
Abstract
Articular cartilage has a limited ability to heal. Mesenchymal stem cells (MSCs) derived from the bone marrow have shown promise as a cell type for cartilage regeneration strategies. In this study, sodium tungstate (Na2WO4), which is an insulin mimetic, was evaluated for the first time as an inductive factor to enhance human MSC chondrogenesis. MSCs were seeded onto three-dimensional electrospun scaffolds in growth medium (GM), complete chondrogenic induction medium (CCM) containing insulin, and CCM without insulin. Na2WO4 was added to the media leading to final concentrations of 0, 0.01, 0.1, and 1 mM. Chondrogenic differentiation was assessed by biochemical analyses, immunostaining, and gene expression. Cytotoxicity using human peripheral blood mononuclear cells (PBMCS) was also investigated. The chondrogenic differentiation of MSCs was enhanced in the presence of low concentrations of Na2WO4 compared to control, without Na2WO4. In the induction medium containing insulin, cells in 0.01 mM Na2WO4 produced significantly higher sulfated glycosaminoglycans, collagen type II, and chondrogenic gene expression than all other groups at day 28. Cells in 0.1 mM Na2WO4 had significantly higher collagen II production and significantly higher sox-9 and aggrecan gene expression compared to control at day 28. Cells in GM and induction medium without insulin containing low concentrations of Na2WO4 also expressed chondrogenic markers. Na2WO4 did not stimulate PBMC proliferation or apoptosis. The results demonstrate that Na2WO4 enhances chondrogenic differentiation of MSCs, does not have a toxic effect, and may be useful for MSC-based approaches for cartilage repair.
Introduction
A
Different approaches have been used to stimulate chondrogenesis and bone growth, such as the use of transforming growth factor beta (TGF-β), bone morphogenetic protein [8,9], and insulin and insulin-like growth factors (IGF) [10]. IGF-1 is involved in MSC chondrogenesis by stimulating proliferation, regulating apoptosis, and inducing differentiation [10]. Insulin is structurally similar to IGF-1, whereby it can bind to the IGF-1 receptor and stimulate extracellular matrix (ECM) production [11]. Previous studies have found systemic insulin treatment increased cell proliferation, soft callus formation/chondrogenesis, biomechanical properties, and callus bone content in diabetes mellitus rats [12]. Furthermore, local administration of insulin has been found to improve healing and bone regeneration in animal models [13,14]. However, insulin is difficult to deliver locally due to its high molecular weight (MW) (51 amino acids and 5,808 Da MW) and stability; insulin goes through hydrolysis/degradation or intermolecular transformation during storage and use [15]. Moreover, increasing the insulin level in normal patients is not an option because of the risk of hypoglycemia. Therefore, insulin mimetics have been sought.
Sodium tungstate (Na2WO4) is a water soluble inorganic compound that can exert an insulin-like effect in diabetes [16,17]. Several studies have shown that oral administration of Na2WO4 into animal models of type 1 and type 2 diabetes normalizes glycemia without hypoglycemia [17 –19]. Dominguez et al. [20] showed that glycogen synthesis and deposition are increased in the presence of Na2WO4. Na2WO4 stimulates insulin secretion and regenerates the β cell population, however, it does not stimulate insulin secretion at low glucose levels, suggesting that Na2WO4 does not induce hypoglycemia [21]. Moreover, Na2WO4 stimulates extracellular signal-regulated kinase (ERK) phosphorylation in different cell types [22,23]. ERK activation is important for MSC chondrogenesis [24,25]. Moreover, the MW of Na2WO4 (293.82 g/mol or Da) is substantially lower than insulin, which may facilitate transport through diffusion to the tissue. This suggests that Na2WO4 could be delivered locally to accelerate MSC-based approaches in cartilage repair.
This study examined the effect of Na2WO4 on MSC chondrogenesis. We hypothesized that Na2WO4 would enhance MSC chondrogenesis. MSC chondrogenic differentiation was studied with varying amounts of Na2WO4 using biochemical analysis, immunostaining, gene expression, and collagen type II production. These studies were conducted on a three-dimensional (3-D) fibrous scaffold with fiber diameters and interfiber spacing in the micron-size range, which has been shown to support chondrogenesis [26]. Tissue engineering scaffolds were used in this study for MSC chondrogenesis because they are widely used for growing cartilage in vitro since they mimic the 3-D structure of the ECM and are used clinically for cartilage repair [27,28]. These studies show for the first time the effect of Na2WO4 on chondrogenic differentiation of human MSCs. Furthermore, the cytotoxicity of Na2WO4 has been poorly investigated. In this study, peripheral blood mononuclear cells (PBMCs) were challenged with different concentrations of Na2WO4. Cell proliferation and viability were used to analyze the effect of Na2WO4 on PBMCs.
Materials and Methods
Scaffold fabrication
15 wt.% poly (ɛ -caprolactone) (PCL, [(CH2)5COO]n−, 80,000 MW, Sigma-Aldrich, Inc., St. Louis, MO) was dissolved in methylene chloride. This solution was electrospun to form a nonwoven fibrous scaffold, as previously described [29]. The electrospinning setup consisted of a syringe pump operating at 5 ml/h, 12 gauge needle, a high voltage source ranging from 20 to 25 kV, and a grounded collector. The electrospun PCL scaffolds were characterized by scanning electron microscopy (SEM) using a LEO 1530 Gemini (Germany) instrument. SEM images were used to determine fiber diameters and interfiber spacing using Image J software (National Institutes of Health), as previously described [29].
