
Abstract
Previous research has indicated that purified perivascular stem cells (PSCs) have increased chondrogenic potential compared to conventional mesenchymal stem cells (MSCs) derived in culture. This study aimed to develop an autologous large animal model for PSC transplantation and to specifically determine if implanted cells are retained in articular cartilage defects. Immunohistochemistry and fluorescence-activated cell sorting were used to ascertain the reactivity of anti-human and anti-ovine antibodies, which were combined and used to identify and isolate pericytes (CD34−CD45−CD146+) and adventitial cells (CD34+CD45−CD146−). The purified cells demonstrated osteogenic, adipogenic, and chondrogenic potential in culture. Autologous ovine PSCs (oPSCs) were isolated, cultured, and efficiently transfected using a green fluorescence protein (GFP) encoding lentivirus. The cells were implanted into articular cartilage defects on the medial femoral condyle using hydrogel and collagen membranes. Four weeks following implantation, the condyle was explanted and confocal laser scanning microscopy demonstrated the presence of oPSCs in the defect repaired with the hydrogel. These data suggest the testability in a large animal of native MSC autologous grafting, thus avoiding possible biases associated with xenotransplantation. Such a setting will be used in priority for indications in orthopedics, at first to model articular cartilage repair.
Introduction
P
To ascertain how well particular cells perform in vivo before human clinical trials, the use of animal models is required [10 –12]. These models allow the efficacy and safety of the proposed methods to be tested in a biological and mechanical environment similar to those that would be encountered in clinical practice.
The International Cartilage Repair Society recommends that while small animal models are useful for initial studies and proof of concept work, large animal studies are required for pivotal studies [12]. A canine model of PSCs has previously been developed and used at the University of California in Los Angeles (Hardy et al., unpublished data). The use of canines in animal research in the United Kingdom is socially unacceptable and unlikely to be approved by the Home Office due to their status as companion animals.
Sheep have successfully been used for implantation of autologous chondrocytes on a collagen matrix [13] and for chondrocytes and MSCs embedded in gels [10 –12,14 –16]. Godoy et al. used sheep to compare stem cells from bone marrow, adipose tissue, and synovium [17]. Their research indicated that MSCs could be harvested from each of these sources and that MSCs from each source shared the ability to from bone, cartilage, and fat as well as sharing a similar immunophenotype based on expression of the marker CD44. These data demonstrated the feasibility of obtaining ovine stem cells.
While pericytes have been identified in the ovine corpus luteum, their perivascular location has not been confirmed in multiple organs as they have in human tissues [18,19]. There have been no reports identifying the role of adventitial cells as a source of stem cells in the ovine model. Neither cell type has been identified using cell surface antigens in a similar manner to human tissue nor have they been isolated using antibody labeling and fluorescence-activated cell sorting (FACS). Without using similar methods used for human PSCs it is harder to state that the ovine PSCs (oPSCs) are truly analogous and therefore an equivalent model.
The ovine stifle joint has been described as a surgical model to represent the human knee joint [20]. The anatomy of the ovine stifle joint has been well described and is considered a reasonable model of the human knee joint [20,21]. It has been used to investigate articular cartilage repair as well as anterior cruciate ligament (ACL) reconstruction [22] and meniscal repair [23] and replacement [24].
Caplan and Correa proposed the idea that MSCs work using a paracrine effect, essentially acting as a “drug store” to aid in tissue regeneration [25]. It is implied that the basis of cell-based repairs must be the cells that are introduced into the defects. There are very few studies that have labeled and tracked the cells that have been placed into articular cartilage defects, raising the question of the fate of the implanted cells. Proposed cell tracking methods include PKH26 [26], iron oxide nanoparticles [27], and DiI and transfection using green fluorescence protein (GFP) [28].
Nakamura et al. used GFP emission to ascertain that injected cells stayed in place following insertion, their DiI-labeled cells remained in the defect at 7 days but were not seen at 1 or 3 months [28]. Miot et al. implanted GFP-labeled human chondrocytes into goats on a Chondro-Gide membrane and found the cells in the defect after 4 weeks [29].
The primary aim of this research was to develop a large animal model of autologous PSCs that could be used to translate laboratory-based research to human clinical trials. The secondary aim was to test the model for articular cartilage repair and to determine if cells were engrafted by surgical implantation.
