2+ H-Ras oncogene-transformed human mammary epithelial cells [73]. Conversely, Woo et al. found that the subpopulation of GD2+ cells in glioblastoma multiforme CSC (Sox2+ and Nestin+), 532T and 464T, did not show greater sphere formation capacity than GD2− subpopulation [76]. Taken together, GD2+ cells may represent a unique subset of breast cancer and MSCs with stemness potential.
Several reports have implicated that GD2 might be essential for the proliferation and invasiveness of tumor cells [77,78], but the precise role of GD2 in tumorigenesis has yet to be elucidated. The molecular mechanisms of GD2 in cancer have been studied by using anti-GD2 mAbs or manipulation of GM2/GD2 synthase, with the caveat that the effects of the latter strategy may not be restricted to GD2. Incubation of GD2+ tumor cells with anti-GD2 mAbs induced cell detachment, followed by cell death [79], inhibited cell invasiveness through suppressing matrix metalloproteinase-2 expression, and its binding to CD166 reducing phosphoinositide 3-kinase activity and phosphorylation of Akt [80]. In addition, anti-GD2 mAb 14G2a has been shown to dampen the expression of Aurora kinase, MYCN, ID1, and TLX2 and upregulate the expression of p53, JUN, SVIL, and RASSF6 [81,82].
Similar findings have been reported for another anti-GD2 antibody 3F8 that inhibited cell growth and induced apoptosis through both activation of caspase 3-, 7-, and 8-dependent pathways and downregulation of antiapoptotic molecules, survivin and cytochrome c [83]. Furthermore, 3F8 was shown to induce Ca++ fluxes in human neuroblastoma SH-SY5Y-TrkB cells by activating Src-family kinases to phosphorylate N-methyl-D-aspartate receptor subunit 2B [84]. The latter may explain neuropathic pain associated with anti-GD2 infusion. The antitumor activities of anti-GD2 are largely recapitulated by downregulation of GM2/GD2 synthase. Such strategy has been reported to reduce c-Met phosphorylation and cell proliferation of the triple-negative (ER−PR−Her2−) breast cancer cell, MDA-MB-231, and enhance anoikis of Renca-v cells [85].
Treatment of human platelets with purified GD2 increased the adhesion of platelets to collagen through upregulating integrin α2β1-mediated tyrosine phosphorylation of focal adhesion kinase [86]. This implies that GD2 might play a role in promotion of tumor metastasis by platelets [87,88]. In addition to platelets, purified GD2 reduced the expression of CD83 and CD86 on CD34+ bone marrow progenitors, suggesting that GD2 may impede differentiation of CD34+ cells into dendritic cells [89]. As for the other immune effector cell functions, Ladisch et al. showed that GD2 suppressed the proliferation of T cells [90]. Along this line, GD2 shed from renal cell carcinoma could be incorporated by circulating T cells to induce apoptosis of the GD2-inserted T cells [91]. However, neoexpression of surface GD2 has been found to cluster with T cell receptor upon activation of CD4+ T cells, which is associated with the induction of the GM2/GD2 synthase [92]. These findings suggest that GD2 may promote tumor growth while modulating immune activation.
Clinical Development of Cancer Immunotherapeutics Targeting GD2
Passive immunotherapy with a variety of GD2-specific mAbs has been the mainstay of GD2-targeting strategies. Several anti-GD2 mAbs, including murine mAbs, 14G2a and 3F8, chimeric mAb ch14.18, humanized mAbs, Hu14.18 and Hu3F8, and mAb fused with cytokine, have been generated and evaluated in clinical trials. In addition, active immunotherapy with synthetic GD2 or GD2-directed anti-idiotypic mAb is undergoing clinical development.