Cells
Peripheral blood was obtained from four healthy subjects, two males and two females (ages 18–30 years old). The use of human subjects was approved by the Rutgers Institutional Review Board, Newark, NJ. PBMCs were isolated by Ficoll Hypaque (Sigma-Aldrich) gradient separation, as previously described [30]. Human MSCs were obtained from human bone marrow aspirates (Lonza, Walkersville, MD) from four healthy subjects, two males and two females, ages 18–30 years old, according to previously published protocols and cryopreserved before use [31]. Human MSCs were characterized for the expression of surface markers CD29, CD44, CD73, CD90, and CD105 and lack expression of CD14, CD34, and CD45 as determined by flow cytometry [32 –34]. The differentiation potency was determined by the differentiation of MSCs into osteoblasts, adipocytes, and chondrocytes in vitro [32,33]. For both PBMCs and MSCs, all experiments described were repeated per donor.
MSC growth
MSCs were cultured on the scaffolds in the presence of Na2WO4 (Na2WO4·2H2O, MW = 329.85 g/mol; Sigma-Aldrich) in the range of 0.01–50 mM to determine which concentrations supported cell growth. Before cell seeding, scaffolds were cut into 6 mm diameter disks using a biopsy punch (Integra Miltex, York, PA), sterilized in 100% ethanol for 20 min, and were air-dried overnight. Cryopreserved MSCs were thawed and expanded in tissue culture-treated polystyrene flasks (Nunc, Thermo Fisher Scientific, Waltham, MA) in growth medium (GM) that comprised Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Grand Island, NY), 10% fetal bovine serum (Hyclone; Thermo Fisher Scientific), and 1% antibiotic-antimycotic (Invitrogen) at 37°C and 5% CO2 until 70%–80% confluent. Cells were trypsinized using 0.25% Trypsin-EDTA (Thermo Fisher Scientific), resuspended in GM, then seeded onto scaffolds at 3.5 × 104 cells/cm2, and cultured for 11 days in GM alone or with Na2WO4. Na2WO4 was added to GM leading to final Na2WO4 concentrations of 0, 0.01, 0.1, 1, 10, and 50 mM. Samples (n = 4) were harvested and analyzed at days 4, 7, and 11 for cell number using the PicoGreen® ds DNA assay (Life Technologies). MSCs of known cell number were used as standards. Standards and samples were prepared by lysing the cells using 0.1% Triton X-100 (Sigma-Aldrich, MO). The cell lysate of samples and standards (n = 4 per group) was mixed with an equal volume of PicoGreen dye, which binds to the double-stranded (ds) DNA. Fluorescence intensity was measured with a microplate reader (FLX800, Biotek Instruments, VT) at 480 nm excitation and 520 nm emission. A standard curve of known cell number was used to determine the sample cell number.
PBMC proliferation assay
The cytotoxicity of Na2WO4 was examined using PBMCs. Cell proliferation was measured using tritiated thymidine (3H-TdR) incorporation as described in Kapoor et al. [35]. PBMCs were resuspended at 106/mL in RPMI 640 (Sigma-Aldrich) and 10% fetal bovine serum (Hyclone; Thermo Fisher Scientific, Waltham, MA). Positive control cells were challenged with 1% phytohemagglutinin (PHA) (Gibco). The experimental groups were stimulated with 0.01, 0.1, and 1 mM Na2WO4. Baseline proliferation was studied in parallel cultures with medium alone or vehicle (1 μL DI H2O). The assay was established with 2 × 105 PBMC in 200 μL volume in a flat-bottom 96-well tissue culture plate. Each experimental group was studied in triplicate cultures. After 48 h at 37°C, each well was pulsed with 1 μCi/mL of 3H-TdR. After 16 h, the cells were harvested on glass fiber filters (Cambridge Technology, Inc.) using a cell harvester (EMD Millipore). The filters were washed with ethanol and then air-dried overnight. The radioactive incorporation of 3H-TdR in the cells was measured using a liquid scintillation counter (Beckman; Fullerton, CA). The stimulation index (SI) was calculated as disintegration per minute (dpm) of treated group/dpm of unstimulated PBMCs (0 mM).
CellTiter-Blue assay
CellTiter-Blue Assay (Promega, Madison, WI) was used to assess the viability of PBMCs. CellTiter-Blue was used to measure the metabolic activity of PBMCs as a surrogate for cell number. Each experimental group was performed in triplicate in 96-well tissue culture plates. After 4 days, the CellTiter-Blue reagent (20 μL/well) was added to each well. The plates were incubated, and after 4 h, the fluorescence intensity was measured with a microplate reader (Synergy, HTX, BioTek, Winooski, VT) at 560/590 nm emission. Percent viability was calculated as the fluorescence reading of treated (stimulated) cells divided by untreated healthy cells, which were considered 100% viable as described in Greco et al. [36].
Chondrogenesis culture and characterization
Chondrogenesis Culture. Cryopreserved MSCs were thawed and expanded in GM until 70%–80% confluent. Cells were trypsinized and then seeded on 6 mm diameter scaffold disks at 1.76 × 105 cells/cm2 and cultured for 28 days in GM, complete chondrogenic induction medium (CCM), or CCM without insulin, according to previously published methods [26]. CCM consisted of DMEM high glucose containing 4 mM
sGAG production and cell number
Production of sGAG and cell number were determined. Samples were digested using the papain (Sigma-Aldrich, MO) buffer overnight at 65°C following the manufacturer's protocol. The sGAG assay is used to measure the total sulfated proteoglycan content using Blyscan sGAG Assay Kit (Biocolor, United Kingdom). Cell lysate of samples (n = 4 per group) and color reagent were combined in a transparent 96-well plate, and the absorbance at 656 nm was measured using a microplate spectrophotometer (EMax, Molecular Devices, Melville, NY). sGAG concentration was determined relative to chondroitin sulfate standard curve. PicoGreen ds DNA assay (Life Technologies) was used to determine the cell number. MSCs of known cell number were used as a standards. Standards were prepared by lysing the cells using papain buffer. The cell lysate of samples and standards (n = 4 per group) was mixed with an equal volume of PicoGreen reagent. Fluorescence intensity was measured with a microplate reader (FLX800; Biotek Instruments, VT) at 480 nm excitation and 520 nm emission. A standard curve of known cell number was used to determine the sample cell number. GAG production was normalized to cell number.