Materials and Methods
Ethical approvals
All animal research was conducted in accordance with the Animals (Scientific Procedures) Act 1986 under Protocol 15 of project license PPL 60/4052. All of the experimental protocols were reviewed by the University of Edinburgh's Named Veterinary Surgeons.
Tissue harvest and cell isolation
Cadaveric tissue was obtained for use for identification, isolation, and characterization of oPSCs before conducting the animal surgeries. Cadaveric tissue was harvested within 1 h of sacrifice and transported to the laboratory in transport media [Dulbecco's modified Eagle's medium (DMEM)/10% fetal bovine serum (FBS)/1% penicillin–streptomycin (PS)].
Tissue samples were mechanically disrupted to ensure they were <1 mm3 and were covered with digestion medium (DMEM/10% FBS, 1% PS, and 0.5% collagenases I, II, and IV). The tissue was digested in a shaking water bath (37°C, 150 rpm) for 50 min. The digested suspension was centrifuged at 1,800 rpm for 10 min. The pellets were resuspended in 25 mL 2% FBS/phosphate-buffered saline [PBS; 5 mM ethylenediaminetetraacetic acid (EDTA)]. The tissue suspensions were successively filtered through a 200-μm nylon mesh and then 100-, 70-, and 40-μm cell strainers. The strained suspensions were centrifuged at 1,500 rpm for 10 min. The supernatant was aspirated and discarded and the pellets resuspended in 1 mL 20% FBS/DMEM/1% PS.
Histology and immunohistochemistry
Tissue samples were resected so that they were <2 cm3 to ensure rapid and complete fixation in 4% formalin. Fixed tissue was placed in Tissue-Tek® cassettes (Sakura Finetek) and processed using a Leica ASP processor using sequential alcohol, xylene, and paraffin wax steps. Tissue was subsequently embedded into molten wax, allowed to solidify on a cold plate, and sectioned 7 μm thick. Sections were incubated at 55°C over night, dewaxed (xylene 5 min three times), and rehydrated (100% ethanol 2 min three times, 95%, 80%, and 50% ethanol 2 min each, and then washed under running water). Slides were stained as per the sections below, then dehydrated (50%, 80%, and 95% ethanol for 30 s each and 100% ethanol for 2 min three times), and mounted using xylene.
Sections were incubated in Harris hematoxylin (Sigma-Aldrich) for 5 min and then washed under running water. They were differentiated in 1% hydrochloric acid in ethanol and transferred to a dish of Scott's tap water substitute for 2 min until the tissue sections turned blue. Slides were counterstained in eosin for 2 min and were washed briefly under running water.
Slides were brought to room temperature (RT) and fresh frozen sections fixed in an ice-cold bath of methanol and acetone (50:50) for 10 min. Slides were air-dried for 5 min at RT before being washed [each washing step consisted of three 5-min washes at 50 rpm on a rotating platform, once with PBS/Tween (0.5 mL/L Tween 20 pH 7.4) and twice with PBS alone]. Slides were incubated with protein block (DAKO) for 1 h at RT and then washed. Slides were incubated with avidin D for 15 min followed by a PBS wash followed by incubation with biotin for 15 min and followed by a further PBS wash. Sections were incubated with the primary antibody in antibody diluent (DAKO) overnight at 4°C. The following day, sections were washed and incubated with a biotinylated secondary antibody against the host species of the primary antibody diluted 1:1,000 in antibody diluent (DAKO) for 1 h at RT. Sections were incubated with streptavidin-AF488 or streptavidin-AF555 (Invitrogen) diluted 1:1,000 in antibody diluent for 1 h at RT. The sections were washed for a final time and mounted with a single drop of 4′,6-diamidino-2-phenylindole (DAPI) mounting medium and then coverslipped. Stained sections were stored at 4°C in the dark until imaged.
For multiple stains, primary antibodies raised in different species were used where possible. The primary and secondary antibodies were incubated at the same time (ie, primary mouse anti-human and rabbit anti-human with secondary goat anti-mouse and goat anti-rabbit).
Histochemical staining was imaged using a brightfield setting and color camera with a Zeiss observer microscope. An Olympus BX61 fluorescence microscope was used to sequentially record the emission from the individual fluorophores used for immunohistochemistry, these were subsequently merged to form a composite image. Controls using isotypes were imaged under identical conditions.
