
Abstract
Dogs have been widely used as a preclinical model for human disease. With the successful generation of canine induced pluripotent stem cells (ciPSCs), the biomedical community has a unique opportunity to study therapeutic interventions using autologous stem cells that can benefit dogs and humans. Unlike mice and human pluripotent cells, which are leukemia inhibitory factor (LIF)– and basic fibroblast growth factor (bFGF)–dependent, respectively, dog iPSCs require both growth factors simultaneously. In an effort to elucidate the role of each factor in the control of ciPSC self-renewal, we performed a series of experiments aiming at understanding the signaling pathways activated by them. We found that bFGF regulates pluripotency by indirectly activating the SMAD2/3 pathway in the presence of feeder cells, exclusively targeting NANOG expression, and inhibiting spontaneous differentiation toward ectoderm and mesoderm. LIF activates the JAK-STAT3 pathway but does not function in the typical manner described in mouse naïve embryonic stem cells. These results show that a unique mechanism for maintenance of pluripotency is present in ciPSC. These findings should be taken into account when establishing stem cell differentiation protocols and may provide more insight into pluripotency regulation in species other than mice and humans.
Introduction
P
Canine induced pluripotent stem cells (ciPSCs) were reported by several groups as well, including ours [6 –9]. We have reported that ciPSCs display pluripotency markers and a differentiation capacity similar to cESCs, and both LIF and bFGF are essential for maintaining ciPSC. LIF but not bFGF is critical for viability of ciPSCs [6 –8]. This dual-growth factor dependency of canine PSCs distinguishes ciPSCs from the two PSC models that seemed to be mutually exclusive: the LIF-dependent naïve ESCs and the bFGF-dependent primed ESCs, both of which are described in mouse and human cells.
Signaling pathways of LIF and bFGF have been studied in mouse and human ESCs (Supplementary Fig. S1; Supplementary Data are available online at
Moreover, both LIF and bFGF can activate the AKT and extracellular signal-regulated kinases 1/2 (ERK1/2) pathways [11,13]. However, bFGF and LIF fail to contribute to pluripotency when applied to mouse ESCs (mESCs) and hESCs, respectively, since bFGF activates ERK1/2 excessively in mESCs and promotes differentiation; whereas LIF cannot support pluripotency of primed hESCs due to low LIF receptor expression [14,15].
The different growth factor requirements between naïve mESC and primed hESC seem to influence the downstream differentiation events. An example is that the SMAD pathway inhibitor induces neural differentiation in mouse naïve ESCs but cardiac differentiation in primed hESCs [16,17]. The dual growth factor requirement of canine PSCs sets themselves apart from naïve and primed ESCs—mouse and human—that can be defined by bFGF and LIF dependency, respectively. Understanding the signaling pathways at the pluripotency stage will better guide protocols for the directed differentiation of cESCs/ciPSCs.
In this article, we hypothesized that both LIF and bFGF are required for maintaining ciPSC pluripotency and survival, and that they exert such function through their canonical regulatory pathways that are defined in mouse (naïve) or human (primed) PSC models. Our results revealed that LIF and bFGF together contribute to pluripotency, inhibition of differentiation, and survival. bFGF regulates pluripotency via a typical mechanism described for primed ESCs; however, LIF does not regulate pluripotency in the typical manner as in naïve ESCs. Moreover, the roles of JAK and STAT3 in ciPSC are described.
Materials and Methods
Cell culture
Mitotically blocked mouse embryonic fibroblasts (MEFs) were used to maintain ciPSCs as previously reported [6]. MEFs were cultured with Dulbecco's modified Eagle's medium (DMEM; Gibco, Carlsbad, CA) containing 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA) at 37°C with 5% CO2. The culture medium was replaced every 24 h. To mitotically block the feeder cells, MEFs were incubated with fibroblast culture medium containing 10 μg/mL mitomycin C for 4 h. The blocked MEFs were seeded at a density of 2 × 104 cells/cm2 overnight as a feeder layer for ciPSCs. ciPSCs were maintained on the feeder layer of mitomycin-treated MEFs with ciPSC medium, which consisted of DMEM/F-12 (Gibco, Carlsbad, CA) supplemented with 15% (v/v) knockout serum (Gibco, Carlsbad, CA), 0.1 mM MEM nonessential amino acid solution (Sigma, St. Louis, MO), 1 mM
Our ciPSC line DI-B3 (over passage 3) expressing the core pluripotency-associated transcription factors, including OCT4, SOX2, and NANOG, was randomly selected among five previously reported ciPSC clones and used throughout this study (Fig. 1A) [6]. This cell line is capable, after 3 weeks of induction, of spontaneous differentiation forming embryoid bodies with cell derivatives from the three germ layers, including class III beta-tubulin (TUJ1, ectoderm marker), vimentin (VIM, mesoderm marker), and alpha fetoprotein (AFP, endoderm marker; Fig. 1A).