Gene expression
Gene expression was evaluated using quantitative reverse transcription (RT) polymerase chain reaction (PCR) analysis. RT-PCR was performed with RNeasy Micro Kit and SYBR Green RT-PCR Kit (Qiagen; Valencia, CA) using the MX4000 detection system (Stratagene) according to the manufacturer's protocol. Relative gene expression of SOX-9, aggrecan, and collagen type II was determined. Samples (n = 4 per group) were harvested and digested using the Lysis Buffer. RNA was isolated from the samples using the RNeasy Micro Kit (Qiagen), including the DNA digestion step (RNase-Free DNase Set; Qiagen). The RNA quantity and integrity were determined by measuring the absorbance at 260 nm and the absorbance ratio of 260/280 nm, respectively, using the NanoDrop™ Spectrophotometer (Thermo Fisher Scientific). Quantitative PCR analysis was performed using QuantiTect SYBR Green Supermix, including the QuantiTect Primer Assays. The RT step ran for 30 min at 50°C, 15 min at 95°C of PCR activation, followed by forty cycles of amplification consisting of 15 s denaturation at 94°C, 30 s of annealing at 55°C, and 30 s of extension at 72°C. A melting curve analysis of the RT-PCR product was included for each reaction. Samples were analyzed in triplicate, and the value of each gene was normalized to the reference gene ribosomal protein, large, P0 (RPLP0) for the same sample (ΔCT). Primers were purchased from Qiagen (Hs_ACAN, QT00001365; Hs_SOX9, QT00001498; Hs_COL2A1, QT00049518; and Hs_RPLP0 QT00075012).
Immunostaining
Immunostaining was used to observe cell morphology and collagen type II deposition at day 28. Samples were fixed using 4% paraformaldehyde for 20 min. Samples were permeabilized with 0.1% Triton-X for 15 min and then nonspecific binding was blocked using 5% donkey serum (Sigma-Aldrich, MO) for 1 hour. Samples were incubated with mouse antihuman collagen type II antibody (EMD Millipore; CA) at 4°C overnight. Samples were then incubated with conjugated northern light 493 secondary antibody (Donkey anti-Mouse IgG, Fisher Scientific, Rockford, IL) and rhodamine phalloidin (Life Technologies) for actin filament for 1 h. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI, Fisher Scientific). The images were taken using a confocal fluorescence microscope (C1-si, Nikon, Japan).
Collagen type II enzyme-linked immunosorbent assay
Collagen type II production was quantified by enzyme-linked immunosorbent assay (ELISA) using a collagen type II detection kit (Chondrex, Inc. Redmond, WA). The ELISA was conducted following the manufacturer's instructions. Briefly, samples (n = 4 per group) were harvested at day 28 and washed with PBS. Collagen was digested by adding pepsin solution (0.1 mg/mL in 0.05 M acetic acid) at 4°C for 2 days with gentle mixing. The digested samples were centrifuged for 3 min, and supernatants were collected. Pancreatic elastase was added to the samples and incubated overnight at 4°C. Supernatants were then collected. The ELISA plate, which was coated with collagen type II antibody, was prepared as described by the manufacturer, and the absorbance at 490 nm was measured using a microplate spectrophotometer (Emax, Molecular Devices). The collagen type II production was determined relative to the standard curve.
Statistical analysis
Statistical analysis was performed using one-way and two-way analysis of variance (ANOVA) and the post hoc Tukey's HSD for statistical differences (using SPSS software). Prior analysis, the Shapiro–Wilk test was used to test the normality and Levene's test for equal variance. Probability (P) values <0.05 were considered statistically significant. Two-way ANOVA was used for analyzing cell number, sGAG, and gene expression, while one-way ANOVA was used for analyzing PBMCs viability and collagen type II ELISA.
Results
Scaffolds
The 3-D fibrous PCL scaffolds had fiber diameters and interfiber spacing in the micron-size range (Fig. 1a). The fiber diameter and interfiber spacing were 5.7 ± 0.9 μm and 48 ± 14 μm, respectively.
