
Abstract
The ability to rationally target disease-causing mutations has been made possible with programmable nucleases with the clustered, regularly interspaced short palindromic repeats/Cas9 system representing a facile platform for individualized gene-based medicine. In this study we employed footprint-free reprogramming of fibroblasts from a patient with mutations to the Fanconi anemia I (FANCI) gene to generate induced pluripotent stem cells (iPSCs). This process was accomplished without gene complementation and the resultant iPSCs were able to be gene corrected in a robust manner using the Cas9 nickase. The self-renewing iPSCs that were maintained under feeder-free conditions were differentiated into cells with characteristics of definitive hematopoiesis. This defined and highly efficient procedure employed small molecule modulation of the hematopoietic differentiation pathway and a vascular induction technique to generate hematopoietic progenitors. In sum, our results demonstrate the ability to induce patient-derived FA cells to pluripotency for patient-specific therapeutic cell derivation.
Introduction
G
FA is a complex disease caused by mutations to one of 15–16 FA pathway genes: FANCM, whose implication in FA is not definitive, and bona fide FA genes: FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCG, FANCI, FANCJ/BRIP1, FANCL, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4, and FANCQ/ERCC4/XPF [4 –7]. Sequence mutations to these genes result in an inability to repair interstrand DNA cross-links causing developmental anomalies, increased rates of tumorigenesis, and bone marrow failure [8]. Treatment by hematopoietic cell transplant (HCT) can be curative for the lethal bone marrow manifestations of the disease; however, the therapeutic benefits do not extend to the physical abnormalities. Furthermore, allogeneic HCT in FA patients is associated with significant risks due to the pre and post-transplant treatment procedures [9] including the possibility for a magnified incidence of tumor occurrence [10]. Toward advancing autologous therapies, and with recognition of potential vector-associated risks, early stage (Phase I/II) gene therapy trials have been implemented. A main advantage of this is the mitigation of transplant-associated complications associated with HCT from an unrelated donor. However, the low numbers of hematopoietic progenitor cells available for modification remains a significant hurdle. One highly desirable solution to this would be to engineer autologous induced pluripotent stem cells (iPSCs) capable of forming engraftable hematopoietic progenitors. To date, however, the poor reprogramming efficiency of FA cells has restricted the realization of this potential [11]. Further, despite intense efforts, a true human iPSC-derived hematopoietic progenitor capable of in vivo engraftment has not yet been demonstrated. Therefore, we established a line of investigation utilizing cells from an FA patient (complementation group FANCI, chosen because of the central position of FANCI in the FA/BRCA pathway) to determine whether genome modification, improved reprogramming methodologies, and advances in cellular engineering could be synergized to address gaps in the fields of FA biology and transplant medicine.
Toward assessing the engineering capacity of FANCI cells, we employed the highly efficient nonintegrating Sendai virus reprogramming methodology [12] to generate iPSCs from primary fibroblasts. These iPSCs were highly permissive to gene editing using the clustered, regularly interspaced short palindromic repeats (CRISPR)/Cas9 platform. This two-component system is comprised of a short guide RNA (gRNA) transcript and the Cas9 protein [13]. Cas9 can function as a double-stranded DNA nuclease or a single-stranded DNA nickase. Of note, we have demonstrated previously that nicking preferentially promotes HDR in FANCC cells [14]. Here too we observed the ability of the Cas9 nickase to mediate FANCI gene correction in patient-derived iPSCs, while the parental fibroblast cells were recalcitrant to gene editing. Using gene-corrected iPSCs we assessed their hematopoietic differentiation capability by performing directed differentiation in vitro. By combining modulation of the fate determinants of primitive and definitive hematopoiesis with a supportive endothelial coculture system, we were able to generate a population of CD34+CD38− cells. This phenotype is consistent with cord blood-derived cells [15] capable of engrafting and collectively represents an advance in cellular engineering and translational application for FA therapy.
Materials and Methods
Research subject line generation and culture
Primary fibroblasts were derived from a 4 mm skin punch biopsy collected from a pediatric patient given the designation FA-28. The cell line was cultured under hypoxic conditions and maintained in complete Dulbecco's modified Eagle's medium with 20% fetal bovine serum (FBS), 0.1 mg/mL each of penicillin and streptomycin, 10 ng/mL each of epidermal growth factor and fibroblast growth factor, 100 U/mL nonessential amino acids, and 1× antioxidant supplement (Sigma-Aldrich, St. Louis, MO). This study was performed in accordance with the principles for research on human subjects set forth by the Declaration of Helsinki and was preceded by parental informed consent and University of Minnesota Institutional Review Board approval.
CRISPR/Cas9 reagent construction
The Cas9 nuclease and nickase plasmids were a gift from Dr George Church (Addgene plasmids #41815 and #41816 [16], Cambridge, MA) and the FANCI-specific gRNAs were synthesized using a 60mer oligonucleotides in the following manner: First, an “empty” SpCas9 gRNA plasmid was constructed by cloning the U6 promoter, SpCas9 sgRNA scaffold sequence, and an intervening EcoRI restriction site into the pCR4 TOPO vector (Invitrogen, Carlsbad, CA). DNA sequences of the 60mer oligos are listed below and contain the 20 base pairs (bp) upstream of EcoRI, the 19 bp FANCI target sequence, and the 20 bp downstream of EcoRI. An additional 20 bp of homology were included on either end by polymerase chain reaction (PCR) amplification of the template with universal primers Fwd (5′-TTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACC-3′) and Rev (5′-ATTTTAACTTGCTATGCTGTTTCCAGCATAGCTCTAAAAC-3′). The 99 bp PCR products were then cloned into the linearized pCR4 U6 gRNA vector by Gibson Assembly [17].
Template oligonucleotide sequences (5′-3′)
s1: GTGGAAAGGACGAAACACCG actgtacaaaggctgctta GTTTTAGAGCTATGCTGGAA
as1: GTGGAAAGGACGAAACACCG cagcctttgtacagtctga GTTTTAGAGCTATGCTGGAA
as2: GTGGAAAGGACGAAACACCG aacaactttgaagaactaa GTTTTAGAGCTATGCTGGAA
1461 donor construction
The left and right donor arms were amplified from the human genome with primers Left Fwd (5′-TTTCTTCTCCCTGCAGCAAT-3′) and Left Rev (5′-TACTGATTCTATATATACTTTTTCTCCAAT-3′) generating a 1,126 bp fragment and Right Fwd (5′-TGCCAAAACTGAGGTTTAACTG-3′) and Right Rev (5′-AATAAAACTCATTCCCCTCAACAA-3′) generating a 1,052 bp fragment. Donor arms were assembled into pcDNA3.1/V5-His plasmid (Invitrogen) containing a floxed PGK-puromycin selection cassette by enzyme digest and T4 DNA ligation (New England Biolabs, Ipswich, MA). A gBlock gene fragment (Integrated DNA Technologies, Coralville, IA) with overlapping homology was added immediately upstream of the right arm by Gibson Assembly for introduction of the corrective base and four silent mutations, including a knockout HindIII site. To facilitate construction of the puromycin and puromycin-free AAV donors, a 196 bp Ultramer (Integrated DNA Technologies) containing left arm 5′ homology and right arm 3′ homology (95 bp each) separated by a unique restriction site (6 bp) was added to pAAV-mCherry-MCS by PCR amplification and T4 DNA ligation. The full left arm→puromycin→right arm sequence was excised as a single fragment from the pcDNA backbone by restriction digest and shuttled into the pAAV vector by way of Gibson Assembly. Similarly, the puromycin-free AAV donor was constructed by Gibson Assembly of PCR-amplified left and right arms and an intervening gBlock with overlapping homology for introduction of the corrective base and 30 silent mutations, including a knockout HindIII site.