For passive immunotherapy, murine mAb 14G2a [93,94] was evaluated in phase I clinical trials, which showed significant dose- and infusion rate-dependent toxicities, including pain, tachycardia, hypotension, hypertension, fever, hyponatremia, and urticaria [95 –97]. Clinical benefits were noted in some patients in these early phase trials. To enhance the antibody-dependent cell-mediated cytotoxicity (ADCC) induced by anti-GD2 mAb, a phase I clinical trial of 14G2a + interleukin 2 (IL-2) demonstrated the feasibility with clinical responses in two patients [98]. To improve the pharmocodynamic properties of 14G2a, a human–mouse chimeric anti-GD2 mAb ch14.18 was generated [99], which displayed ADCC activities 50–100 times greater than the murine 14G2a [100]. As expected, the half-life of ch14.18 (66.6 ± 27.4 h) was longer than 14G2a (18.3 ± 11.8 h), but the toxicity profiles were similar to 14G2a [101 –103].
Since granulocyte–macrophage colony-stimulating factor (GM-CSF) also stimulates ADCC activities of granulocytes [104 –106] while promoting the production of monocytes, eosinophils, and neutrophils [107], a phase II study of ch14.18 + GM-CSF was conducted in neuroblastoma and objective clinical responses were noted in 10/49 patients [108,109]. During these early phase trials, most responders had relatively small disease burden, especially those patients with ≤10% tumor cells in the bone marrow. This led to the subsequent development of anti-GD2 immunotherapy to target minimal residual disease in neuroblastoma.
Patients with high-risk neuroblastoma who achieved at least PR to induction therapy and completed stem cell transplantation and radiotherapy were randomly assigned to standard therapy with six cycles of isotretinoin or immunotherapy, which consists of isotretinoin and five concomitant cycles of ch14.18 with alternating GM-CSF and IL-2 (NCT00026312). Analysis of the 226 randomized patients showed that 2-year EFS (66% ± 5% vs. 46% ± 5%, P = 0.01) and OS (86% ± 4% vs. 75% ± 5%, P = 0.02) were significantly higher for patients randomized to immunotherapy [43]. This major breakthrough formed the basis for the subsequent regulatory approval of ch14.18 (Dinutuximab, Unituxin®) in United States and Europe in 2015 and immunotherapy with Dinutuximab has since been considered as standard of care for high-risk neuroblastoma. It also marks the first successful immunotherapy to target a glycan antigen.
To further improve the efficacy, humanized 14.18 (hu14.18) antibody was generated by CDR grafting of 14.18 V regions [110]. In addition, to ameliorate the neuropathic pain induced by anti-GD2 mAb, a K322A mutation of the Fc region was introduced to limit the ability of complement fixation by C5a [111], which reduced allodynia, while maintaining its ADCC activity [111]. A phase I clinical trial showed higher maximum tolerated dose of hu14.18K322A than ch14.18. In addition, ongoing clinical trials of combining chemotherapy with ch14.18 or hu14.18K322A have yielded impressive clinical responses in patients with relapsed/refractory neuroblastoma [112,113], prompting the development of incorporating immunotherapy to frontline chemotherapy for newly diagnosed high-risk neuroblastoma.
Another anti-GD2 antibody 3F8a, murine IgG3 mAb, has been extensively evaluated in a series of nonrandomized clinical trials, which showed similar side effects and antitumor activities as 14G2a [68]. Subsequently, a series of sequential phase II studies of mAb3F8 showed an overall 5-year EFS of 62% for stage 4 patients in first remission who received 3F8 + GM-CSF + cis-retinoic acid [114] and a correlation of a better outcome for patients with the FCGR2A (R/R) genotype [115]. Recently, humanized 3F8 (hu3F8) has been generated [116], which is now undergoing phase I clinical trial. Another strategy to enhance the antitumor efficacy of antibody is to link antibody with cytokine so as to achieve high cytokine concentrations in the tumor microenvironment. Hu14.18-IL-2, a fusion protein (hu14.18 and IL-2), was found to have similar toxicities as anti-GD2 combined with IL-2. Evidence of antitumor activity for neuroblastoma and melanoma was noted in phase I and II studies [117 –119], and patients with KIR-ligand mismatch seemed to be associated with better clinical response [120,121].