Fluorescence-activated cell sorting
The cells were blocked in 10% mouse serum in PBS and left at RT for 20 min, 100 μL for analysis and 500 μL for cell sorting. Antibodies were added to the cell suspension in the dark at a dilution of 1:100 unless otherwise stated. All antibodies were tested using the stromal vascular fraction (SVF) from multiple tissues; all samples were compared to their corresponding isotype except the anti-VWF-FITC, which used an unstained sample as its negative control.
The cells were incubated for 20 min on ice in the dark. The cells were washed in 2 mL of PBS and centrifuged at 1,000 rpm for 5 min. The supernatant was aspirated and discarded and the pellet resuspended in 2% FBS/PBS, 300 μL for analysis and 500 μL for cell sorting.
For each antibody, a compensation tube was set up using a single drop of positive compensation beads (BD Biosciences) diluted with 70 μL 2% FBS/PBS and stained in an identical manner to the cells. These were resuspended in a final volume of 270 μL and a drop of negative control beads added just before FACS analysis or sorting. Gates were set using negative controls to determine the level of true-positive staining.
Cell culture
FACS sorted cells were collected in 300 μL endothelial cell growth medium (EGM-2; Lonza) as pericytes (CD34−CD45−CD146+) and adventitial cells (CD34+CD45−CD146−). Wells and flasks were precoated with 0.2% (w/v) gelatin (Calbiochem) for 10 min at 4°C. Cells were plated at a density of ∼4 × 104/cm2 in EGM-2 and cultured at 37°C, 5% CO2. The EGM-2 medium was changed after 7 days and then every 4 days after that until the cells reached 90%–100% confluency. From passage 1 (P1) onward, all cells were cultured in DMEM/20% FBS/1% PS. Cells were washed twice in PBS and then incubated in 0.05% trypsin-EDTA at 37°C, 5% CO2 for 3–5 min until >90% of the cells were mobilized. Complete medium was added to the plate/flask and the cell suspension transferred to a 15-mL centrifuge tube. The cells were pelleted by centrifuging at 1,000 rpm for 5 min. The supernatant was aspirated and discarded and the pellet resuspended for further culture expansion, differentiation, or flow cytometry. Freshly isolated SVF was used for identifying the reactivity of FACS antibodies, and cell characterization and differentiation were undertaken with passage 6 cells.
Immunocytochemistry
For immunocytochemistry, PBS was removed and the cells permeabilized with 0.05% Triton-PBS for 3 min at RT. The cells were washed three times with PBS and then blocked with Protein Block (DAKO) for 1 h at RT. Cells were washed in PBS and then incubated with the primary antibody overnight at 4°C. The following morning, the wells were washed three times for 5 min, once with PBS-Tween and twice with PBS. Cells were incubated with the secondary antibody for 1 h at RT in the dark. After further three washes, the coverslips were removed and any excess fluid removed. The coverslips were mounted onto slides using DAPI Fluoromount and allowed to air dry for 1 h in the dark before being fixed in place with an adhesive.
Mesenchymal differentiation and functional histochemical staining
Monolayer differentiation was undertaken using 10 wells of a 24-well plate, 5 wells were used for growth medium (DMEM/10% FBS/1% PS) and 5 for differentiation medium (HyClone AdvanceSTEM osteogenic and adipogenic media; GE Life Sciences). The cells were diluted to a concentration of 2 × 104/mL and 1 mL added to each well. Plates were incubated at 37°C, 5% CO2 until they were 50%–70% confluent at which point they were considered to be at day 0 of the differentiation course. Plates were taken at day 0 as controls. The medium was removed from those plates undergoing differentiation and replaced with 1 mL of either growth (DMEM/10% FBS/1% PS) or differentiation medium. The media were changed twice weekly for 21 days.
Three-dimensional pellet culture was used for chondrogenic differentiation. After cell counting, the cells were resuspended in either chondrogenic medium (HyClone AdvanceSTEM; GE Life Sciences) or DMEM/10% FBS/1% PS at a concentration of 6 × 105 cells/mL and 500 μL pipetted into an individual well of a 96-well v-bottom plate. Cells were pelleted by centrifugation at 800 rpm for 5 min followed by incubation at 37°C, 5% CO2. Three pellets were used for growth medium controls and three for differentiation medium. The medium was changed twice weekly by centrifuging the plates at 800 rpm for 5 min, the medium was aspirated, discarded, and replaced with a fresh medium. After medium changes, the pellets were gently agitated to ensure that they were not adherent.