iPSC generation
Uncorrected fibroblasts were reprogrammed into iPSCs as described [18,19], using Sendai virus for delivery of reprogramming factors [12]. As part of Quality Assurance/Quality Control evaluations, karyotype, gene expression, and immunofluorescence were performed as previously described [18 –21]. iPSCs were cultured in feeder-free mTeSR1 (STEMCELL Technologies, Vancouver, BC) on Geltrex-coated (Thermo Fisher Scientific, Waltham, MA) tissue culture plates at 37°C under hypoxic conditions. Sendai virus clearance [22] was assessed on reverse transcribed RNA using the following forward (F) and reverse (R) primers: b-actinF: GGCACCCAGCACAATGAAGATCAA b-ActinR: AGGATGGCAAGGGACTTCCTGTAA, (Sendai virus genome: SeV) SeVF: GGATCACTAGGTGATATCGAGC, SeVR: ACCAGACAAGAGTTTAAGAGATATGTA, KlfF: TTCCTGCATGCCAGAGGAGCCC, KlfR: AATGTATCGAAGGTGTCTCAA. mycF: TAACTGACTAGCAGGCTTGTCG and mycR: TCCACATACAGTCCTGGATGATGATG, (Sendai Klf, Oct3/4, Sox2; KOS) KOSF: ATGCACCGCTACGACGTGAGCGC, KOSR: ACCTTGACAATCCTGATGTGG. PCR conditions were as follows: 95°C × 2 min and 35 cycles of 95°C × 1 min, 58°C × 40 s, and 68°C for 1 min using AccuPrime DNA polymerase (ThermoFisher Scientific, Waltham, MA).
Gene transfer
For 293T transfections, 500 ng each of Cas9 nuclease and gRNA were delivered with Lipofectamine 2000 (Invitrogen). For all other cell types 10 μg of the 1461 pcDNA puromycin plasmid donor was delivered with 1 μg each of Cas9 nickase and gRNA for Neon electroporation (1,500 V, 20 ms pulse width, and a single pulse, Invitrogen, Valencia, CA). Gene correction of iPSCs was achieved by Neon transfection using 1,100 V, 20 ms pulse width, and a single pulse. Cas9 nickase and gRNA were delivered at 500 ng each with either 5 μg of 1461 AAV puromycin plasmid donor or 5 μg of 1461 AAV puromycin-free plasmid donor. For 12–24 h before gene transfer, iPSCs were treated with 10 μM Rho-associated protein kinase (ROCK) inhibitor, Y-27632 (Selleck Chemicals, Houston, TX). All cells were incubated at 30° C for 48 h following gene transfer, excluding iPSCs that were consistently maintained at 37°C under hypoxic conditions.
Surveyor nuclease
293T genomic DNA was isolated 72 h following gRNA and Cas9 nuclease or nickase transfection and was PCR amplified with 1461 87F: GCAGTGTAATCGGTTTCTTCC-3′) and 1461 764R: AACTGGTTGCAAATGCCTCT-3′) or 3058F: TCCCACACCTTTCTGATAACC and 3058R: TTTCTACCTCCACCCCCAGT for on target assessment. Off-target sites were chosen using the MIT CRISPR Design Tool (
PCR conditions were as follows: 95°C × 3 min followed by 35 cycles of 95°C × 1 min, 58°C × 40 s, and 68°C × 1 min with AccuPrime DNA polymerase (ThermoFisher Scientific, Waltham, MA). PCR products were incubated with Surveyor nuclease (Integrated DNA Technologies) and resolved on a 10% TBE PAGE gel (Invitrogen) [14,23]. Rates of activity were quantified by densitometry using the ImageJ application [23,24]. All gel images used the same exposure times.
Selection
Cells were selected in bulk in 0.3 μg/mL puromycin (EMD Millipore, Billerica, MA) 4 and 6 days after gene transfer, respectively. Resistant fibroblasts were plated at low density for monoclonal expansion as previously described [14,20]. Bulk iPSCs receiving the 1461 AAV puromycin plasmid donor were selected 7 days after gene transfer in increasing amounts of puromycin escalated at each passage starting at 0.3 μg/mL. Bulk iPSCs transfected with the 1461 AAV puromycin-free plasmid donor were subjected to three progressive rounds of mitomycin C selection lasting 48 h each and beginning 16 days after gene transfer. The concentrations used were 10, 14, and 16 nM mitomycin C (Sigma-Aldrich).
Cell correction molecular screening
To screen for correction by puromycin donor, the donor-genome junction was amplified using internal primer F39 (5′-AGCCTCGACTGTGCCTTCTA-3′) and external primer R2339 (5′-TCACTGAGGCGCAATGATTA-3′) downstream of the right donor arm within the endogenous locus. For correction by puromycin-free donor, R2339 was used along with internal 7F (5′-GAGGCCTTCGACTACCTGAG-3′) to prime off the corrective base and 6 and of the 30 silent mutations. PCR products were TOPO cloned into the pCR4 vector for confirmation of HDR by Sanger sequencing. cDNA correction was detected by amplification with Exon 15 Fwd (5′-ACCTGCTTTCAAATATCGTCATGT-3′) and Exon 17 Rev (5′-CTGACTGACACTGAGAGACTGA-3′), followed by HindIII restriction digest. HindIII-resistant amplicons were cloned and sequenced as described above.