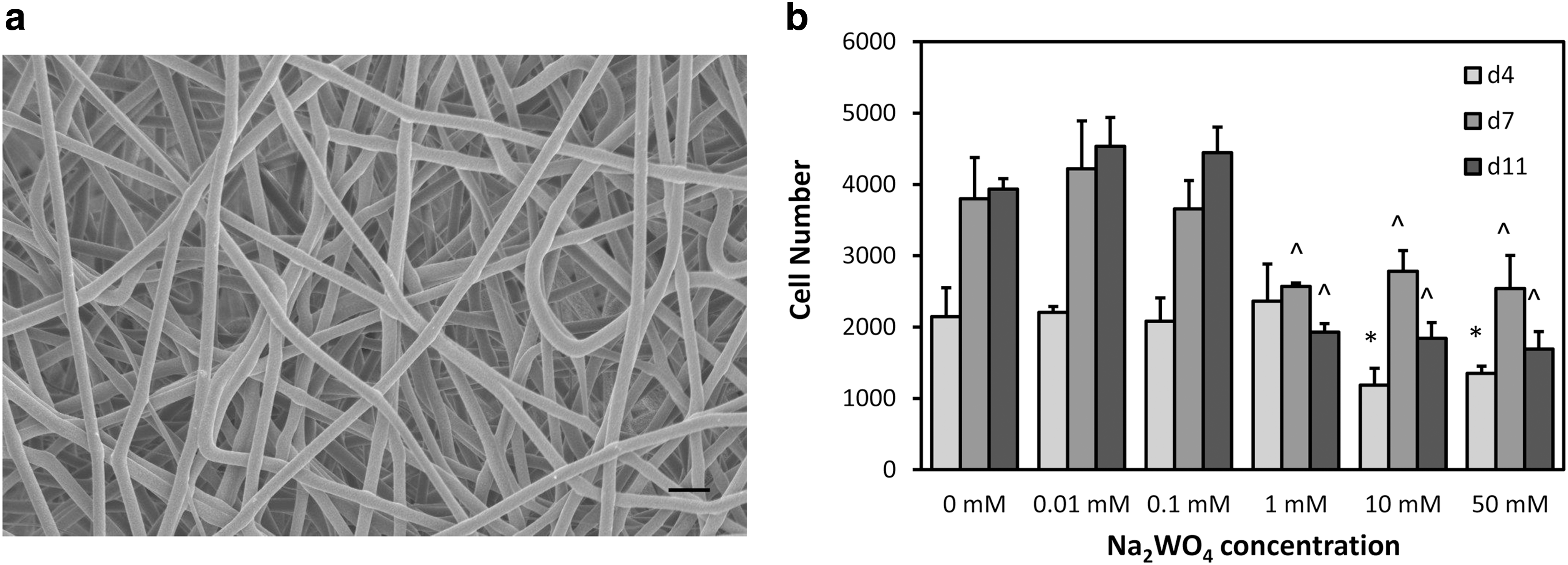
Scanning electron microscope (SEM) image of the scaffold and mesenchymal stem cell (MSC) number at days 4, 7, and 11 in culture.
MSC growth
To determine which concentrations of Na2WO4 could support cell growth, MSCs were cultured on the scaffolds in GM. 0, 0.01, and 0.1 mM Na2WO4 groups significantly increased in cell numbers from days 4 to 7 (P < 0.05) (Fig. 1b). 1, 10, and 50 mM Na2WO4 groups had significantly lower cell numbers compared to the other groups at days 7 and 11 (P < 0.05 and P < 0.001, respectively). 0.01, 0.1, and 1 mM Na2WO4 were chosen for further investigation.
PBMC proliferation and viability
The cytotoxicity of Na2WO4 was evaluated using PBMCs as a surrogate for untoward effects on the immune system. PBMC proliferation and viability studies were performed by stimulating with varying amounts of Na2WO4 (0.01, 0.1, and 1 mM). The experimental system was evaluated by stimulating with a T-cell mitogen, PHA. The results indicated no significant difference in cell proliferation with different concentrations of Na2WO4. The SIs, which were calculated as the proliferation of stimulated PBMCs over unstimulated PBMCs, were ∼1 for all Na2WO4 groups (Fig. 2a). The PBMC proliferation in the presence of Na2WO4 was