For active immunotherapy, Jerne's idiotypic network hypothesis provides a strategy to produce antigen mimetics [122]. An anti-idiotype antibody, 1A7, mimicking GD2 antigen was generated by immunizing mice with 14G2a [123]. Active immunotherapy with mAb1A7 in patients with advanced melanoma and neuroblastoma was well tolerated with only local reactions [124]. Vaccination with mAb1A7 induced significant IgG anti-1A7 titers, which could bind to GD2-expressing tumor cells and display CDC and ADCC activities [124 –126]. Thus, active immunotherapy with anti-idiotypic antibody-based GD2 vaccine may offer the advantage over passive immunotherapy with reduced infusion-related toxicities, but its therapeutic efficacy awaits verification in randomized clinical trials.
Using purified GD2 as a vaccine to activate the immune system is another strategy for active immunotherapy, but it is hampered by the poor immunogenicity of GD2. Many strategies are employed to enhance the immunogenicity of GD2 vaccine, such as chemical conjugation of GD2 to a highly immunogenic protein scaffold and addition of immune stimulating agent as adjuvant. Based on these strategies, synthetic GD2 was conjugated to immunogenic protein KLH, and monophosphoryl lipid A (MPL-A) was used as an adjuvant. The GD2-KLH + MPL-A vaccine was well-tolerated without adverse effects in patients with gliomas, but it failed to induce anti-GD2 antibody or clinical response [127]. Another phase I clinical trial of GD2-KLH using QS-21 combined with oral beta-glucan as adjuvants induced anti-GD2 antibody in a majority of patients, and more importantly, clearance of minimal residual disease in some patients [128]. Similarly, GM2-KLH + GD2-KLH + QS-21 vaccine induced anti-GM2 (97%) and anti-GD2 (73%) IgG/IgM Abs in patients with melanoma or sarcoma in phase I trial [129]. These promising results suggest that carrier protein and adjuvants may enhance the immunogenicity of GD2-based vaccines or other carbohydrate-based vaccines.