Differentiation of iPSCs to CD34
Human iPSC lines were maintained on Matrigel or Geltrex coated plasticware with feeder free, serum free culture conditions in mTeSR™1 media (STEMCELL Technologies, Vancouver, BC). iPSC and derivative cells were cultured under hypoxic conditions (2% O2). Unless otherwise noted, all cytokines were from R&D Systems (Minneapolis, MN). For embryoid body (EB) generation, hiPSCs were cultured at 80–90 % confluency, followed by EB formation as follows: undifferentiated hiPSCs were dissociated with Accutase (STEMCELL Technologies, Vancouver, BC) and aggregates were resuspended in APEL medium (STEMCELL Technologies, Vancouver, BC), supplemented with bone morphogenic protein-4 (BMP-4; 25 ng/mL) and basic fibroblast growth factor (bFGF; 5 ng/mL) and replated on non-tissue culture treated 10 cm2 dishes. At 42–48 hours, developing EBs were collected and resuspended in APEL-differentiation media containing BMP-4 and bFGF, at the same concentration as above, along with 3 μM CHIR99021 (Stemgent, Lexington, MA) and 6 μM SB431542 (Selleck Chemicals, Houston, TX). Twenty-four hours later the EBs were collected and resuspended in 40% APEL + 60% STEMSPAN II (STEMCELL Technologies, Vancouver, BC) media containing vascular endothelial growth factor (20 ng/mL), bFGF (5 ng/mL), IL-3 (20 ng/mL), Flt3L (20 ng/mL) and stem cell factor (SCF; 100 ng/mL) and cultured for a further 5–6 days. At day 8/9 EBs were harvested, washed once with PBS and dissociated using Accutase (Innovative Cell Technologies, San Diego, CA) and 0.25% Trypsin-ethylenediaminetetraacetic acid (EDTA; ThermoFisher Scientific, Waltham, MA) until no visible clumps remained. CD34+ cells were isolated using the EasySepTM CD34+ isolation kit (STEMCELL Technologies, Vancouver, BC).
Differentiation of CD34 to T cell progenitors
Dissociated CD34 cells from EBs were plated on OP-9 DL9 stromal cells that were at ∼70% confluency in alpha-MEM media supplemented with SCF (100 ng/mL), IL7 (5 ng/mL), and Flt3 (10 ng/mL) for 7 days, at which point the cells were passed to new OP9 without SCF. The cells were maintained until flow cytometric analysis.
Differentiation of CD34 to hematopoietic progenitors
Single cell CD34 cells at a density of 1 × 106 were plated on VeraVec™ cells (Angiocrine Bioscience, New York, NY) that were at 80% confluency in StemSpan II media supplemented with: IGF-1 (25 ng/mL), BMP-4 (10 ng/mL), SCF (200 ng/mL), Flt3 L (10 ng/mL), IL-6 (10 ng/mL), thrombopoietin (30 ng/mL), IL-11 (5 ng/mL), bFGF (5 ng/mL), VEGF (5 ng/mL), IL-3 (30 ng/mL), and erythropoietin (2 IU/mL). Cocultures were maintained for 9 days, at which point they were analyzed by flow cytometry.
Colony forming unit assay
Cells were placed in MethoCult according to the manufacturer's instructions (STEMCELL Technologies). Colonies were enumerated by an experienced analyst using light microscopy at low magnification (4 × objective).
Flow cytometry
All antibodies were acquired from eBiosciences (San Diego, CA), and FACS was performed on a FACSAria instrument (Beckton-Dickinson, Franklin Lakes, NJ).
Cellular isolation
Antigen-specific cell isolation was performed using the EasySep magnetic separation system (STEMCELL Technologies).
Results
FANCI gene, CRISPR/Cas9 targeting, and experimental approach
The FANCI gene is located at 15q26.1 (Fig. 1A) that encompasses ∼75 kb of genomic sequence and encodes a 150 kd protein that associates with FANCD2 to form a complex that localizes to sites of DNA damage [7,25]. Two FANCI compound heterozygous mutations were present in cells acquired from a male patient: the c.1461 T > A mutation in exon 15 causing a premature stop codon and the c.3058 + 4A>G intron mutation that, similar to other FA genes [26], likely causes a splicing abnormality that prevents functional protein production.
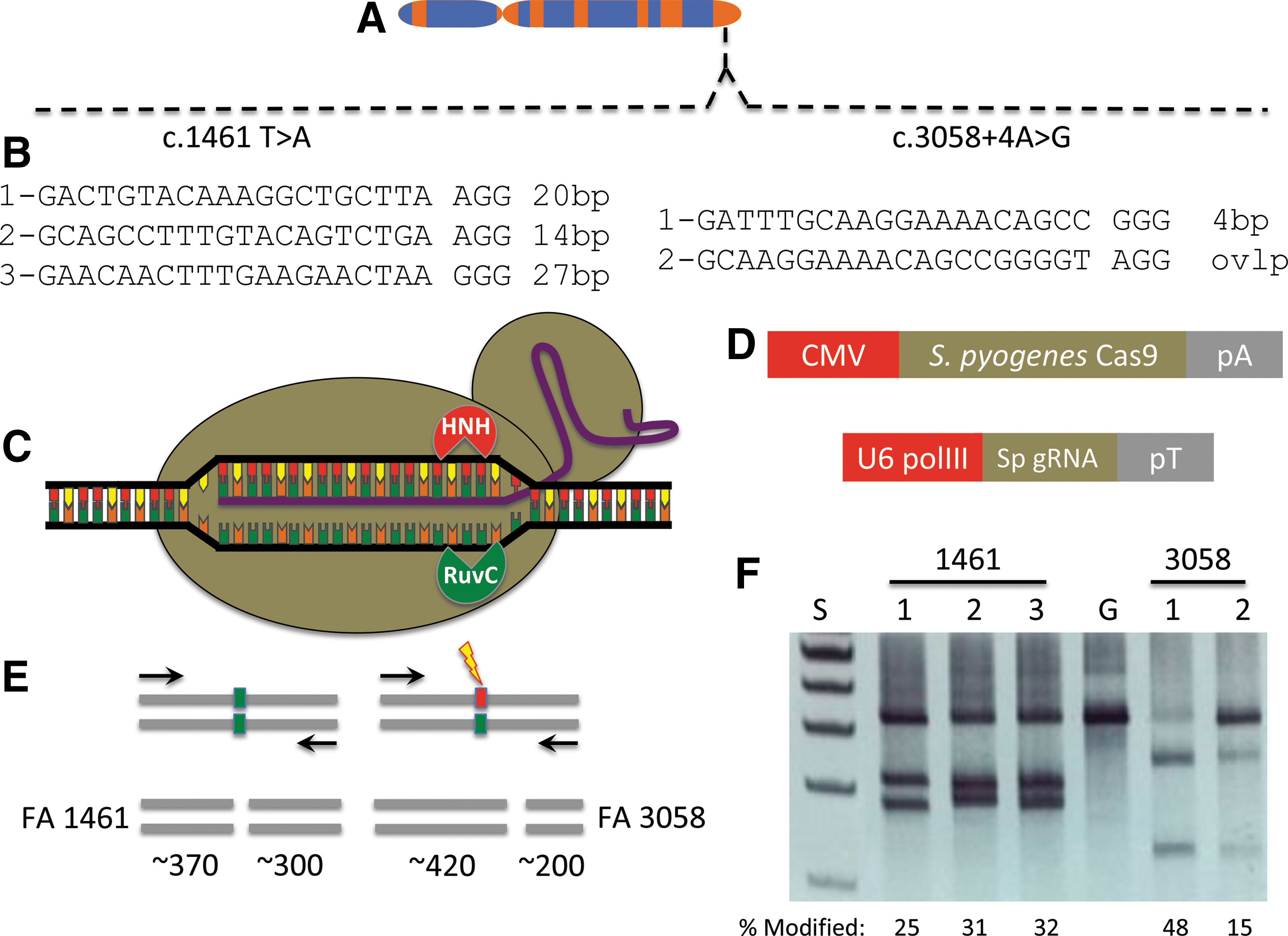
FANCI gene and CRISPR/Cas9 targeting.