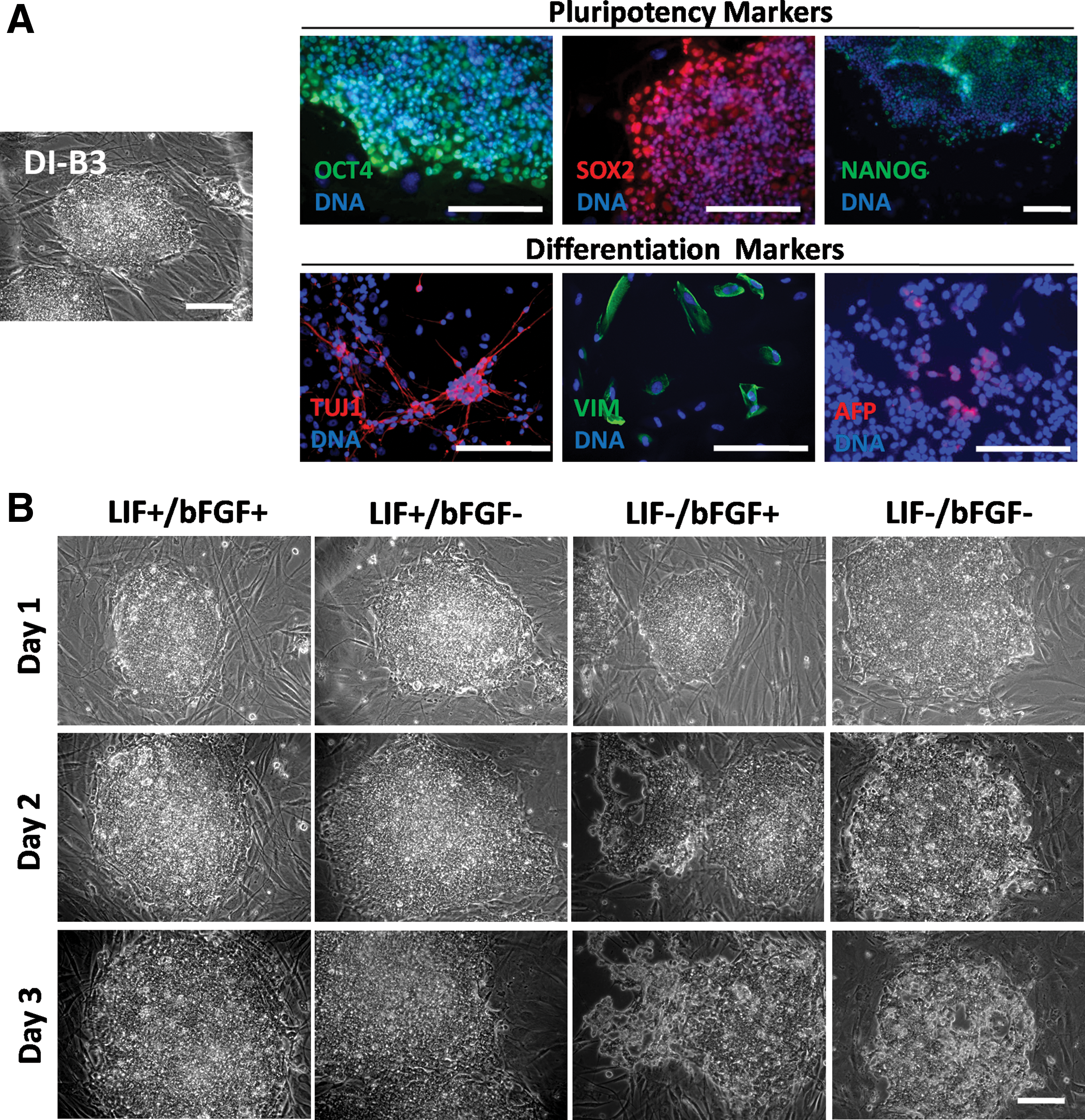
ciPSCs require bFGF and LIF to maintain pluripotency.
Immunocytochemistry assay
The immunocytochemistry (ICC) assays were performed according to the protocols previously described [6]. Supplementary Table S1 lists the use of primary and secondary antibodies. After washing the cells with phosphate-buffered saline (PBS), we stained the nuclei by rinsing the cells with PBS containing Hoechst 33342 (1 μg/mL; Sigma, St. Louis, MO) for 15 min. ICC was repeated for at least three biological replicates for each sample and each protein marker.
RNA extraction and quantitative reverse transcription–polymerase chain reaction analysis
RNA was extracted and purified using the NucleoSpin RNA XS Total RNA Isolation Kit (Macherey-Nagle, Bethlehem, PA). We performed quantitative RT-PCR reactions as previously described [18,19]. Supplementary Table S2 lists the primers designed for canine endogenous genes. Three biological replicates and two technical replicates were used for each experiment, and the results were analyzed using PROC GLM from SAS.
Western blotting assay
ciPSCs were cultured in different media or treated with different small-molecule inhibitors and harvested as indicated in the experimental design in the Results section. All ciPSCs for western blotting assays were cultured on Matrigel-coated plates and maintained in culture medium that had been earlier conditioned by the feeder cells for 24 h. Cell samples were collected mechanically using the Corning Costar cell scraper (Sigma, St. Louis, MO). Cells were lysed in RIPA buffer and kept at–80°C until use. Protein concentration was determined using a BCA assay according to the manufacturer's instructions (Thermo Scientific, Rockford, IL). Thawed samples were boiled for 5 min and loaded into 10% SDS-PAGE for protein electrophoresis, and the resolved polypeptides were transferred onto PVDF membranes (Millipore, Billerica, MA). The membranes were blocked in 5% nonfat dry milk in PBS (Sigma Aldrich, St. Louis, MO) with 0.1% Tween (Sigma, St. Louis, MO) for 30 min at room temperature and incubated overnight at 4°C with the primary antibody. On the second day, the membranes were incubated for 1 h with a horseradish peroxidase-labeled secondary antibody. Immunoreactivity was detected by the Amersham ECL western blotting detection system according to the manufacturer's instructions (GE Healthcare, Buckinghamshire, United Kingdom) and developed using Amersham Hyperfilm™ MP (GE Healthcare, Buchinghamshire, United Kingdom). Three biological replicates were performed per protein analyzed. See Supplementary Table S3 for the complete list of antibodies used.
In vitro differentiation of ciPSCs
ciPSC colonies were subcultured from the MEFs into low-attachment Petri dishes without bFGF or human LIF. After 5 days in suspension, budding embryoid bodies (EB) were placed on tissue culture dishes that were coated with 0.1% gelatin (Sigma, St. Louis, MO), and they were cultured using the same medium with 5% FBS (Gemini, West Sacramento, CA) and 10% serum replacement but without growth factors. The culture medium for suspension and subsequent spontaneous differentiation was partially changed daily. Attached EBs were cultured in the differentiation media for at least 3 weeks.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling assay
ciPSCs were cultured and harvested as indicated in the experimental designs in the Results section. To collect each sample, ciPSCs were dissociated into single cells by trypsin treatment, pelleted, washed with PBS, and fixed in 4% paraformaldehyde for 15 min. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays were performed according to the In Situ Cell Death Detection Kit (Roche Applied Science, Indianapolis, IN) following the manufacturer's instructions. As a positive control, cells were treated with DNase (Promega, Madison). After washing in PBS, we counterstained all nuclei with propidium iodide (PI; 50 μg/mL) for 30 min at 37°C. The stained cells were stored at 4°C until they were subjected to flow cytometry assay to quantify the percentage of TUNEL-positive cells. Three biological replicates were tested for each treatment.
PI staining for unfixed cells
ciPSCs were cultured and stained with PI, as indicated in the experimental design in the Results section. To stain the ciPSCs while in culture, we added 4 mg/mL of PI to the medium for 5 min. Then, the cells were immediately washed once with PBS, trypsinized, washed again with PBS, and immediately subjected to a flow cytometry assay to calculate the percentage of PI-positive cells. As a control, we used cells that were exposed to 5 mM of hydroxyl peroxide (H2O2) for 24 h. Three biological replicates were completed for each treatment. Stained cells were immediately subjected to a flow cytometry assay to count PI-positive cells. Three biological replicates were tested for each treatment.