Globo H-Targeted Immunotherapy
Globo H, a hexasaccharide (Table 1), was initially identified in the human breast cancer cell line, MCF-7 [30], and subsequently found in a variety of epithelial cancers, including breast, colon, ovarian, gastric, pancreatic, lung, and prostate cancers [130,131]. We further showed the expression of Globo H and its precursor, SSEA-3, in 61% (25/41) and 77.5% (31/40), respectively, of breast cancer tissues and 20% (8/40) and 62.5% (25/40), respectively, in breast CSCs [44]. The immunosuppressive effects of Globo H-ceramide were first demonstrated by our group [45]. Globo H-ceramides shed from tumor cells were taken up by T and B lymphocytes (Fig 4), with consequent inhibition of their proliferation and cytokine/immunoglobin production (Fig 5). Consistent with this, tumor lymphocytes in Globo H-expressing tumor cells showed positive staining for Globo H (Fig 4).
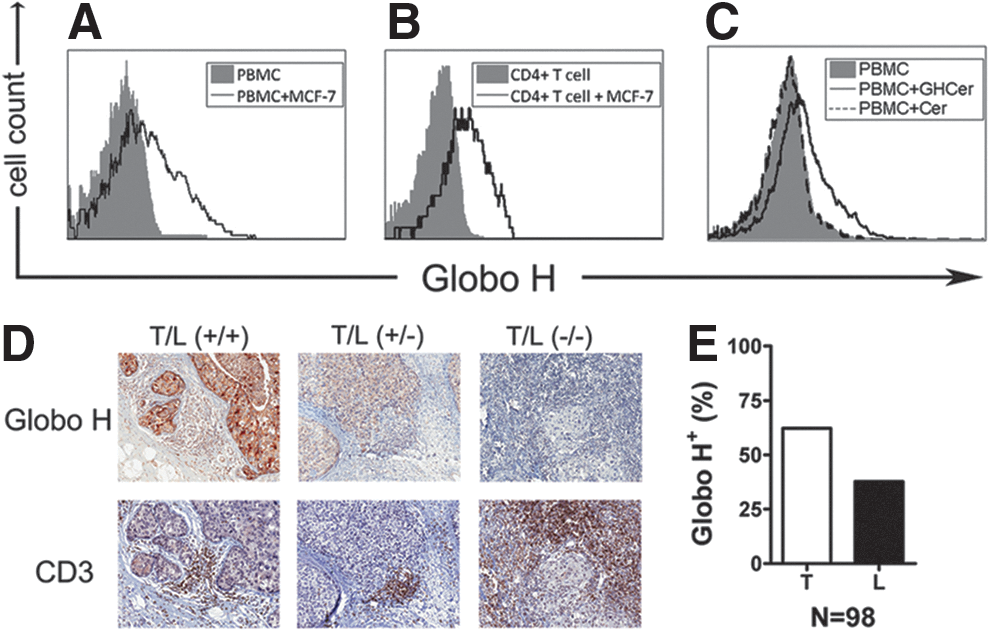
Uptake of Globo H by lymphocytes in vitro and in vivo. Human PBMCs were cultured with/without MCF-7 cells, and then the expression of Globo H on peripheral blood mononuclear cell (PBMC)
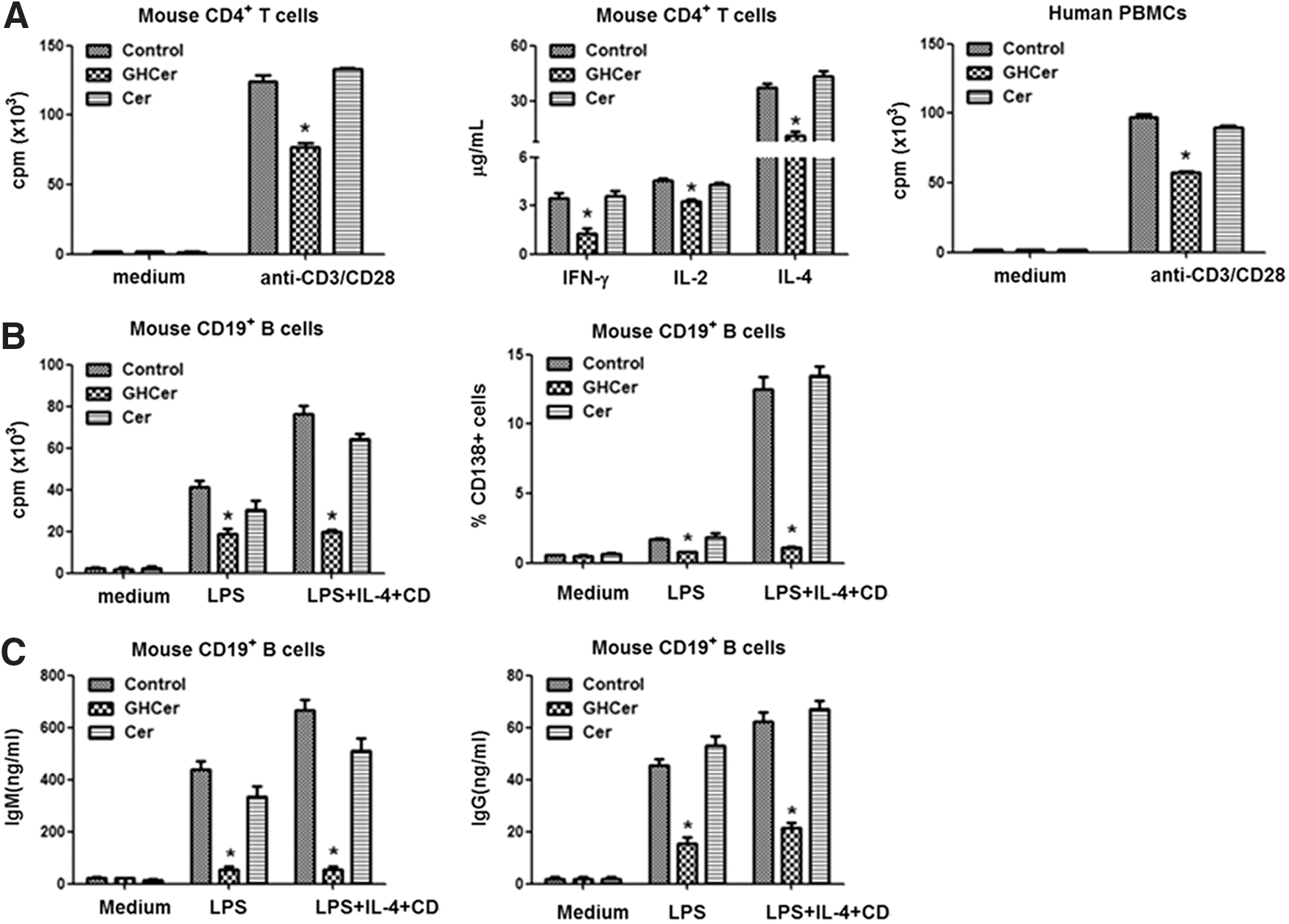
Effects of Globo H-ceramide on the proliferation of immune cells and production of cytokines and immunoglobulins. The indicated immune cells were incubated with Globo-H-Cer, ceramide, or medium control and then activated by anti-mouse or human CD3 and CD28 monoclonal antibodies