The gene sequence proximal to each mutation was analyzed for permissive target sites for the CRISPR/Cas9 system derived from Streptococcus pyogenes [27]. S. pyogenes Cas9 recognizes sequences in the context of a three bp–NGG sequence termed a protospacer adjacent motif (PAM) [13] and targeting sites proximal to the respective mutations were identified (Fig. 1B). Five gRNA candidates were constructed for testing with the Cas9 nuclease that possesses helicase activity and has two functional domains (HNH and RuvC), each responsible for cleaving one strand of DNA (Fig. 1C). The Cas9 plasmid and a second plasmid encoding the specific gRNA (Fig. 1D) were introduced into 293T cells and analyzed for functionality using the Surveyor nuclease method [23]. This assay detects target-specific activity by virtue of Cas9 nuclease-mediated insertions and deletions resulting in amplicon cleavage by the Surveyor enzyme (Fig. 1E) [23] with all five candidates showing robust FANCI gene targeting activity (Fig. 1F). Based on these data, we implemented an experimental procedure summarized in Fig. 2 to correct the FANCI gene mutation in primary cells. A primary fibroblast cell line was derived from a skin punch biopsy (Fig. 2A) and this population of cells served as an experimental template for CRISPR/Cas9 gene correction (Fig. 2B) using a plasmid-based donor (Fig. 2C).
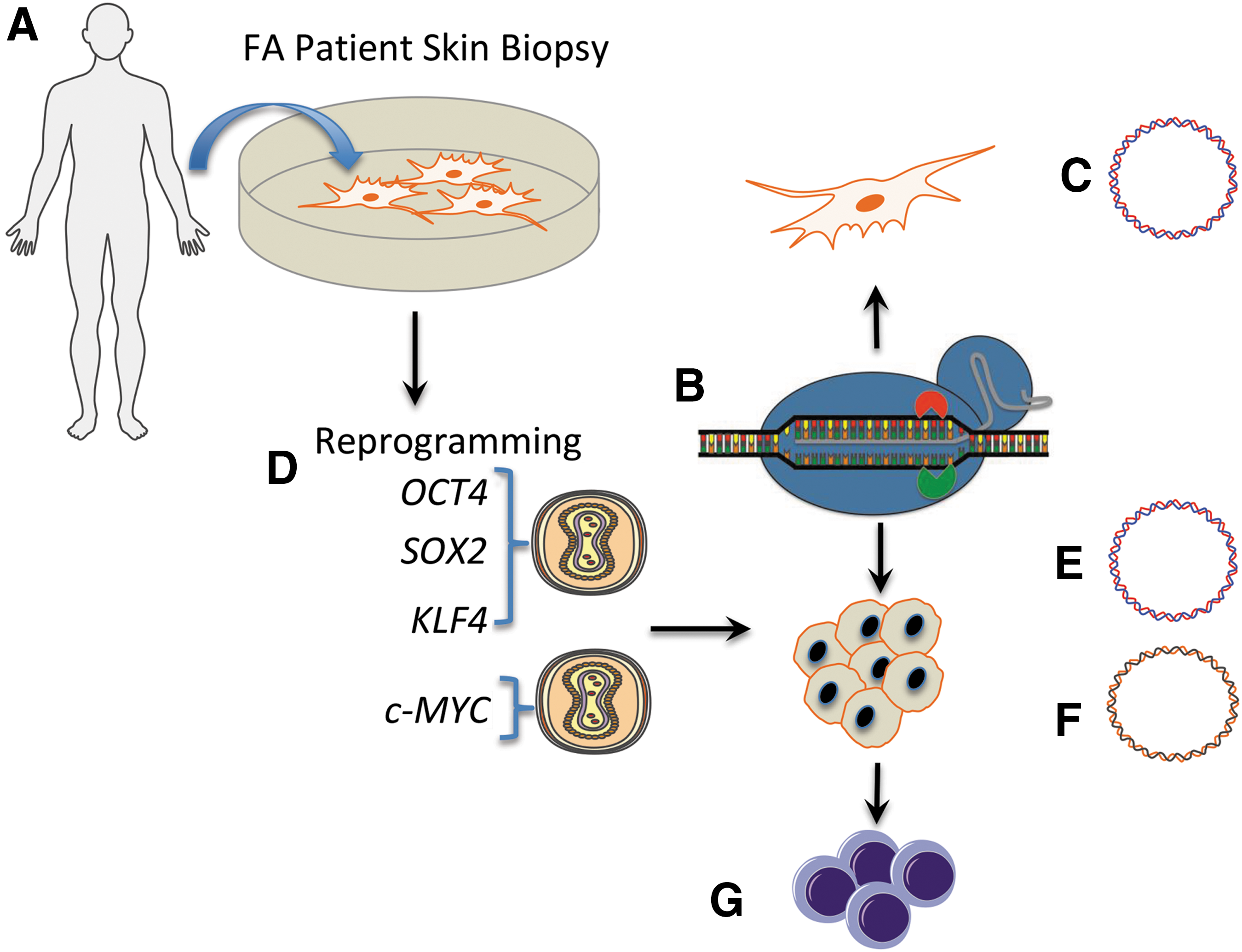
Experimental schema.
The unmanipulated primary fibroblasts were also reprogrammed to pluripotency using Sendai virus gene transfer of a four factor cocktail of OCT4, SOX2, KLF4, and c-MYC (OSKM) that were delivered in trans (Fig. 2D). Plasmid donor templates allowing for puromycin selection (Fig. 2E) or an exogenous marker sequence free template reliant on phenotypic correction-mediated resistance to mitomycin C (MMC; Fig. 2F) were assessed for their ability to modify cells. FANCI genotypically corrected were then utilized for the design and optimization of hematopoietic cell phenotype engineering (Fig. 2G).