Osteogenic differentiation was determined by using Alizarin red to form a birefringent complex with calcium deposits. Alizarin red solution was made by adding 2 g Alizarin red S (C.I. 58005) to 100 mL distilled water (dH2O). This was thoroughly mixed and the pH altered to 4.1–4.3 with 10% ammonium hydroxide. The wells were stained with 1 mL Alizarin red solution for ∼30 min at RT to produce red-orange staining with calcium. Excess stain was removed by washing four times with dH2O.
Adipogenic differentiation was assessed using Oil Red O. A stock solution was prepared by mixing 0.7 g Oil Red O (Sigma O-062) with 200 mL isopropanol. This was stirred overnight and then passed through a 0.2-μm filter. The stock solution was stored at 4°C. The working solution was made by adding six parts stock solution to four parts dH2O. This was mixed and left at RT for 20 min before being passed through a 0.2-μm filter. The wells were washed twice with dH2O and then dehydrated with 60% isopropanol for 5 min at RT. The isopropanol was removed and the wells dried without washing. The cells were stained with 1 mL Oil Red O solution for 10 min at RT. Excess stain was removed by washing four times with dH2O.
Chondrogenic differentiation was indicated by Alcian blue staining for proteoglycans. Slides or wells were incubated with acetic acid for 3 min at RT followed by Alcian blue for 30 min at RT. Excess stain was removed using acetic acid and the slides were washed three times with dH2O.
Animal surgery
Sheep were induced with propofol (5–6 mg/kg) and maintained on 2%–3% isoflurane (Abbot Laboratories Ltd.). Vetergesic (Buprenorphine; Reckitt Benckiser Healthcare Ltd.) was used for postoperative analgesia (14 μg/kg) for 3–4 days. Streptacare (procaine penicillin, 20% dihydrostreptomycin; Animalcare Ltd.) was given as prophylactic antibiotics (1 mL/25 kg) for 5–7 days.
The sheep were placed in a supine position, and for unilateral surgery, the contralateral limb was secured away from the surgical field. The wool was sheared and the skin prepared with a Betadine/alcohol solution. The hoof was placed in a sterile bag and the limb draped.
The stifle joints were opened using a midline incision followed by a medial parapatellar arthrotomy. The infrapatellar fat pad (IFP) was excised with a scalpel and placed in DMEM/10% FBS/1% PS for transport to the laboratory. Thorough hemostasis was carried out using bipolar diathermy. The medial femoral condyle (MFC) was exposed by flexion of the stifle joint and a circular lesion made in the cartilage using a punch biopsy. The cartilage was then excised using a 4 mm ring curette.
The arthrotomy was closed with a continuous 1 Vicryl suture and the subcutaneous fat with a running 2/0 Vicryl suture. The skin was closed with deep dermal and subcuticular 2/0 Vicryl sutures. The wound was cleaned with Betadine spray and left without a dressing.
Lentivirus transfection
An emerald green fluorescent protein (eGFP) lentivirus under the control of a cytomegalovirus promoter was used. The virus was stored at −80°C and the final virus titer was calculated to be 4.35 × 106 infectious units/μL. PSCs were transfected at ∼80% confluence. The culture medium was aspirated and discarded and the cells washed twice in PBS. Serum-free medium (5 mL) was mixed with 1 μL virus for every 0.8–1.0 × 106 cells, giving a multiplicity of infection of approximately five. The cells were incubated at 37°C for 4–5 h. DMEM supplemented with 20% FBS/1% PS (5 mL) was added and the cells incubated overnight. The following morning the cells were washed twice in PBS and the medium replaced.
Cell loading
The cell-loaded collagen membranes were prepared using two separate methods to duplicate techniques currently used in clinical practice. In both techniques, cells were grown ex vivo, trypsinized, pelleted, and resuspended in 1 mL DMEM for counting.
The first method mimicked Genzyme's method of delivering the cells for surgery preseeded on the membrane. This was done by pipetting the cells onto the Chondro-Gide membrane at a density of 1 × 106 cells per cm2. The membranes were left to incubate in a 12-well plate for 4 h before adding additional culture media. The cell membranes were left for 48–72 h before reinsertion. During surgery, the cartilage defect was debrided back to a stable rim and a template made to fill the defect. The membrane was then cut to shape and glued into place using Tisseel fibrin glue (Baxter).