Globo H-ceramide-induced immunosuppression could not be attributed to lymphocyte apoptosis or regulatory T cell expansion. However, the expression of Notch, which is crucial for T cell activation, was diminished in Globo H-treated cells, while the expression of its regulatory molecules, inhibitor of DNA-binding protein 3 (id3), casitas B-lineage lymphoma proto-oncogene b (cbl-b), and itchy E3 ubiquitin protein (itch), was upregulated. The latter was preceded by the upregulation of early growth response 2/3 (egr2/3) [45]. These results provide the first evidence that Globo H-ceramide acts as an immune checkpoint molecule, allowing cancer cells to escape from immune surveillance.
In addition to immunosuppressive effects of Globo H-ceramide, we also provided the first evidence for its angiogenic activity [46]. Globo H-ceramide induced tube formation of endothelial cells in vitro and angiogenesis in Matrigel plug assay in vivo. In line with this study, Globo Hhi subpopulation of MCF-7 tumor cells grew faster with greater vessel density than Globo Hlow tumor cells in vivo. This was recapitulated by the observation of higher vessel density in Globo H+ than Globo H- breast cancer specimens. Mechanistic investigations linked the angiogenic effects of Globo H-ceramide to its endocytosis and binding to translin-associated factor X (TRAX), which was identified as Globo H interactive protein in endothelial cells. Upon binding of Globo H-ceramide to TRAX, phospholipase C beta 1 (PLCβ1) is released from TRAX to trigger Ca2+ mobilization and induce angiogenesis (Fig 6). This is the first globoside shown to display angiogenic activity, along with elucidation of its mechanisms.
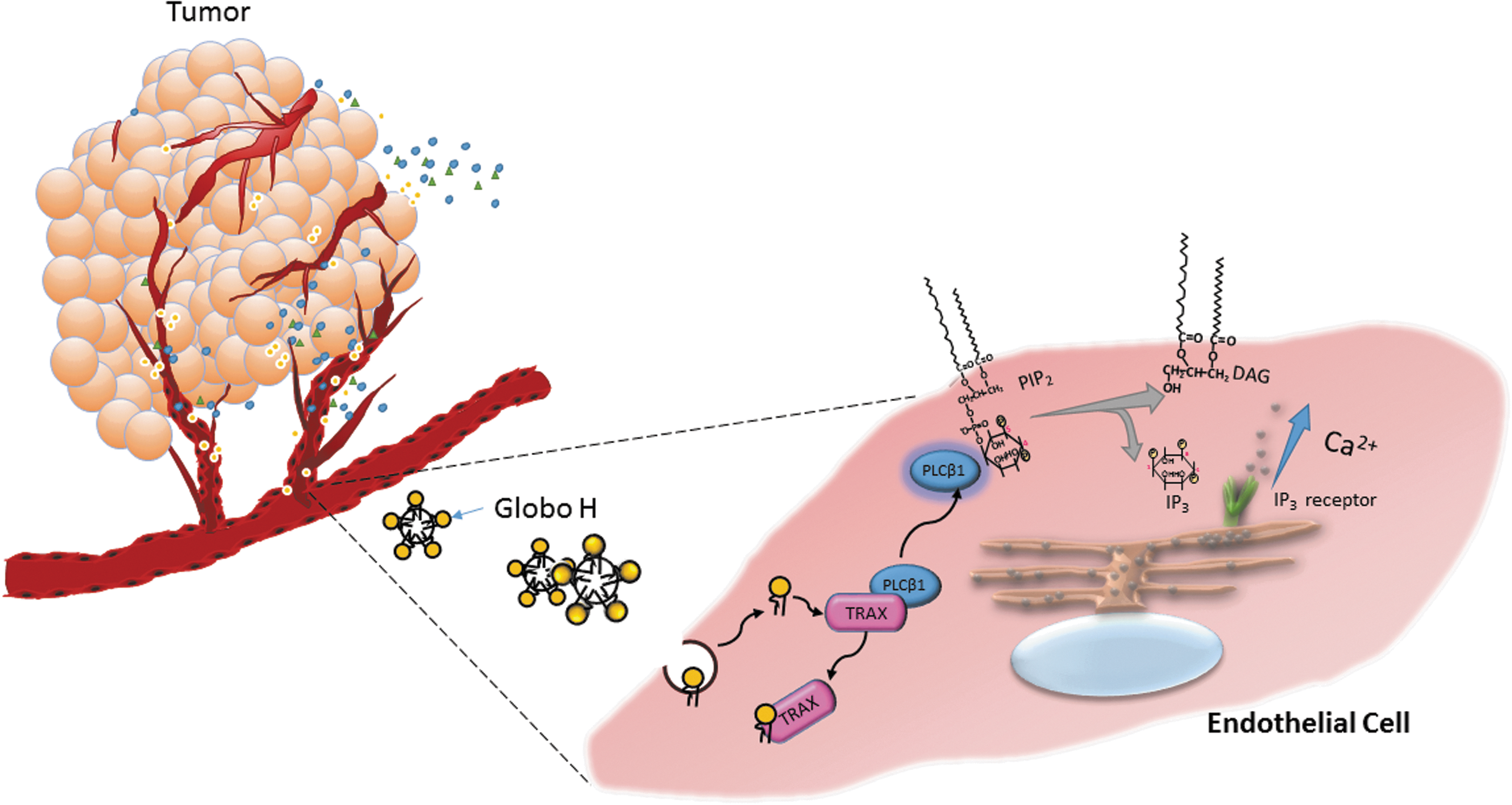