Cellular reprogramming and FANCI gene correction
Previous studies have shown that FA cells are highly refractory to reprogramming to pluripotency using retroviral vectors expressing the OSKM factors [11,28]. One study observed better efficiency using lentiviral vectors and maximal rates when the FA phenotype was corrected [11]. In subsequent studies, Liu et al. successfully reprogrammed FANCA fibroblasts that were not complemented using nonintegrating reprogramming five factor delivery and p53 inhibition [29]. In this study we employed the Sendai virus platform to determine whether reprogramming FANCI fibroblasts to iPSCs could be accomplished. We observed expression of the hallmark pluripotency proteins: TRA-1-60, NANOG, SSEA-3, TRA-1-81, SSEA-4, and Oct3/4 by immunofluorescence microscopy (Fig. 3A and B). Implantation of iPSCs into immune-deficient animals resulted in teratoma development with tissues derived from all three germ layers (Fig. 3C). Importantly, both pre- and postreprogramming cell populations maintained normal karyotypes, and derivative iPSCs showed hallmark iPSC gene and epigenetic profiles and loss of Sendai virus genome particles (Supplementary Figs. S1–S3; Supplementary Data are available online at

iPSC characterization.
We next compared the ability of primary fibroblasts and iPSC-derived cells to undergo CRISPR/Cas9 gene editing. For this we focused on the c.1461 T > A mutation using gRNA candidate 2 (Figs. 1B and 4A) as the c.3058 + 4A>G locus showed a window of short interspersed nuclear elements that can complicate gene targeting and as heterozygous gene correction is sufficient for FA phenotype rescue. For HDR-based correction we constructed two donors: one with donor arms proximal to the mutation site flanking a floxed PGK-puromycin cassette and a second, puromycin marker-free template (Fig. 4B). As an important design consideration to prevent retargeting of the modified sequence, and to provide signature sequences for unambiguous detection, the donor templates each contained polymorphic substitutions (Supplementary Fig. S4). The repair templates, Cas9 nickase, and gRNA were delivered as plasmid DNA molecules by electroporation-based gene transfer into fibroblasts and iPSCs. HDR in a polyclonal population of bulk puromycin-selected fibroblasts was observed (Supplementary Fig. S5); however clonal cellular expansion was not possible due to senescence of the isolates (Fig. 4C). In contradistinction, the iPSCs treated similarly showed that 66% of the postselection analyzed clones had undergone HDR (Fig. 4C).

FANCI cell treatment and screening.
Because the FA phenotype is characterized by sensitivity to alkylating agents such as MMC, we also constructed a donor plasmid suitable for gene correction and subsequent selection by MMC. Primary fibroblasts showed a high degree of senescence when MMC was applied (unpublished observations), and we were unable to demonstrate HDR in this treatment group (Fig. 4D). In contrast, MMC addition to treated iPSCs showed selective outgrowth of cells, and 70% of the post-selection clones showed HDR (Fig. 4D). Taken together these results show the ability of the CRISPR/Cas9 system to mediate HDR in clonogenic iPSCs using plasmid-based donors that allow selective amplification of corrected cells (Fig. 4 and Supplementary Figs. S6 and S7).
Off-target analysis
An important consideration for the employment of CRISPR/Cas9 reagents for genome modification is the possibility for off-target activity at other loci that contain sequence homology to the intended target site. We analyzed the FANCI reagent using predictive in silico software (MIT CRISPR Design Tool) [27] and identified intragenic off-target candidates (Table 1) that were assessed by the Surveyor method. We observed no off-target effects at any of the predicted sites, suggesting a highly specific reagent at the resolution of the Surveyor assay (Table 1 and Supplementary Fig. S8).
Intragenic off-target sites were identified by the CRISPR Design Tool that are shown at left with mismatches between the FANCI site in bold and underlined and genes and their functions are shown. At right are the results of the Surveyor analysis in Cas9 nuclease (“nuc”) or Cas9 nickase (“nick”) indicated by a +/− sign in regards to whether locus modification (ie, cleavage products generated by the Surveyor enzyme) was observed or not.
iPSC-directed differentiation
Hematopoiesis occurs in two waves: primitive and definitive, with the latter giving rise to long-term repopulating hematopoietic stem/progenitor cells (HSPCs) [30]. Efforts to date employing embryonic stem cells or iPSCs for HSPC generation have resulted in cells resembling the primitive hematopoietic fate [31]. More recent studies have shown the importance of the Wnt-β catenin pathway in driving mesodermal commitment to the definitive wave, while primitive hematopoiesis is reliant upon activin-nodal signaling [32]. As such, we hypothesized that EB formation under previously described conditions with mesodermal induction driven by addition of BMP-4 and bFGF could be augmented with the activin-nodal inhibitor SB431542 and the GS3Kβ inhibitor CHIR99021 [32 –35] to drive the cells toward a definitive phenotype (Fig. 5A).