comparable to that achieved in the absence of Na2WO4. While activating the PBMCs with mitogen, PHA increased the SI significantly (11.5 ± 1.2) (Fig. 2a). The cell viability assay was used to ensure the cells did not undergo apoptosis. No significant differences in cell viability between groups with Na2WO4 and without (0 mM) were detected (Fig. 2b).
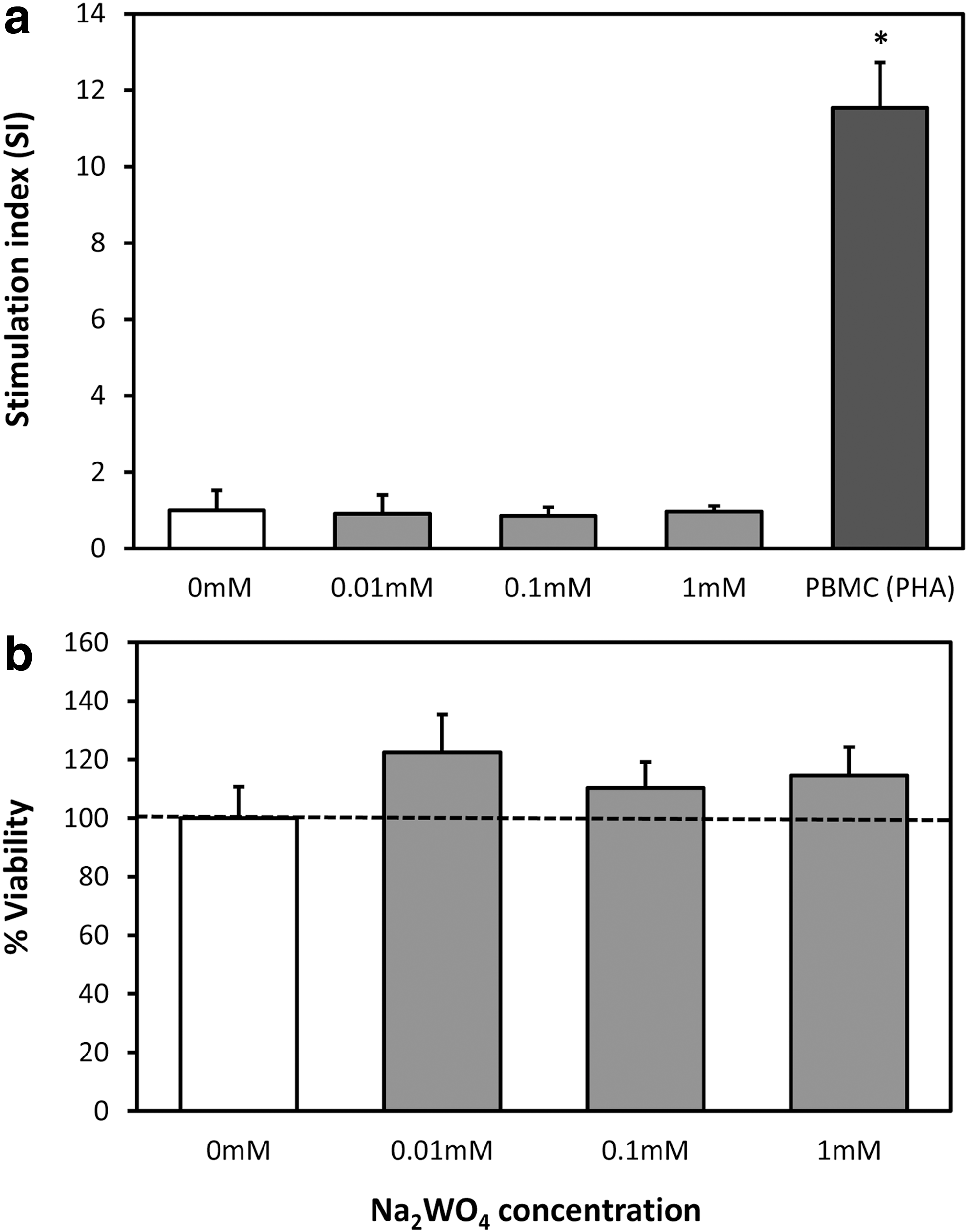
Proliferative response of Na2WO4 on peripheral blood mononuclear cells (PBMCs).
Chondrogenic differentiation
To determine the effect of Na2WO4 on chondrogenic differentiation of MSCs, sGAG production was determined at days 14 and 28 as shown in Fig. 3a. Cells in 0.01 mM Na2WO4 group had higher sGAG production than all other groups at day 28 (P < 0.005) in CCM. In addition, a significant increase in sGAG production occurred between days 14 and 28 for 0.01 mM Na2WO4 group in CCM. Overall, sGAG production in GM was less than in CCM, however, it was significantly higher in 0.1 mM Na2WO4 in GM compared to GM alone at day 14 (Supplementary Fig. S1; Supplementary Data are available online at
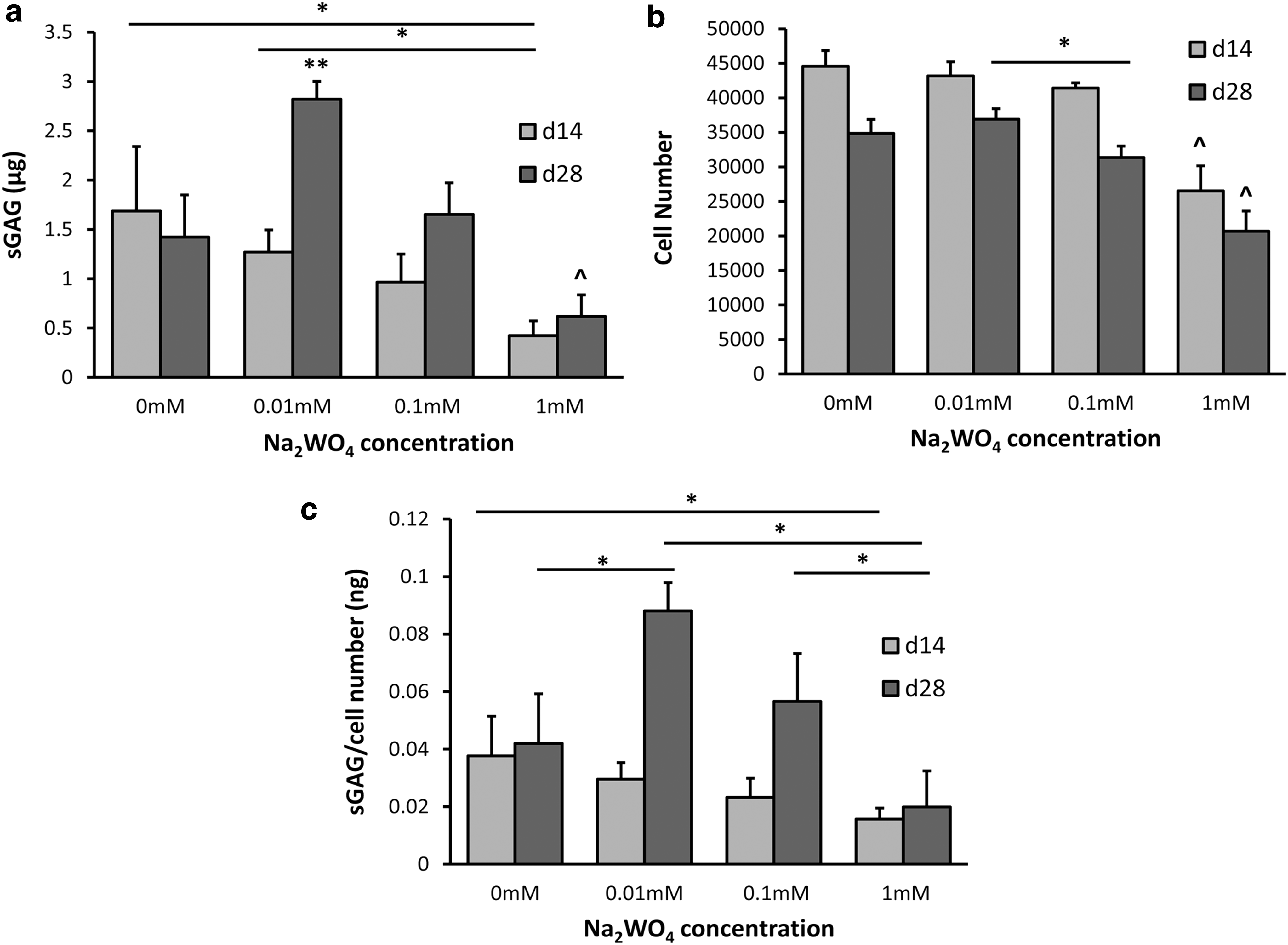
Sulfated glycosaminoglycan (sGAG) production and MSC number on varying concentrations of Na2WO4 in the chondrogenic induction medium (CCM) at days 14 and 28.