Shedding of Globo H-ceramide from tumor promotes angiogenesis in the microenvironment. Endothelial cells in the tumor microenvironment incorporate the Globo H-ceramide (Globo H-Cer) in microvesicles shed by tumor cells. Globo H-Cer interacts with translin-associated factor X (TRAX) to dissociate the phospholipase C beta 1 (PLCβ1) from the TRAX-PLCβ1 complex, leading to hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to form inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). The IP3 interacts with the IP3 receptor on endoplasmic reticulum (ER) and induces calcium influx to trigger angiogenesis.
On the other hand, Globo H was reported to bind to human RNase 1, facilitating internalization of RNase 1, which induced cell death. Blocking the interaction of Globo H and RNase 1 with anti-Globo H antibody partially rescued the cells from RNase-induced cell death [132]. These findings suggested multifaceted roles of Globo H in tumor biology.
The expression of Globo H in undifferentiated ES cells disappeared after differentiation at gastrulation. Overexpression of Globo H in cancer with limited expression in normal tissues thus makes Globo H-ceramide a potential target for cancer immunotherapy. This is further supported by our demonstration of triple roles of Globo H-ceramide in cancer: a stem cell marker [44], an immune checkpoint molecule [45], and an angiogenic factor [46]. These findings provide further impetus for clinical development of Globo H-targeted immunotherapy [133]. In an animal study, injection of Globo H-KLH mixed with α-GalCer induced antibodies reactive with Globo H and SSEA-3, suggesting that a Globo H-based vaccine will target tumor cells expressing Globo H or SSEA-3, including breast CSCs [44]. Like Globo H, SSEA-3 expression in normal tissues was limited to the secretory borders of epithelium, where access to the immune system is restricted.