Flow cytometry assay
The cells were trypsinized and transferred to a flow buffer (PBS). All assays were performed using an LSRII Flow cytometer (BD Biosciences, San Jose, CA) and analyzed using Diva v.6 (BD Biosciences, San Jose, CA) with the assistance of Dr. Louis King at the flow cytometry core of Michigan State University.
Statistical analysis
All statistical analyses were completed using SAS software. Two-tailed Student's t-test was used to determine the significance of difference between the controls and the experimental groups. PROC GLM was used for a comparison among multiple groups. A P value less than 0.05 was considered significant. Numerical data were reported in the format of mean ± standard error of mean from independent experiments containing three biological replicates (different batches of ciPSC) and two technical replicates.
Results
Both LIF and bFGF are indispensable for maintaining ciPSCs
ciPSCs that demonstrated pluripotency marker expression and differentiation capacity were cultured in media containing different growth factor combinations (LIF+/bFGF+, LIF+/bFGF−, LIF−/bFGF+, and LIF−/bFGF−) for 3 days (Fig. 1). Removal of either growth factor caused the loss of colonial morphologies, as a sign of spontaneous differentiation (Fig. 1B). In contrast to ciPSCs maintained in standard medium (LIF+/bFGF+), the absence of LIF (LIF−/bFGF+ and LIF−/bFGF−) but not the absence of bFGF (LIF+/bFGF−) triggered cell death, as indicated by the emergence of phase-contrast bright cells and holes in the colonies.
We then measured the gene expression levels of OCT4, SOX2, NANOG, and c-MYC when ciPSC were cultured under standard conditions (LIF+/bFGF+) or in media lacking either LIF or bFGF for 4 days (Fig. 2A). The absence of LIF but not bFGF decreased the expression of OCT4. There was no significant change in SOX2 expression when either LIF or bFGF was absent. Interestingly, LIF absence induced a dramatic upregulation of NANOG expression—more than 2.5-fold—whereas bFGF absence severely repressed NANOG expression. Absence of bFGF increased the transcription level of c-MYC close to 1.5-fold, and LIF removal reduced KLF4 expression (Supplementary Fig. S2). Expression of the ICM marker REX1 was decreased in ciPSCs when either growth factor was removed (Supplementary Fig. S2).
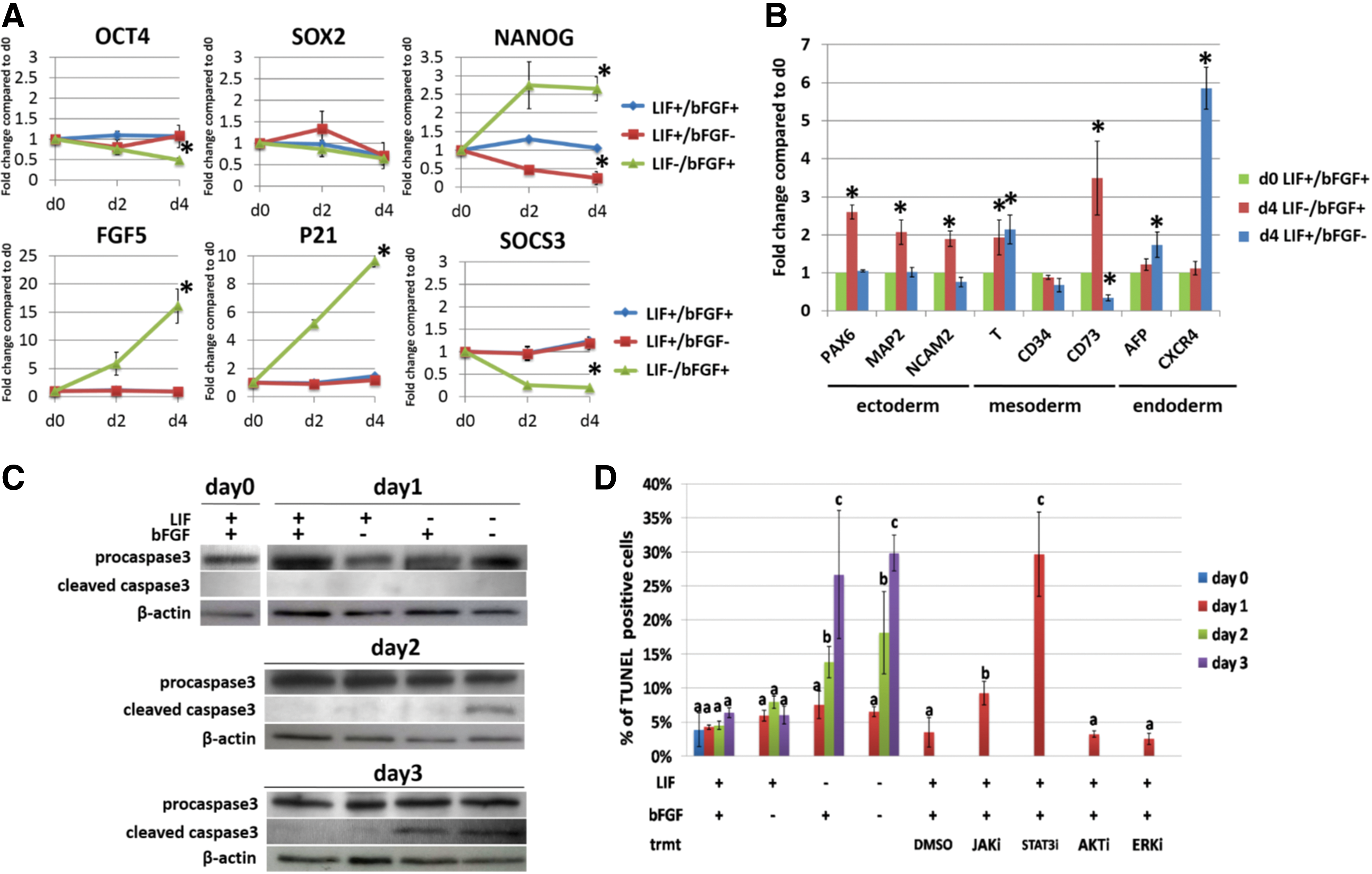
Impact of bFGF or LIF removal from culture medium on ciPSC pluripotency, differentiation, and survival.
We also found that the removal of LIF, in the presence of bFGF for 4 days, strongly enhanced the expression of epiblast stem cell marker FGF5 (Fig. 2A). The absence of LIF increased the expression of cyclin-dependent kinase inhibitor P21 in ciPSCs up to 10-fold. As an indicator of STAT3 activity, the expression of the STAT3-targeted gene SOCS3 was downregulated in ciPSCs when LIF was absent. These results provide evidence that both LIF and bFGF are required for the expression of critical genes that are related to pluripotency and self-renewal.