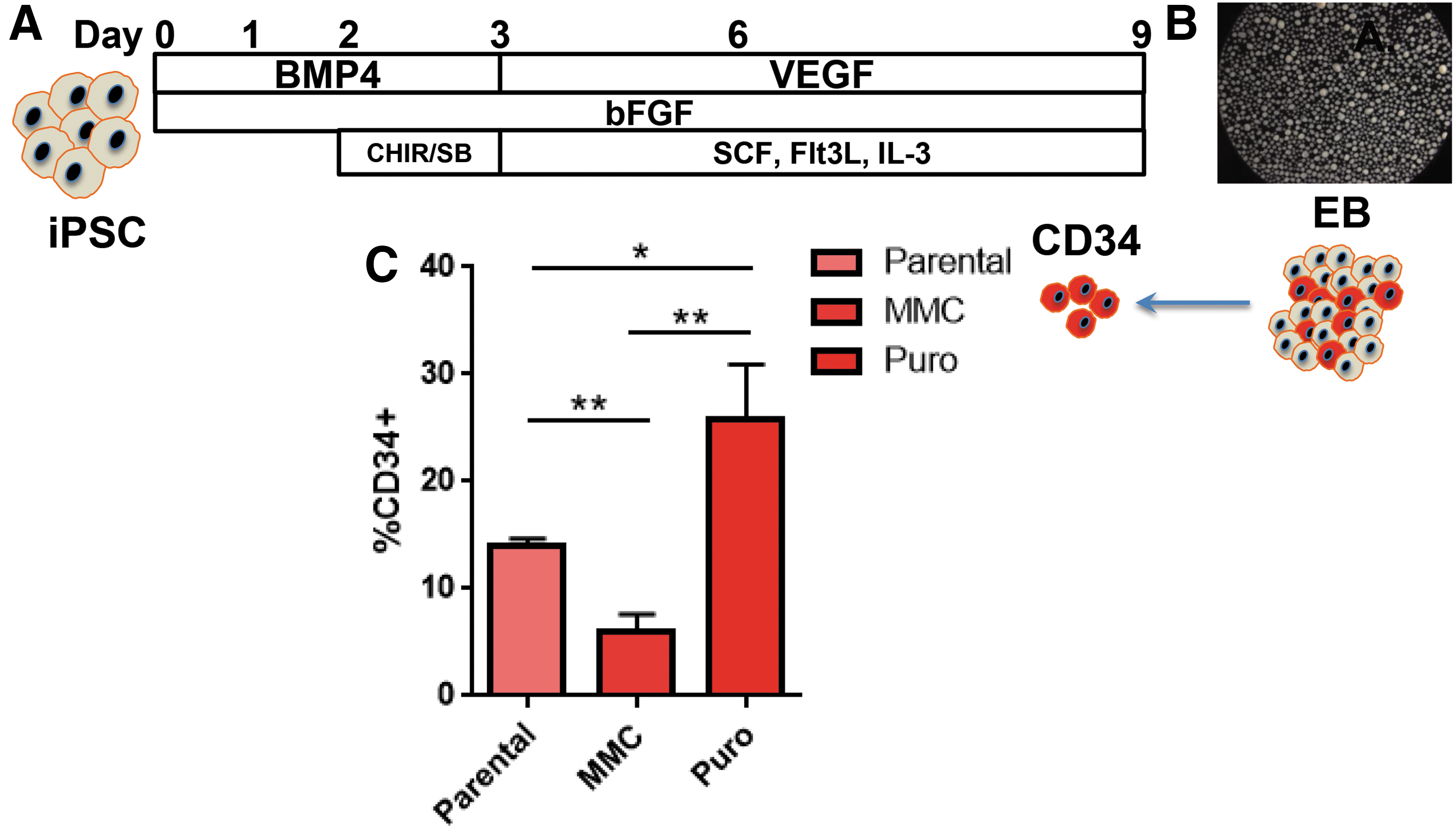
EB CD34 cell generation from gene corrected FANCI iPSC.
Hematopoietic commitment and progenitor expansion within embryoid bodies (EBs) was subsequently mediated by exposure to VEGF, SCF, Flt-3 ligand, and IL-3 (Fig. 5A). EB formation (Fig. 5B) under these conditions resulted in CD34 cell production with a noted (∼2-fold) increase when gene-corrected cells that had been selected with puromycin were employed (Fig. 5C). These data suggested that MMC may negatively impact EB and/or CD34 formation, and to test this we treated iPSCs derived from a patient with an intact FA DNA repair pathway with MMC for 48 h before EB formation. We observed a marked decrease in EB formation in the MMC group with diminished cellularity as determined by trypan blue exclusion (mean MMC group = 1.2 × 106 ± 3 × 105 and mean untreated group = 5 × 106 ± 1 × 106). As a result, fewer CD34 cells, representing <10% of the EB cellular population, were able to be isolated (data not shown).
To test the colony-forming unit (CFU) potential of gene-corrected cells, mixed and pure populations of cells were placed in methylcellulose for CFU assays (Fig. 6A). Bone marrow aspirates containing populations of progenitors and stroma showed higher levels of CFU in normal than in FA patients (Fig. 6A). Similarly, uncorrected FA cells as EBs containing CD34 positive and negative cells, and pure populations of CD34+ EB-derived cells, showed diminished CFU potential compared to a representative clone of corrected cells obtained using a puromycin-based HDR donor (Fig. 6A). In addition, the CFU morphology of uncorrected parental cells (Fig. 6B) was consistently smaller than corrected cells (Fig. 6C).
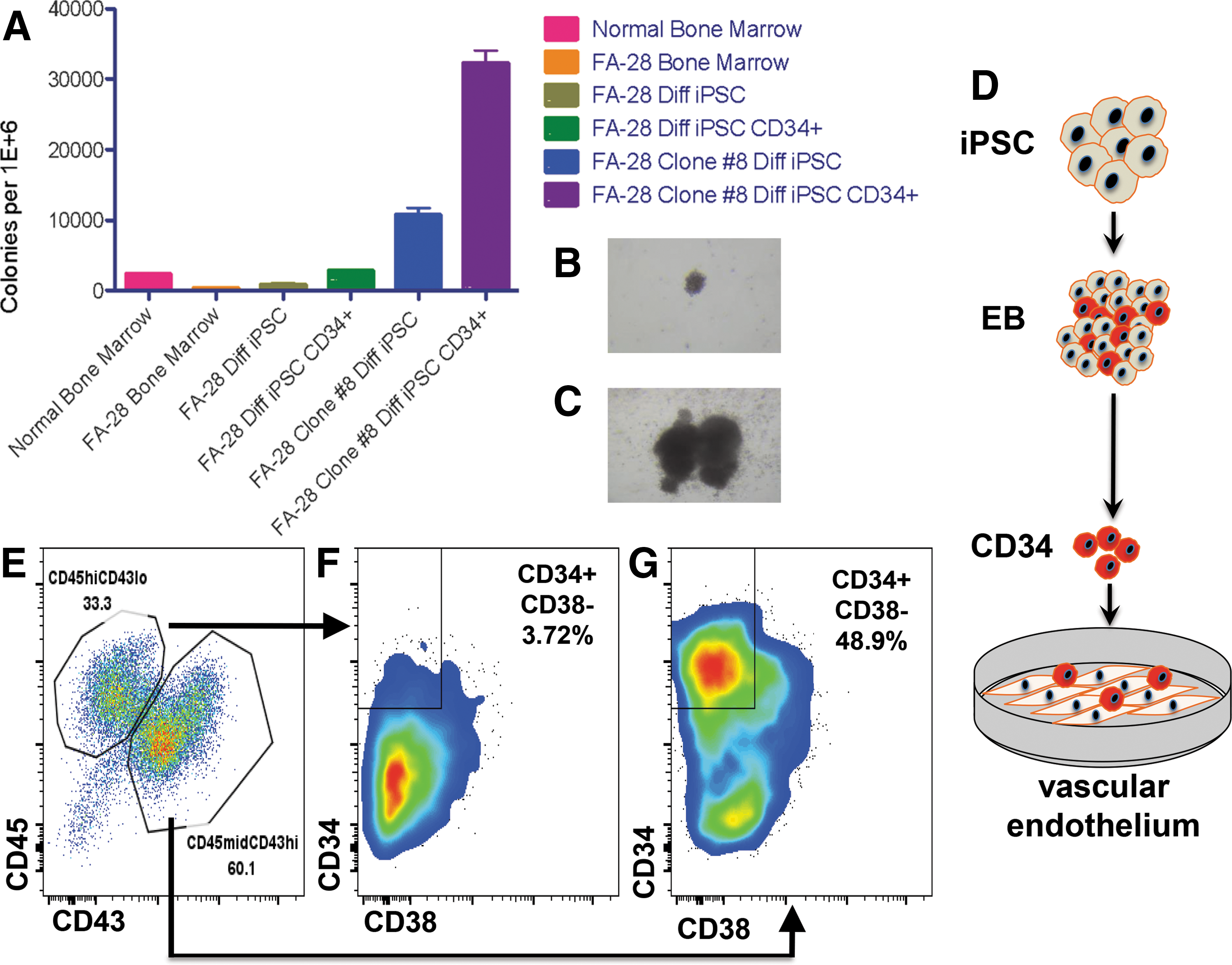
Hematopoietic potential of gene corrected FA iPSC.