Two phase I clinical trials of Globo H-KLH/QS-21 vaccine in patients with relapsed prostate cancer (n = 18) [134] and metastatic breast cancer (n = 27), respectively [135], demonstrated the safety of the vaccine, along with induction of humoral antibody responses. Recently, a multinational randomized phase II/III clinical trial of Globo H-KLH vaccine versus placebo in patients with metastatic breast cancer has completed accrual of 349 patients. The Globo H-KLH vaccine significantly improved progression-free survival and OS in those patients who mounted IgG anti-Globo H responses [47]. Based on these promising results, a global phase III trial is underway. Another phase II clinical trial of this vaccine in ovarian cancer is ongoing.
Furthermore, to overcome the hurdles over the regulatory issues and supplies of the carrier protein KLH and the adjuvant OPT-821 used in the current Globo H-KLH + QS-21 vaccine, a new generation of Globo H vaccine has been developed. In a preclinical study, three carrier proteins, including bovine serum albumin, tetanus toxoid, and genetically modified cross-reacting material of diphtheria toxoid (CRM197 DT), in combination with various adjuvants, including α-GalCer and its analogs 7DW8-5, C34 and C17, alum, and MF59, were evaluated. Immunization with the Globo H-CRM197 DT + C34 vaccine induced the greatest level of anti-Globo H IgG and IgM, equivalent to Globo H-KLH + QS-21 vaccine [136]. Thus, Globo H-CRM197 DT + C34 vaccine is now under early phase clinical development.
Conclusions
GSLs are membrane lipids consisting of a polar oligosaccharide chain attached to a hydrophobic sphingosine-containing ceramide lipid moiety. Antibodies against several GSLs designated as SSEAs have been widely used to characterize the undifferentiated state of ES cells. Since the cross-reactivities of these antibodies with multiple glycans can yield misleading interpretations and conclusions, a few laboratories, including ours, have employed advanced MS technologies to define the dynamic changes of surface GSLs upon ES differentiation. With MALDI-MS and tandem MS/MS analyses, we delineate changes in GSLs systematically during differentiation of human ES and iPS cells into various derivatives. In addition to the well-known human ES markers, SSEA-3 and SSEA-4, several previously undisclosed globo- and lacto-series GSLs (Gb4Cer, Lc4Cer, fucosyl Lc4Cer, Globo H, and disialyl Gb5Cer) were identified in the undifferentiated human ES and iPS cells. A unique switching of GSL core structures from globo- and lacto- to ganglio-series during human ES differentiation was revealed, consistent with the observed changes in GSL expression during murine embryonic development.
Lineage-specific differentiation was also marked by alterations of specific profiling of GSLs. During differentiation into neural progenitors, core structures shifted to primarily ganglio-series dominated by GD3. During endodermal differentiation, GSL patterns shifted to prominent expression of Gb4Cer with little SSEA-3 and −4 or GD3. These dynamic changes in GSL profiles were associated with altered expression of key GTs in GSL biosynthetic pathways and were clearly linked to lineage specificity and differentiation of human ES cells. Finally, with better understanding of the relevance of GSL signatures in ES cells to cancer, the new anticancer immunotherapy targeting tumor-associated surface glycans, which are expressed in ES cells or progenitors, is gaining international attention. Until recently, all approved cancer immunotherapies target proteins, but not glycans. While GD2-targeted immunotherapy of neuroblastoma marks the first new agent targeting a GSL molecule to receive FDA approval for standard medical care, Globo H-targeted immunotherapy of breast cancer has generated promising results in the ongoing multinational clinical trials. These advances in the studies of glycolipids point to the need for further mechanistic investigations to provide scientific rationales for targeting glycans in cancer immunotherapy.
Footnotes
Acknowledgments
This work was supported by grants of the Chang Gung Medical Foundation and the Ministry of Science and Technology for A.L.Y. (OMRPG3C0013, MOST104-2321-B-182A-002, and MOST104-2321-B-182A-003), J.-T.H. (CMRPG3D1492), M.-Y.H. (CMRPG3D1592 and MOST104-2321-B-001-030), and J.Y. (OMRPG3C0043, MOST104-2321-B-182A-001, and MOST104-2321-B-182A-004).
Author Disclosure Statement
No competing financial interests exist.