In terms of differentiation capability, culturing ciPSC for 4 days without bFGF but in the presence of LIF was conducive to the upregulation of marker genes for neuroectoderm (PAX6, MAP2, and NCAM2) and mesoderm (T and CD73) (Fig. 2B). LIF absence significantly enhanced the expression of mesodermal marker T and endodermal marker CXCR4 (Fig. 2B). Surprisingly, the expression of mesenchymal marker CD73 was downregulated in ciPSCs that were cultured in the absence of LIF (Fig. 2B).
We then evaluated the effects of bFGF and LIF on cell viability. As previously indicated and according to earlier results, ciPSCs demonstrated signs of cell death only when cultured without LIF [6]. To assess cell death, ciPSCs were cultured in the four conditions with different growth factor supplementation for 3 days, and they were harvested on days 0–3. Samples were subjected to western blotting for caspase-3 cleavage, TUNEL assay for apoptosis, and the unfixed cell PI staining assay for necrosis. Evaluation of caspase-3 activation and TUNEL assay revealed that ciPSCs partially underwent apoptosis only when LIF was withdrawn (Fig. 2C, D). However, growth factor depletion did not cause significant necrosis (Supplementary Fig. S3). These results suggest that the absence of LIF, but not bFGF, plays an essential role in protecting ciPSCs from apoptosis.
Efficient differentiation of PSCs requires the removal of growth factors that support pluripotency. However, attempts to differentiate ciPSCs for 14 days by removing bFGF failed to robustly upregulate the expression of differentiation marker genes (Supplementary Fig. S4). Specifically, the expression of alpha-fetoprotein (AFP, an early hepatocyte differentiation marker) and nestin (NES, a marker of early neural differentiation) was either unaffected or significantly downregulated. This result suggests that LIF may block the differentiation of ciPSCs toward specific lineages.
bFGF plays an essential role in maintaining pluripotency in ciPSCs by maintaining the expression of NANOG
The pluripotency of primed ESCs or epiblast stem cells is dependent on bFGF. In hESCs, bFGF regulates pluripotency via activating the ERK1/2 pathway and eventually targeting the expression of pluripotency-associated genes [19]. More importantly, when hESCs are co-cultured with MEFs, bFGF indirectly activates the activinA-SMAD2/3 and exclusively maintains the expression of NANOG but not OCT4 or SOX2 [11]. In ciPSCs, we noticed that the absence of bFGF dramatically downregulated the expression of NANOG but not OCT4 or SOX2, a response similar to that described for hESCs, suggesting that in ciPSC bFGF is supporting pluripotency through a pathway similar to that of primed hESCs (Fig. 2A).
To test our hypothesis, ciPSCs were cultured under conditions providing different growth factors (LIF+/bFGF+, LIF+/bFGF−, LIF−/bFGF+, and LIF−/bFGF−) for 3 days, and the activation of the bFGF-associated pathways was assessed by evaluating the phosphorylation of SMAD2/3 and ERK1/2. Activation of SMAD2/3 was not significantly different between groups until day 3: In the absence of bFGF (LIF+/bFGF− and LIF−/bFGF−), the SMAD2/3 was significantly inactivated (Fig. 3A, Supplementary Fig. S5). Moreover, no change was observed in ERK1/2 between groups (Fig. 3A, Supplementary Fig. S5). These data indicate that activation of SMAD2/3, but not ERK1/2, is associated with the presence of bFGF in ciPSCs.
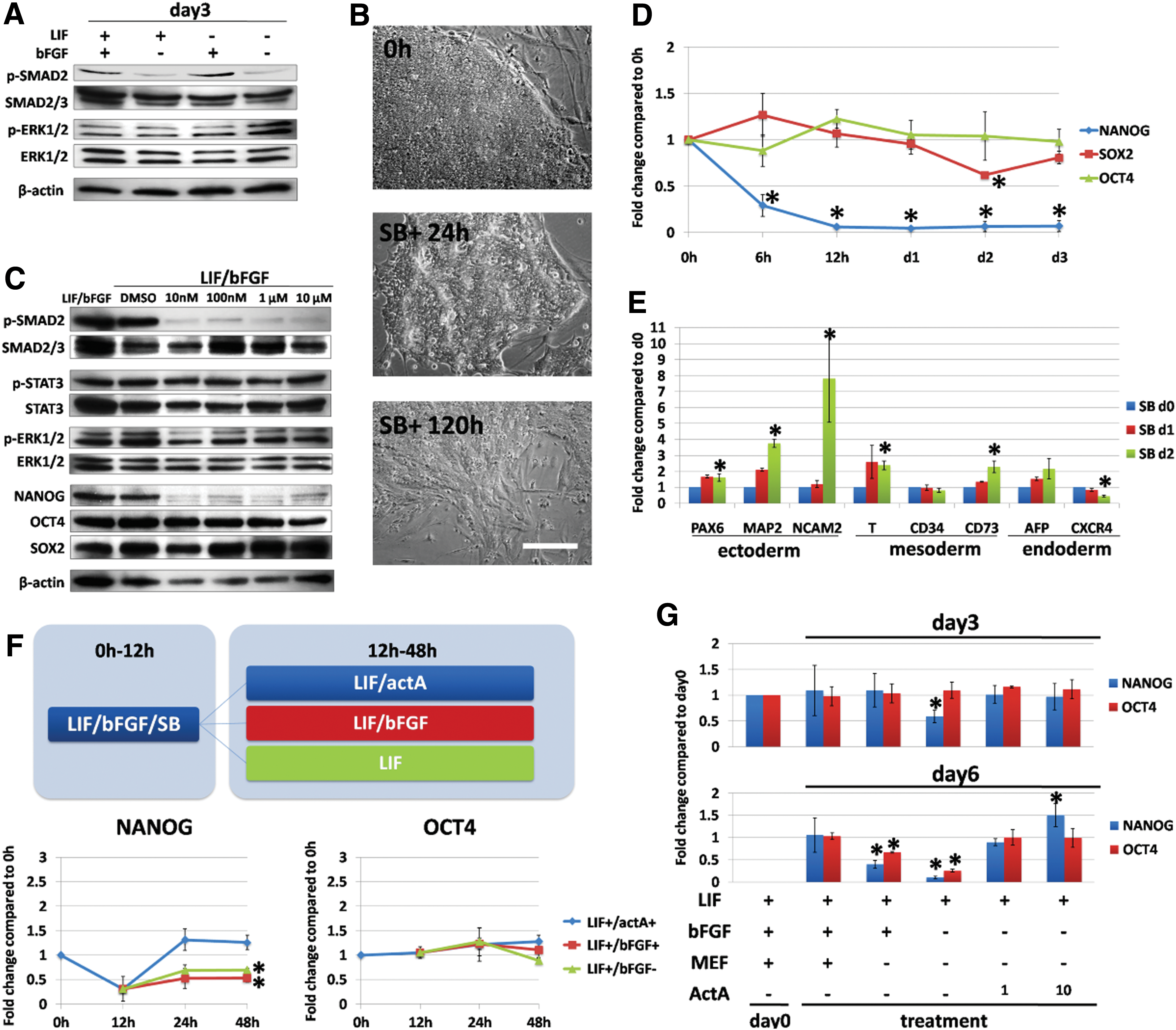
bFGF regulates ciPSC pluripotency and differentiation through the activin-A-SMAD2/3-NANOG pathway in the presence of MEFs.