Gene expression for chondrogenic markers was evaluated at days 14 and 28. Aggrecan gene expression was significantly higher in the 0.1 mM Na2WO4 group compared to all groups at days 14 and 28 in CCM (P < 0.05 and P < 0.001, respectively; Fig. 4a). Cells in 0.01 mM Na2WO4 exhibited lower aggrecan expression compared with 0 mM at day 14 (P < 0.05). Cells in 0.1 mM Na2WO4 had the highest SOX-9 gene expression at day 14 (P < 0.05; Fig. 4b). In addition, cells in 0.01 and 0.1 mM expressed significantly higher SOX-9 compared with 0 mM Na2WO4 (P < 0.05 and P < 0.01, respectively) at day 28. Cells in 0.01 and 0.1 mM Na2WO4 expressed significantly higher collagen type II compared with 0 mM at day 14 (P < 0.001 and P < 0.001, respectively; Fig. 4c). By day 28, collagen type II gene expression was significantly higher for 0.01 mM Na2WO4 compared to all groups in CCM (P < 0.001). A significant increase in collagen type II expression was detected between days 14 and 28 for cells in 0.01 mM in CCM. Cells in 1 mM Na2WO4 exhibited the lowest gene expression for all genes in CCM. There was no significant upregulation of chondrogenic genes in GM with Na2WO4 (Supplementary Fig. S2). For cells cultured in CCM without insulin, gene expression for aggrecan and collagen type II was significantly higher in 0.01 and 0.1 mM Na2WO4 compared with 0 and 1 mM groups at days 14 and 28 (Supplementary Fig. S3). In addition, SOX-9 gene expression was significantly higher in 0.01 mM compared to 0 mM at day 14 in CCM without insulin (Supplementary Fig. S3).
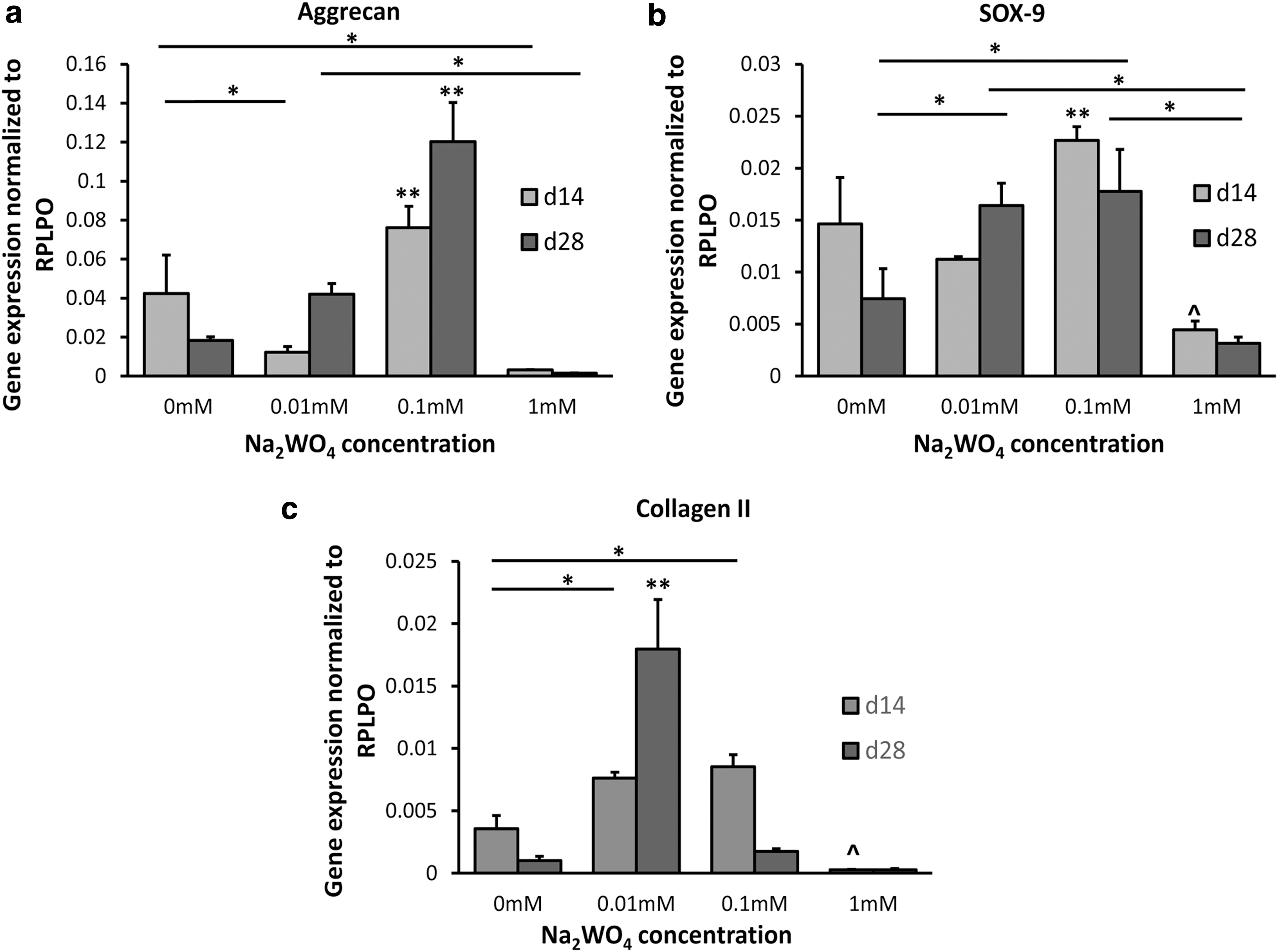
Gene expression for MSCs on scaffolds with varying amounts of Na2WO4 in CCM at days 14 and 28.