The second method was based on TiGenix ChondroCelect procedure. Cells were transported to surgery in 100 μL phenol-free DMEM as this is used as the transport medium for matrix-assisted chondrocyte implantation (MACI) membranes provided by Genzyme. The articular cartilage defect was debrided back to a stable rim and the membrane cut to fit the defect. Cells were then pipetted onto the membrane at an approximate density of 1 × 106 cells per cm2. The membrane was then placed into the defect and the edges sealed with Tisseel fibrin glue.
In situ crosslinking of cells encapsulated in a hydrogel was performed using the HyStem®-C Hydrogel Kit (GS313; Esi Bio) as per the manufacturer's instructions. Approximately 1 × 106 cells were re-suspended in 100 μL inactivated hydrogel, during surgery the hydrogel the cross-linker was added. Once the viscosity was high enough to allow the hydrogel to remain in the defect, it was pipetted in slowly and left for at least 30 min to crosslink in situ.
Ex vivo cell tracking
A Zeiss Axioskop 510 (Carl Zeiss Ltd., Welwyn Garden City, United Kingdom) Confocal Laser Scanning Microscope (CLSM) was used to image cell cytoplasm [5-chloromethylfluorescein diacetate (CMFDA)] of viable cells and the cell nuclei of nonviable cells [propidium iodide (PI)] on MACI membranes. The × 10 objective was used to obtain images for cell counting and the × 63 objective for higher magnification to observe cell phenotype. The CMFDA was excited using an Argon laser at 488 nm and the emission detected using a bandpass filter at 525/50 nm. PI was excited using a helium–neon laser at 543 nm and detected with a long-pass filter at >560 nm. An average of two readings was used for each pixel to reduce background signal. The gain and amplifier settings were adjusted to allow the maximal use of the detectable range. Axial images were collected throughout the entire fluorescently visible z-axis at intervals of 5.7 μm for the × 10 objective and 0.75 μm for the × 63 objective.
eGFP-transfected cells were imaged using the same acquisition protocol used for CMFDA with excitation at 488 nm and a peak emission at 507 nm (525/50 nm bandpass filter). For the hydrogel encapsulated cells, optical sections were taken at 5.7 μm throughout the z-axis. The implanted cells were identified using the × 10 objective and imaged using optical slices every 5.7 μm. Higher magnification images acquired optical sections 0.75 μm apart.
Results
Histology and immunohistochemistry of multiple ovine tissues
Histology of ovine adipose sections demonstrated the same microscopic structure as human tissue with the majority of the tissue being pale where the processing has removed the adipose tissue leaving a mesh of cell membranes interspersed with blood vessels of varying calibers, from capillaries to arterioles (Fig. 1A). Sections from skeletal muscle had blood vessels running in between the myofibers (Fig. 1B). A section taken through the synovial border of the IFP demonstrated a large concentration of cells and blood vessels adjacent to the synovium with adipose tissue lying underneath (Fig. 1C) with the same structure as subcutaneous fat (Fig. 1A).