We then tested whether bFGF activates SMAD2/3 via the TGF-beta receptor. ciPSCs were treated by small-molecule inhibitor SB431542 (SB) in the presence of bFGF and LIF. Morphologically, ciPSCs reduced colonial compactness after 24 h of SB treatment, and they even displayed a fibroblastic morphology after 5 days of treatment (Fig. 3B). We found that SMAD2/3 was efficiently dephosphorylated at very low doses of SB treatment within 24 h. However, the activation of other pathways, such as STAT3 and ERK1/2, was not significantly affected (Fig. 3C). The expression of OCT4 and SOX2 was not affected by SB treatment, whereas the expression of NANOG was dramatically reduced by SB treatment (Fig. 3C). Similar results were seen by measuring gene expression levels of these genes after 3 days of SB treatment (Fig. 3D). mRNA expression levels of c-MYC, P21, REX1, and FGF5 were also analyzed in SB-treated ciPSCs (Supplementary Fig. S6). The expression of FGF5 decreased, whereas other genes remained unchanged.
To assess the trend of spontaneous differentiation of ciPSCs after SB inhibitor treatment, we collected SB-treated ciPSCs on days 0–2 and subjected them to a quantitative reverse transcription–polymerase chain reaction (qRT-PCR) assay (Fig. 3E). Similar to ciPSCs cultured without bFGF, the genes for neuroectoderm (PAX6, MAP2, and NCAM) and mesoderm (T and CD73) were upregulated in ciPSCs within 2 days of SB treatment. However, the expression of the endodermal marker AFP was enhanced after SB treatment. These results suggest that SMAD2/3 activation is critical for maintaining pluripotency of ciPSCs and for blocking differentiation in by promoting expression of NANOG.
In hESC culture, bFGF stimulates MEFs to synthesize activin-A, which directly triggers the activation of SMAD2/3 of hESCs [11]. To test whether activin-A plays a role in ciPSC pluripotency, first, ciPSCs were cultured in standard condition (LIF+/bFGF+) plus 10 μM SB for 12 h to block SMAD2/3 activation and, as a consequence, suppressed NANOG expression. We then cultured the ciPSCs for 36 h in different conditions: standard medium with LIF and bFGF; medium with LIF and 10 ng/mL activin-A; and medium with LIF only, as illustrated in Fig. 3F. ciPSCs were collected at 0, 12, 24, and 48 h from three groups, and expression levels of OCT4 and NANOG were evaluated (Fig. 3F). Results indicated that only the group of LIF plus 10 ng/mL activin-A robustly reinforced NANOG expression; the rest displayed a decrease in the expression of NANOG. OCT4 expression did not change significantly among groups.
We then tested whether activin-A alone, without bFGF or MEFs, could maintain the expression of pluripotency genes. We cultured ciPSCs for 6 days with or without bFGF, MEFs (ciPSCs without MEF support were maintained in a Matrigel-coated dish), and/or activin-A. The samples were collected on days 0, 3, and 6 to evaluate the expression of OCT4 and NANOG. We noticed that the absence of MEF did not change OCT4 and NANOG expression on day 3 but significantly decreased their expression on day 6, and the absence of both bFGF and MEF triggered a more dramatic decrease of both NANOG and OCT4 expression (Fig. 3G). In comparison, the groups with activin-A treatment successfully maintained the expression of OCT4 and NANOG in the absence of MEF and bFGF. NANOG, but not OCT4 expression in ciPSCs cultured with 10 ng/mL of activin-A, was more robust than those cultured with 1 ng/mL of activin-A (Fig. 3G). These data indicated that bFGF regulates ciPSC pluripotency by activating the activin-A-SMAD2/3-NANOG axis in the presence of MEFs.
LIF plays a divergent role in ciPSCs in maintaining pluripotency and survival
In naïve ESCs, LIF activates the JAK-STAT3 pathway, resulting in the expression of various pluripotency and survival-associated genes [13]. In addition, AKT and ERK1/2 cascades can be activated in naïve mESCs to facilitate pluripotency and survival [13]. Therefore, we hypothesized that LIF plays a role in ciPSCs, similar to mouse naive ESCs, in regulating pluripotency and survival.
First, we evaluated the activation of LIF-associated signaling pathways when ciPSCs were cultured with or without LIF. The candidates included STAT3, AKT, and ERK1/2. ciPSCs were cultured in conditions with different growth factor supplementation (LIF+/bFGF+, LIF+/bFGF−, LIF−/bFGF+, and LIF−/bFGF−) for 3 days. The phosphorylation of STAT3, AKT, and ERK1/2 in ciPSCs collected on days 0–3 was assessed. Removal of LIF and bFGF (LIF−/bFGF−) immediately triggered the STAT3 inactivation on day 1, and removal of LIF only caused the STAT3 dephosphorylation since day 2 (Fig. 4A and Supplementary Fig. S5). However, AKT and ERK1/2 were constantly activated in all groups (Fig. 3A, and Supplementary Fig. S5). These data indicate that the JAK-STAT3 pathway, but not AKT or ERK1/2, is downregulated when LIF is removed from the media in ciPSCs.