The cell morphology and collagen type II immunostaining on scaffolds with different concentrations of Na2WO4 were evaluated using confocal microscopy at day 28 as shown in Fig. 5a. Na2WO4 groups in CCM appeared to show more intense collagen type II staining compared with 0 mM. Collagen type II staining appeared to exhibit a punctate staining pattern, which is suggestive of intracellular localization. All groups had intense actin filament staining as an indicator of attachment. Moreover, collagen type II staining appeared to be more intense in 0.01 and 0.1 mM Na2WO4 in GM compared with 0 mM, where there was no staining (data not shown). CCM without insulin groups, except for 1 mM Na2WO4, showed collagen type II staining and intense actin staining (images not shown).
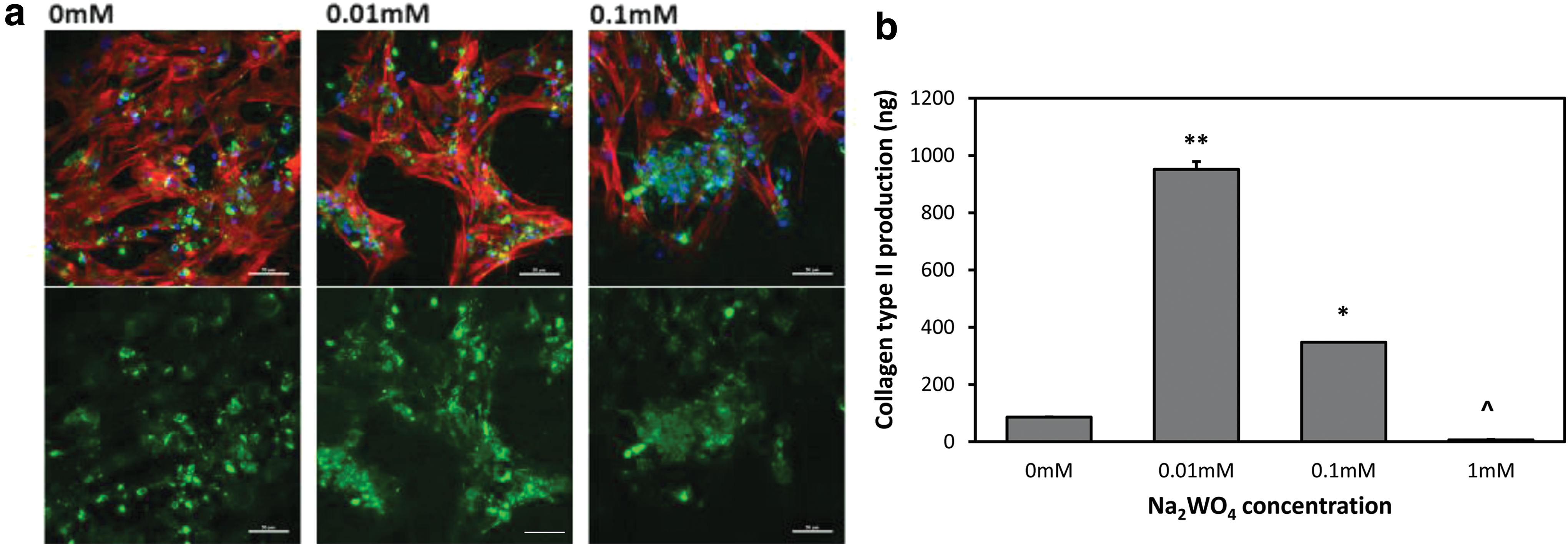
Confocal microscopy images of MSCs seeded on scaffolds with varying concentrations of Na2WO4 and collagen type II production as detected by enzyme-linked immunosorbent assay using varying concentrations of Na2WO4 in CCM at day 28.