To demonstrate that FA iPSCs under these conditions can form definitive cells of the hematopoietic compartment, we utilized a stringent procedure with parental cells to assess whether T progenitor cell formation was possible utilizing a coculture method on OP9 DL4 stromal cells (Supplementary Fig. S9). Toward optimizing conditions further, we extended our studies to include a vascular induction step that has been shown to promote multilineage progenitor production [36]. To accomplish this we generated CD34+ cells from EBs as above and placed them on a vascular endothelial cell feeder layer (Fig. 6D). This engineered cell line (termed VeraVec) was generated by adenoviral E4ORF1 transduction and provides supportive cues to developing cells [36]. Seven-day coculture of CD34+ cells and VeraVec cells resulted in two cell populations: CD45high/CD43low and CD45mid/CD43high (Fig. 6E). These cells were fractionated based on these parameters for analysis of CD34 and CD38 expression and we observed an emergent population of CD34+CD38− cells (Fig. 6F and G). This finding is highly significant as this cell subpopulation is prevalent in the definitive hematopoietic cells of the bone marrow [15] and these results demonstrate an efficient and robust methodology for iPSC-derived multilineage HSPCs.
Discussion
We present the following key findings: the ability to reprogram FANCI fibroblasts to pluripotency using Sendai viral, nonintegrating gene delivery; CRISPR/Cas9 mediated in situ gene correction; and the generation of a population of cells with characteristics of HSPCs using a novel differentiation methodology.
The reprogramming efficiency of FA cells is a significant hurdle in the field and was first reported by Raya et al. [28] and followed by Muller et al. [11]. Each study highlighted the requirement of an intact FA DNA repair pathway to accomplish iPSC generation and, of note, the reprogramming factors were delivered on integrating retro or lentiviral constructs. The highest reprogramming frequencies were observed using lentiviral transgenesis [11]. Following these studies Liu et al. demonstrated, using nonintegrating plasmid-based five factor reprograming factor delivery, that they could reprogram noncomplemented FANCA cells that also required p53 inhibition [29]. The reprogramming frequency was 10-fold higher by Liu et al. than other reports and the reason for this is unclear. One hypothesis is that viral vector gene expression in a stable fashion is incumbent on integration, a process that employs cellular DNA repair using endogenous factors. These factors can be differential with regard to the individual platforms [37,38] potentially indicating that in the DNA damage repair defective FA phenotype integrating vectors are suboptimal. Our results show that the nonintegrating Sendai virus was able to reprogram FANCI cells without complementation (Figs. 2 and 3) and while further experimentation directly assessing the platforms and complementation group deficiencies in a head-to-head manner are required, the generation of iPSCs proved crucial to effective gene repair in our study.
Programmable nucleases exist in multiple formats: zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases, meganucleases, and the CRISPR/Cas9 system. Toward addressing the most commonly occurring FA gene mutation in the FANCA gene, Rio et al. utilized a ZFN pair to insert the FANCA cDNA by HDR into the AAVS1 safe harbor locus [6]. Therapeutic gene expression was mediated by the PGK promoter, resulting in sustained transgene expression representing a platform that would be applicable to any patient with FANCA gene mutations [6]. Previously, the present group has utilized the CRISPR/Cas9 D10A nickase derived from S. pyogenes to correct a prevalent FANCC gene mutation [14]. However, common to each study was the requirement of hTERT gene expression to facilitate gene repair in patient fibroblasts [6,14]. Gene-corrected FANCA fibroblasts were amenable to iPSC reprogramming by the Rio group while the FANCC cells were unable to be reprogrammed [6,14]. These studies differed in a key area related to the temporal aspects of gene correction. As stated, the Rio study generated cells with constitutive FANCA expression while we, both here and previously, targeted the disease-causing mutation in situ so that the endogenous command and control systems are retained (Figs. 1 and 4) [14]. Given that external DNA-damaging stimuli can result in FANCI gene upregulation [39], we consider this a critical and clinically relevant consideration. A potential downside to an individualized mutation targeting approach is that the highly heterogeneous FA mutation profile may make portions of this or other FA genes inaccessible to precision, site-specific nuclease design requirements. For the S. pyogenes Cas9, this design requirement is a target sequence containing an–NGG PAM, where “N” is any nucleotide [13]. Analysis of the compound heterozygote gene mutations present in this FANCI patient identified multiple candidates for our experimentation (Fig. 1). All of the candidates showed activity when delivered as plasmid DNA expression cassettes (Fig. 1F) and demonstrated the potent activity we have consistently observed with the S. pyogenes Cas9 [14,40]. Moreover, it shows that despite the putative recognition restrictions of Cas9, we were able to target sequence proximal to the mutation, an important consideration as HDR efficiency decreases as the distance from break site to target modification locus increases [41]. Relevant to this, the targeting capacity of the CRISPR/Cas9 system has been expanded with the discovery of alternate PAM variants through direct engineering or bacterial strain discovery [42 –44], making larger regions of the genome accessible for genome editing.
We attempted to correct fibroblasts and iPSCs using CRISPR/Cas9 D10A plasmids with an inactive HNH domain (Figs. 1C and 2B) capable of nicking a single strand of DNA [45]. Further, we tested two donor platforms: one based on artificial (ie, puromycin) drug resistance and selection and a second MMC-based approach requiring correction of the cellular phenotype (Fig. 4B). The puromycin plasmid template showed demonstrable HDR in bulk populations of fibroblasts and iPSCs (Fig. 4C, D and Supplementary Figs. S5–S7). Clonal derivation of corrected fibroblasts was not possible; however, puromycin selection allowed recovery of 12 corrected iPSC clones (Fig. 4C). In non-puromycin donor treated fibroblasts we noted an inability to select modified cells using MMC as it triggered senescence (unpublished observations and Fig. 4D). In iPSCs we isolated 17 MMC-resistant clones that contained donor sequence modification (Fig. 4D and Supplementary Fig. S6). In total these results demonstrate the difficulty in mediating gene editing-based repair in primary, non-hTERT transformed FA fibroblasts; this is consistent with our previous observations and those of Rio et al. [6,14]. Conversely, iPSC gene-corrected clones were readily obtained when selective pressure was applied (Fig. 4). This consideration is important, as in the absence of selection, HDR rates in bulk-treated cells were sufficiently low to evade detection by our PCR strategy (data not shown). Thus, selection can streamline the obtainment of gene-corrected cells without extended screening of individual colonies. Further, given that cell lines in which the variables of gene transfer and poor ability to be clonally derived are removed, HDR rates can be <10% [14,46], making selection strategies important in the context of a chosen line of investigation.