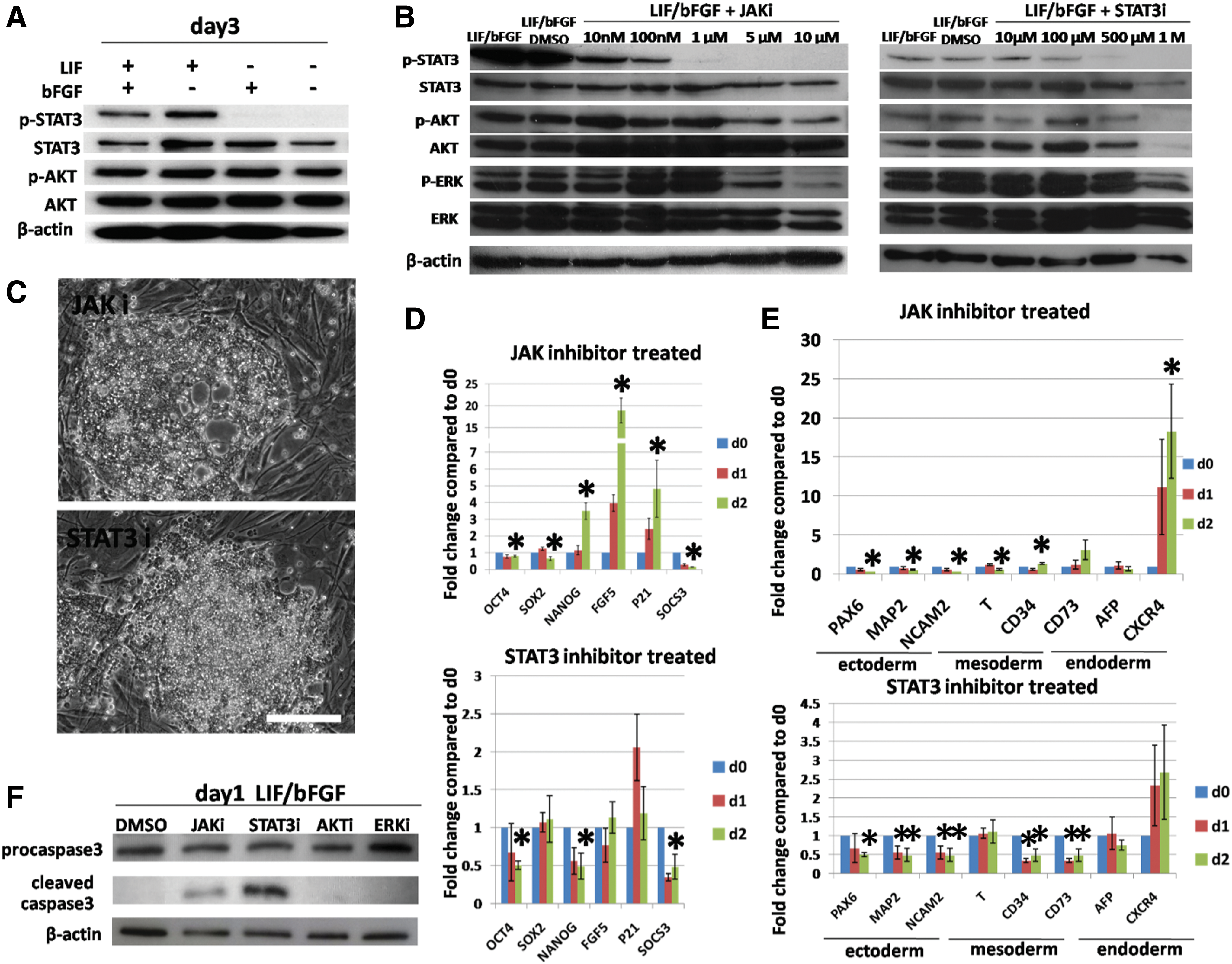
LIF maintains ciPSC pluripotency, differentiation, and survival through the JAK-STAT3 pathway.
To further understand how LIF regulates pluripotency and survival, small-molecule inhibitors were applied to inhibit LIF-associated signaling transduction in ciPSCs. We treated the ciPSCs cultured under standard condition (LIF+/bFGF+) with inhibitor targeting JAK1/2 (JAK inhibitor 1, JAKi) or STAT3 (NSC74859, STAT3i) to mimic the inactivation of the JAK-STAT3 pathway when LIF is absent, as indicated by previous results. ciPSCs were also treated with inhibitors blocking the signal transducers in non-specific LIF-associated pathways, including AKT (MK2206, AKTi) and ERK1/2 (PD184352, ERKi). A dose–response curve was completed to determine the working concentrations of each inhibitor. We chose the minimum dose for each inhibitor that was capable of specifically inducing the dephosphorylation of the targeted protein without affecting the phosphorylation of unrelated proteins. Briefly, ciPSCs were cultured for 24 h under plain standard conditions (LIF+/bFGF+), with DMSO (vehicle for the inhibitors), or with small-molecule inhibitors at a range of concentrations. ciPSC samples were collected and subjected to western blotting assays to evaluate the phosphorylation of STAT3, AKT, and ERK1/2 (Fig. 4B and Supplementary Fig. S7). Based on these results, the concentrations chosen were as follows: 1 μM JAKi, 500 μM STAT3i, 10 μM AKTi, and 1 μM ERKi for subsequent experiments.
ciPSCs were cultured in standard ciPSC medium with LIF and bFGF and treated with a small-molecule inhibitor at the determined dose for 2 days. ciPSCs were assessed on days 0–2 for morphological changes, expression of pluripotency and differentiation genes, and survival. First, treatment with JAKi or STAT3i induced more phase-contrast bright cells and decreased the compactness of colonies, which appeared as a sign of cell death and spontaneous differentiation, similar to ciPSCs cultured without LIF (Fig. 4C). However, neither AKTi nor ERKi induced any signs of cell death (Supplementary Fig. S8).
Second, we collected ciPSCs treated with JAKi or STAT3i to assess the expression patterns of genes for pluripotency and differentiation (Fig. 4D, E). When treated with JAKi, the expression of OCT4 and SOX2 was slightly decreased with statistical significance. However, similar to ciPSCs cultured without LIF, the expression of NANOG, P21, and FGF5 was robustly increased. Moreover, the STAT3-targeted gene SOCS3 displayed decreased expression in ciPSCs treated with JAKi due to inactivation of STAT3 [13]. In terms of expression of differentiation-associated genes, JAKi treatment induced a minor increase of CD34 expression, and similar to ciPSCs cultured without LIF, a dramatic upregulation of CXCR4 expression was found. However, the expression of all other genes (PAX6, MAP2, NCAM2, T, CD73, and AFP) tested was either unchanged or decreased when compared with the untreated ciPSCs. Nevertheless, when treated with STAT3i, ciPSCs displayed an entirely distinct profile of gene expression, that is, the expression of OCT4, NANOG, and FGF5 was decreased to ∼0.5-fold after 48 h compared with untreated ciPSCs, but SOX2 expression remained unchanged. P21 expression was elevated twofold in ciPSCs after 24 h of STAT3i treatment, but it fell back close to onefold soon thereafter. Similar to what was observed after JAKi treatment, SOCS3 mRNA levels declined after STAT3i treatment in ciPSCs. The expression of all differentiation markers was maintained, without a significant change after STAT3i treatment. These data suggest that the role of LIF in ciPSCs in maintaining pluripotency is different from that in mESCs, in which LIF deprivation dramatically reduces the expression of pluripotency genes, including OCT4, SOX2, and NANOG. Moreover, JAK and STAT3 play distinct roles in regulating ciPSC pluripotency [13].