Collagen type II production was determined for all groups at day 28 in CCM by ELISA (Fig. 5b). Cells cultured in 0.01 mM Na2WO4 produced the highest collagen type II (P < 0.001) compared to all other groups. Cells cultured in 0.1 mM Na2WO4 demonstrated significantly higher collagen type II production than without Na2WO4 (0 mM) and 1 mM Na2WO4 (P < 0.001), and production of collagen type II was the lowest in the 1 mM group (P < 0.001). Furthermore, cells in the 0.01 mM group in GM produced significantly higher collagen type II compared to the 0 mM group (P < 0.05) (Supplementary Fig. S4). Collagen type II production was not detected for CCM without insulin groups using the ELISA assay.
Discussion
The effect of Na2WO4 on MSC chondrogenesis was investigated for the first time in this study. Low concentrations of Na2WO4 enhanced chondrogenic differentiation of MSCs in vitro, as indicated by higher expression of chondrogenic markers in comparison to cultures without the use of Na2WO4. Low concentrations of Na2WO4 also enhanced MSC growth and did not provoke a proliferative response in PBMCs, indicating Na2WO4 could be a potential therapeutic agent for MSC approaches in stimulating cartilage formation.
Na2WO4 has been investigated as an insulin mimetic [37]. Insulin and IGF play important roles in cartilage and chondrogenic differentiation of MSCs. Insulin is an essential stimulator for chondrocyte proliferation [38] as well as MSC chondrogenesis [39]. Mueller et al. [39] demonstrated that chondrogenic differentiation of MSCs was not induced without insulin, and chondrogenesis markers, mainly GAG and collagen, were increased by insulin in a dose-dependent manner. Local insulin delivery in diabetic bone fracture enhanced bone repair [40]. However, insulin treatment could lead to hypoglycemia. Moreover, it is difficult to control insulin delivery since it is a high MW protein. Na2WO4 as a stable, low MW material and an insulin mimetic may overcome these challenges.
The mechanism of action, safety, and efficacy of Na2WO4 has been demonstrated in preclinical animal diabetic models [37]. Similar to insulin, Na2WO4 can increase glycogen synthesis through activation of the ERK pathway [20]. However, Zafra et al. showed that Na2WO4 activates the ERK pathway, independent of the insulin receptor using G-protein [41]. Na2WO4 also does not activate the IGF receptor and has not shown hypoglycemic action in preclinical or clinical studies [37]. ERK activation is involved in MSC chondrogenesis [24] and may be a part of the mechanism for the Na2WO4 effect on MSC chondrogenesis in this study.
MSC chondrogenesis was enhanced for low concentrations of Na2WO4 in complete induction medium containing insulin. Chondrogenic gene expression, aggrecan, sox-9, and collagen type II significantly increased compared to control (without Na2WO4). Collagen type II and sGAG production were highest in the 0.01 mM group. Moreover, Na2WO4 enhanced the expression of chondrogenic markers in cultures without insulin, both in GM and induction medium without insulin. Its effect was less pronounced than when combined with insulin, suggesting insulin may have a synergistic effect. For therapeutic strategies, the concentration of Na2WO4 may need to be optimized by examining additional concentrations of Na2WO4 between 0 to 0.1 mM and using media with lower concentrations of insulin that more closely mimic physiological levels [42]. Na2WO4 combined with low levels of insulin to avoid hypoglycemia may be an effective approach to promote MSC-based cartilage repair.
Studies have been performed to investigate the toxicity of tungsten or Na2WO4 [43,44]. The concentration of Na2WO4 used to evaluate the antidiabetic effect was around 6 mM [18,19]. Toxicity was not reported in animal studies [17,18,45] or in vitro [20] as an antidiabetic agent. Moreover, Na2WO4 was studied in obese nondiabetic patients without any toxicity effect [46]. Our results demonstrate that low concentrations of Na2WO4 enhanced MSC proliferation, while high concentrations of Na2WO4 significantly lowered MSC numbers. Moreover, low doses of Na2WO4 did not stimulate PBMC proliferation or apoptosis, which suggests that low doses of Na2WO4 do not have a toxic effect. Our studies compliment findings by Osterburg et al., which investigated the effect of Na2WO4 on human peripheral blood lymphocytes in vitro. They demonstrated that at 1 mM dose, significant toxicity occurred, while 0.01 and 0.1 mM Na2WO4 did not stimulate an immune response [47].
Na2WO4 as a water soluble small molecule could be readily delivered to sites of injury through direct injection or incorporated in drug delivery or scaffolding devices. Metallic ions or compounds would be stable during implant processing, allowing for a range of solvents, temperatures, or pressures to be used [48]. They also are low cost in comparison to recombinant proteins and may have a higher safety [48]. Metallic ions can interact with other ions that can change cellular functions, activate signaling pathways or ion channels, and bind to macromolecules such as enzymes or nucleic acids [49]. Increasing evidence has demonstrated the biological benefit of metallic ions releasing from implants. A biodegradable magnesium alloy has been studied for orthopedic applications [50] and has been shown to facilitate bone healing in clinical trials [51]. Furthermore, local delivery of vanadium and manganese chloride, both are insulin mimetics, has been shown to enhance bone fracture repair [52,53].
This study demonstrated the potential of Na2WO4 as an inductive agent for MSC chondrogenesis for cartilage tissue engineering applications. Na2WO4 may be used with MSC-based approaches to induce cartilage tissue formation. Na2WO4 could be combined with scaffolds to control the release of this compound or it could be delivered locally through direct injection to enhance chondrogenesis. Future studies will further optimize the concentration of Na2WO4 or combination with insulin for promoting chondrogenesis and characterize the regenerative response in in vivo conditions.
Footnotes
Acknowledgment
The authors would like to thank the National Science Foundation (NSF) DMR-1006510 for their support.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.