Bone marrow failure is a life-threatening manifestation of FA, making gene modification of hematopoietic cells highly desirable. Direct HSPC gene correction has been demonstrated [47,48]; however, the depleted numbers of HSPCs in FA patients make them a difficult clinical and research tool to acquire. Further, the clinical status of individual patients will be highly variable, making standardized genome engineering in this context increasingly difficult. As such, the ability to engineer the genome of cells with direct or acquired therapeutic properties holds great promise for FA. Toward realizing this promise Raya et al. showed that FA iPSCs could be differentiated into CD34+CD45+ cells with a peak expression of ∼7% and ∼1% respectively, with an ability to form CFUs in vitro [28]. Likewise, the study by Rio et al. showed enhanced CFU potential from gene-corrected FA iPSCs and the following hematopoietic population frequencies: 12% CD34+CD43+, ∼7% CD34+CD45+, and ∼12% CD34−CD45+, respectively [6]. A study in the same year by Liu et al. demonstrated ∼9% CD34+CD43low, a marker for multipotent progenitors [49] in gene-corrected FANCA iPSCs [29]. In these studies Raya and Liu employed OP9 cells, a mouse stromal cell with the capacity to support hematopoiesis [50], to drive EB-derived CD34s to a hematopoietic phenotype [28,29]. More recently, Rio et al. used supplemental recombinant Wnt-3a and Wnt-11 protein in their differentiation schema [6]. Accumulating evidence has shown that the governance of fate determination between primitive and definitive hematopoiesis is modulated by activin-nodal and Wnt-β catenin signaling during the mesodermal phase of development [32]. As such, the inhibition of activin-nodal signaling and/or augmentation of the Wnt pathway results in enhanced definitive hematopoietic progenitor development from iPSCs [32]. Once derivative cells are specified for definitive lineage commitment, their subsequent phenotype can be directed by coculturing with cells providing niche developmental cues. For instance, T lymphoid generation is possible due to the instructive signals provided by the OP9 stromal cell layer expressing the Notch ligand Delta-like (OP9-DL4) [51,52], and we show acquisition of the CD5 and CD7 pan-T cell markers using a T cell biased differentiation protocol (Supplementary Fig. S9). To extend these foundational findings, we applied frontline differentiation procedures using, for the first time, tandem activin-nodal and Wnt signaling modulation with SB431542 and CHIR99021 (Figs. 5 and 6). The resultant CD34 cells were then cocultured on a supportive matrix of VeraVec E4ORF1 transgenic vascular endothelial cells [36]. This population of cells aids in recapitulating the instructive vascular niche and has allowed the acquisition of engraftment potential of human vascular endothelial cells [36]. After 1 week of coculture two populations of cells were observed: CD45highCD43low and CD45midCD43high (Fig. 6E). These findings are in agreement with previous data by Kennedy et al. using OP9 coculture methodologies that showed definitive CD43+CD45+ cells [52]. We further characterized the CD45midCD43high population and observed that they possessed properties consistent with definitive multipotent progenitors with engraftment properties as evidenced by a CD34+CD38− phenotype (Fig. 6G) [53]. As such, these data show that the acquisition of multilineage hematopoietic progenitors is possible using an in vitro vascular induction methodology. Our data are highly relevant and complement other studies utilizing VeraVec coculture to promote in vitro conditions for the specification of alternate cell types to transplantable and engraftable HSPCs and are additive to the cellular engineering field [36,54].
An additional finding we observed that is of importance to the FA field is the decreased efficiency of EB formation with an associated decrease in CD34 generation and poor ability to form CFUs when cells were selected with MMC (Fig. 5C and 6A). By using MMC pretreatment in iPSCs with an intact FA DNA repair pathway, we also observed diminished cellularity of the EBs (data referenced above) suggesting that MMC may impact the differentiation capabilities of iPSCs. Further experimentation to define the mechanism and related temporal effects of MMC on iPSCs is required and will further aid in selection strategies that allow isolation of gene-corrected cells with a high degree of plasticity able to serve as a foundation for directed therapeutic differentiation.
Finally, a crucial consideration, particularly in the genomic instability phenotype of FA, is the specificity of the candidate gene-editing reagent. Promiscuous cutting at off-target sites resulting in multiple DNA breaks can result in translocations and/or disruption of unintended sequences [40,55,56]. To mitigate the potential of off-target effects, we employed the Cas9 D10A nickase that has been shown to promote higher rates of HDR with minimal mutagenic nonhomologous endjoining (NHEJ) [14,46]. Indeed, treatment of cells with a Cas9 nuclease showed higher rates of NHEJ than did nickase-treated cells for the FANCI gene target (Table 1 and Supplementary Fig. S8). By assigning putative off-target sites using a predictive algorithm, we analyzed 10 off-target candidates contained within genes and did not observe appreciable modification at any of the sites (Table 1 and Supplementary Fig. S8). These findings suggest a specific reagent whose activity profile is restricted to the FANCI target. This fact, coupled with the use of a nicking version of Cas9, represents a maximally safe approach for FANCI genome editing.
In summary, these data highlight the robust nature in which iPSCs derived from a FANCI individual can be modified at the genome level to serve as an engineering template for therapeutic cell development. This stable and renewable population of cells holds tremendous potential for the entire spectrum of FA disease and its associated pathological manifestations as part of an autologous regenerative medicine approach.
Footnotes
Acknowledgments
We are grateful to Nancy Griggs Morgan and Kayla Cook for expert assistance in article preparation and editing. We are also thankful for the generosity of the Kidz1stFund, the Fanconi Anemia Research Fund, the Children's Cancer Research Fund, the Lindahl Family and the Corrigan Family. MJO is supported by 8UL1TR000114-02. JT is supported in part by R01 AR063070 and P01 CA065493. BRW is supported by NIH T32- HL007062. Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114 (MJO). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. DP is a major contributor along with BRW and MJO to conceiving, designing, and establishing the protocol to generate CD34+ hematopoietic cells. The authors are grateful to Chong Jai Kim, MD, PhD and the present work was partly supported by research funds from the National Research Foundation of Korea (NRF-2015K1A4A3046807).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.