In an effort to determine the role of LIF in ciPSC survival, we evaluated the levels of apoptosis and necrosis after the inhibition of LIF-associated pathways. The results of the TUNEL assay showed that treatment with JAKi or STAT3i significantly increased the TUNEL-positive cell rate in the ciPSC population (Fig. 2D). In agreement with the TUNEL results, only treatment with JAKi or STAT3i caused the cleavage of caspase-3 in ciPSCs (Fig. 4F). All other inhibitors tested did not affect cell membrane integrity (Fig. 2D). These data suggest that inhibition of the LIF-JAK-STAT3 pathway in ciPSCs cultured under standard conditions (LIF+/bFGF+) triggers apoptosis.
Discussion
Dogs have been used in human preclinical trials for more than 30 years. This is, at least in part, due to advantages such as an appropriate body size, age, biochemical and physiological features, and genetic diversity [3,20]. Autologous iPSC-based therapeutic strategies are primarily tested on rodents, and though providing valuable indications as to the efficacy and safety of a given approach, large animal models such as the dog are urgently needed to validate therapeutic interventions before reaching humans. Recently, reports from our laboratory and others have shown that canine iPSCs with bona fide pluripotency features can be generated [1,6 –8,21]. Canine ESCs and ciPSCs express SSEA-3, SSEA-4, TRA-1-61, and TRA-1-80 [6]. This profile of surface marker expression is highly similar to that of primed hESCs and mouse epiblast stem cells, but it is distinct from naïve mESCs that express only SSEA-1.
A major factor known to influence the development of robust differentiation protocols is to take into account the molecular mechanism of pluripotency maintenance of the cells to be used. Dog pluripotent cells, different from the naïve mouse or primed human PSC model, which only depend on LIF or bFGF, respectively, are dependent on the presence of both LIF and bFGF.
In this study, we focused on understanding the roles of LIF and bFGF in regulating ciPSC pluripotency, differentiation capability, and survival. We found that bFGF controls ciPSC pluripotency through a mechanism that is highly similar to that described for primed epiblast stem cells. When cultured without bFGF, ciPSCs significantly decreased the expression of NANOG, but not that of OCT4 or SOX2, a finding that was also observed when the SMAD2/3 pathway was inhibited. The absence of bFGF—or the inhibition of the SMAD2/3 pathway—prompts the bias of spontaneous differentiation of ciPSCs toward neuroectoderm and mesoderm. Supplementation with activin-A can replace the effect of bFGF and MEFs and maintain NANOG expression. The roles of bFGF, activin-A, and the activation of the SMAD2/3 pathway in ciPSCs are consistent with those reported for primed hESCs [11]. Our results strongly suggest that activin-A could maintain self-renewal and pluripotency, and it should be considered when designing new culture media for ciPSCs (Fig. 3F). Interestingly, the absence of either LIF or bFGF did not change the activation status of ERK1/2 or AKT, likely due to the presence of other growth factors such as LIF, insulin growth factor, or epidermal growth factor, which may be secreted by the feeder cells [13]. It has been previously reported that, similar to primed hESCs but distinct from naïve mESCs, the sequence alignment of the promoter region of NANOG in canine cells contains the SMAD2/3 binding sequence [22]. In all, bFGF maintains ciPSC pluripotency by indirectly regulating NANOG expression, which is a similar signaling mechanism to that of primed epiblast stem cells.
In terms of the role of LIF in ciPSCs, we found that JAK-STAT3 was specifically activated by LIF but, unlike naïve mESCs, when LIF was absent or specific inhibitors of STAT3 activity were employed, pluripotency gene expression was maintained, and we were unable to detect major escalation of expression of differentiation genes. Moreover, when the STAT3 pathway in ciPSCs was inactivated, by LIF withdrawal or treatment of the JAK inhibitor, we observed an increased expression of NANOG, P21, and FGF5. In addition, JAK inhibition did not have an impact on OCT4 expression. In contrast, direct STAT3 inhibition decreased the expression of OCT4 and NANOG, but it did not affect the expression of P21 or FGF5. In terms of differentiation markers, the inactivation of the LIF-JAK-STAT3 signaling axis did not increase the expression levels of major differentiation genes, with the exception of CD73 and CXCR4. Similarly, STAT3 inhibition alone had no impact on differentiation induction, with the only exception of increased expression of CXCR4. A previous study showed that inactivation of JAK1/2 by mutant JAK2 V617F upregulates NANOG expression level through the phosphorylation of histone 4 lysine 51 in mESCs [23]. Our result indicates that this regulatory system is possibly conserved in ciPSCs. On the other hand, our data show that STAT3i treatment partially influences the expression of OCT4 and NANOG in ciPSCs. However, STAT3i treatment did not increase the expression level of differentiation genes. These results suggest that, unlike the role of STAT3 in naïve mESCs, STAT3 in ciPSCs does not seem to have much of an impact on pluripotency maintenance.
Nonetheless, our data do show a critical role for LIF in ciPSC survival. We found that LIF removal or the inhibition of the JAK-STAT3 pathway induced apoptosis in ciPSCs. The JAK inhibition induced a lower TUNEL-positive cell rate (average, 9.27%) than the STAT3 inhibition (average, 29.67%). The same pattern was observed when we examined caspase-3 activation. In naïve mESCs, NANOG overexpression can benefit cell survival by inducing expression of HSPA1A [24]. It is possible that the increase of NANOG expression caused by inhibition of JAK activity helped in tapering down apoptosis in ciPSCs. Another anti-apoptosis effect that may be caused by JAK inactivation is a decrease in the levels of P21. As a cell cyclin inhibitor, P21 induces cell cycle arrest in the G1/S phase to ensure normal DNA replication and overexpression of P21 can induce cell death [25 –27]. It is possible that death of ciPSCs is caused by P21 expression. The role of STAT3 in ciPSC survival was indicated by the rapid and dramatic induction of apoptosis when ciPSCs were treated with STAT3i. It has been reported that LIF withdrawal during naive-mESC culture may induce the inactivation of STAT3, which, subsequently, fails to inhibit the stress-induced activity of p38 MAP kinase and triggers an apoptosis rate of more than 30% that is indicated by the rate of TUNEL-positive cells [28,29]. It is quite likely that the current culture conditions for ciPSC are not optimal and therefore, while promoting their expansion, they can induce stress in some cells. The activation of the JAK-STAT3 pathway by LIF may compensate some of the detrimental effects caused by such stress. Apoptosis is a common response to stress during in vitro culture. It was reported that ESCs are hypersensitive to apoptosis triggered by DNA damage due to mismatch repair, as a mechanism that may contribute to reduction of the mutational load in the progenitor population [30]. The upregulation of P21 when ciPSCs were cultured without LIF indicates that ciPSCs may be prone to DNA damage, which, in turn, may trigger the expression of P21.
Since ciPSCs stressors are yet to be properly identified, the addition of small molecules that are shown to enhance survival must be considered, for example, inhibition of glycogen synthase kinase 3, shown to be critical to enhance survival of somatic cells during reprogramming, could complement or replace LIF altogether [31]. In addition, considering the potential complexity of pluripotency regulation in large animal PSCs, the differences of donor cell type and reprogramming system may influence the pluripotency regulation of ciPSCs. Our results were based on a single pre-characterized ciPSC line derived from single-donor origin; therefore, more studies are needed to comprehensively test the roles of growth factors in ciPSCs or canine ESCs derived from different reprogramming or isolation systems.
Taken together, our results support the hypothesis in which bFGF maintains pluripotency of ciPSCs by exclusively activating endogenous NANOG, triggering the activity of the activinA-SMAD2/3 signaling pathway in the presence of MEFs, as seen in epiblast stem cells. We also found that LIF—unlike naïve mESCs—has a lesser role in maintaining pluripotency of ciPSC while being required to support ciPSC survival (Fig. 5A, B). Previous studies in human, rabbit, swine, canine, and bovine PSCs have reported the use of LIF and bFGF simultaneously to keep the cells pluripotent under what seems to be a mutually exclusive growth factor combination, but they fell short from explaining the underlying signaling pathways controlling pluripotency [32 –36]. Our present study provides the basis for the development of new protocols to derive lineage-specific cell derivatives, which can be subsequently applied in cell or tissue transplantation, tissue engineering, and drug screening.
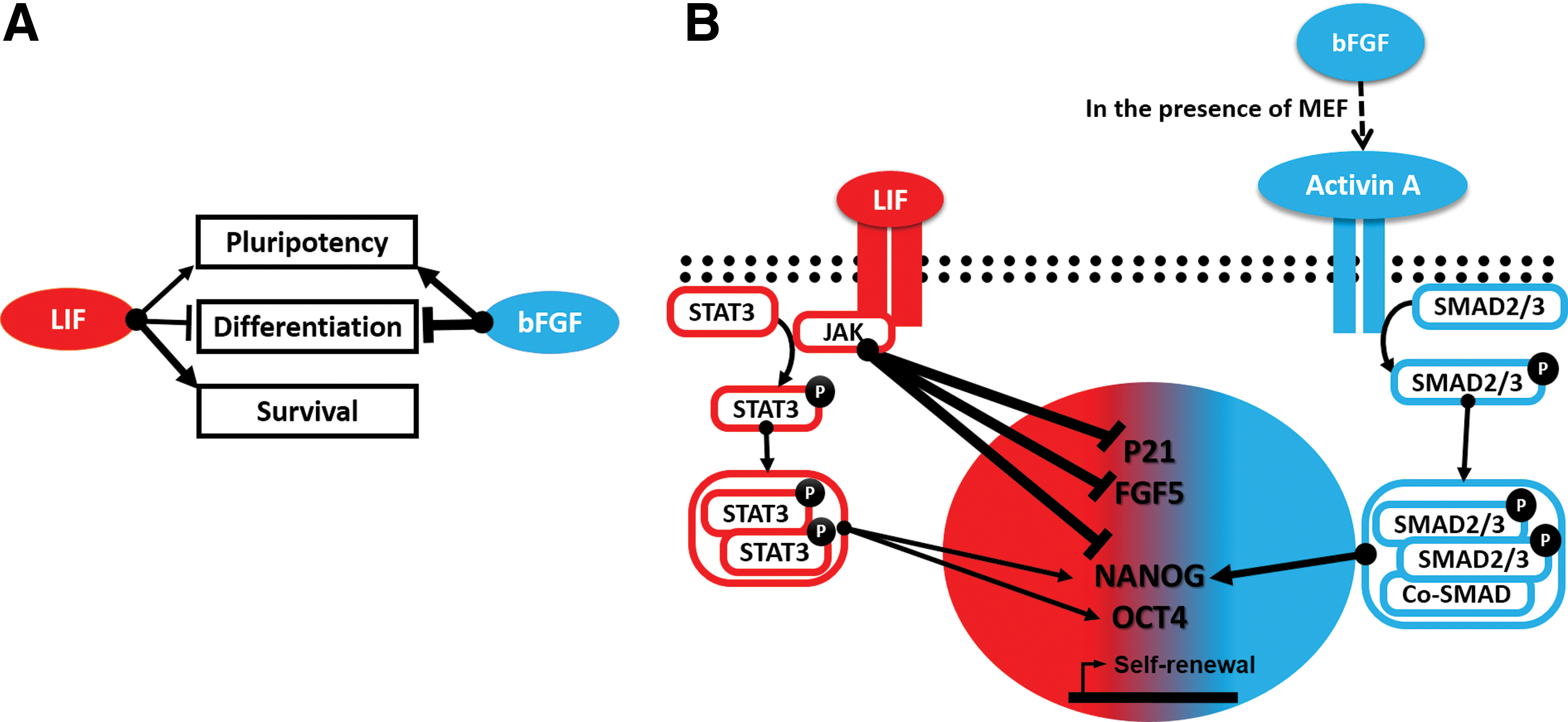
Proposed models illustrating the collaborative roles of LIF, bFGF, and the associated signaling pathways that maintain self-renewal in ciPSC.
Footnotes
Acknowledgments
The authors are grateful for the support rendered by Dr. Steven Suhr, Dr. Gloria Perez, and Dr. Eun-Ah Chang. This study was supported by grant D12CA-066 from the Morris Animal Foundation. The authors appreciate the support from the Michigan State University (MSU) Foundation, MSU's Vice President of Research, and Michigan AgBioresearch.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.