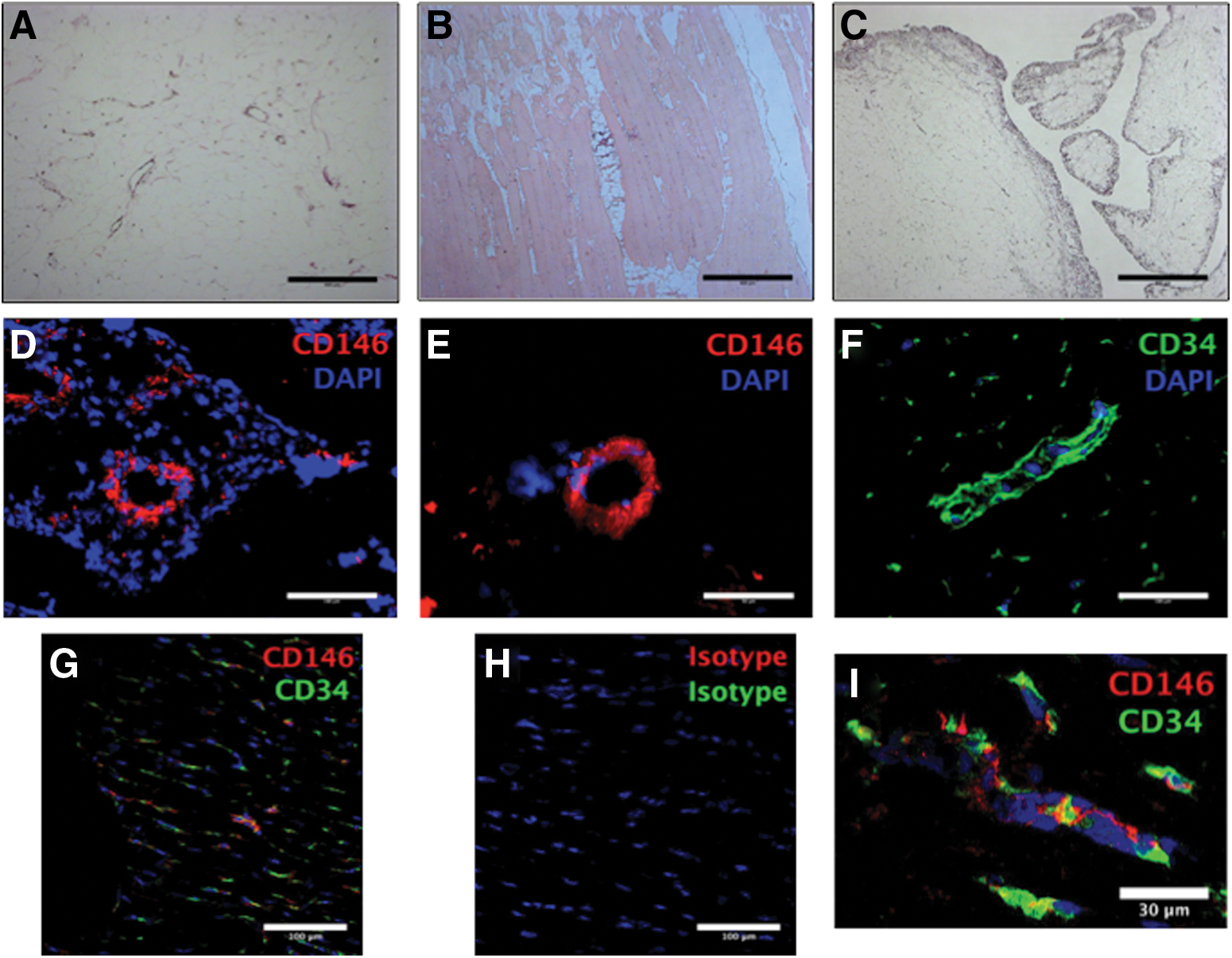
Histology and immunohistochemistry of ovine tissue sections. Subcutaneous fat
Anti-human CD146 antibodies positively identified small-caliber blood vessels in multiple tissues. Sections from placenta and myocardium are shown in Fig. 1D and E, respectively. An anti-ovine CD34 antibody was confirmed to stain in a perivascular location (Fig. 1F) and this was confirmed to share a perivascular location with CD146 (Fig. 1G, I). Isotype controls were used for all sections to determine the presence of any false-positive staining; the isotype control for the dual stains in Fig. 1G and I is shown in Fig. 1H. Multiple anti-human and anti-ovine CD31 antibodies were used to try and identify an endothelial marker but no positive staining was seen compared to isotype controls (data not shown).
Reactivity and cross-reactivity of antibodies to identify oPSCs
Flow cytometry antibodies raised against human and ovine tissues were tested for their cross-reactivity and reactivity with freshly isolated ovine SVF (Table 1). The majority of the antibodies did not demonstrate a positive signal with ovine cells, including the anti-ovine anti-CD31 antibody (data not shown). Figure 2 shows the results for the anti-human CD146 antibody as well as the anti-ovine CD34, CD45, and PDGFR-β antibodies. While there was not a large peak when the staining with the anti-human CD146 antibody and isotype was overlaid (Fig. 2A, left-hand plot), the density plots clearly identified a population of CD146+ cells when compared to the isotype control. Similar results were seen for the anti-CD34 and anti-PDGFR-β antibodies. Only the anti-CD45 antibody demonstrated a large peak when the histograms were overlaid. These data meant that oPSCs could be identified and hematopoietic stem cells excluded.
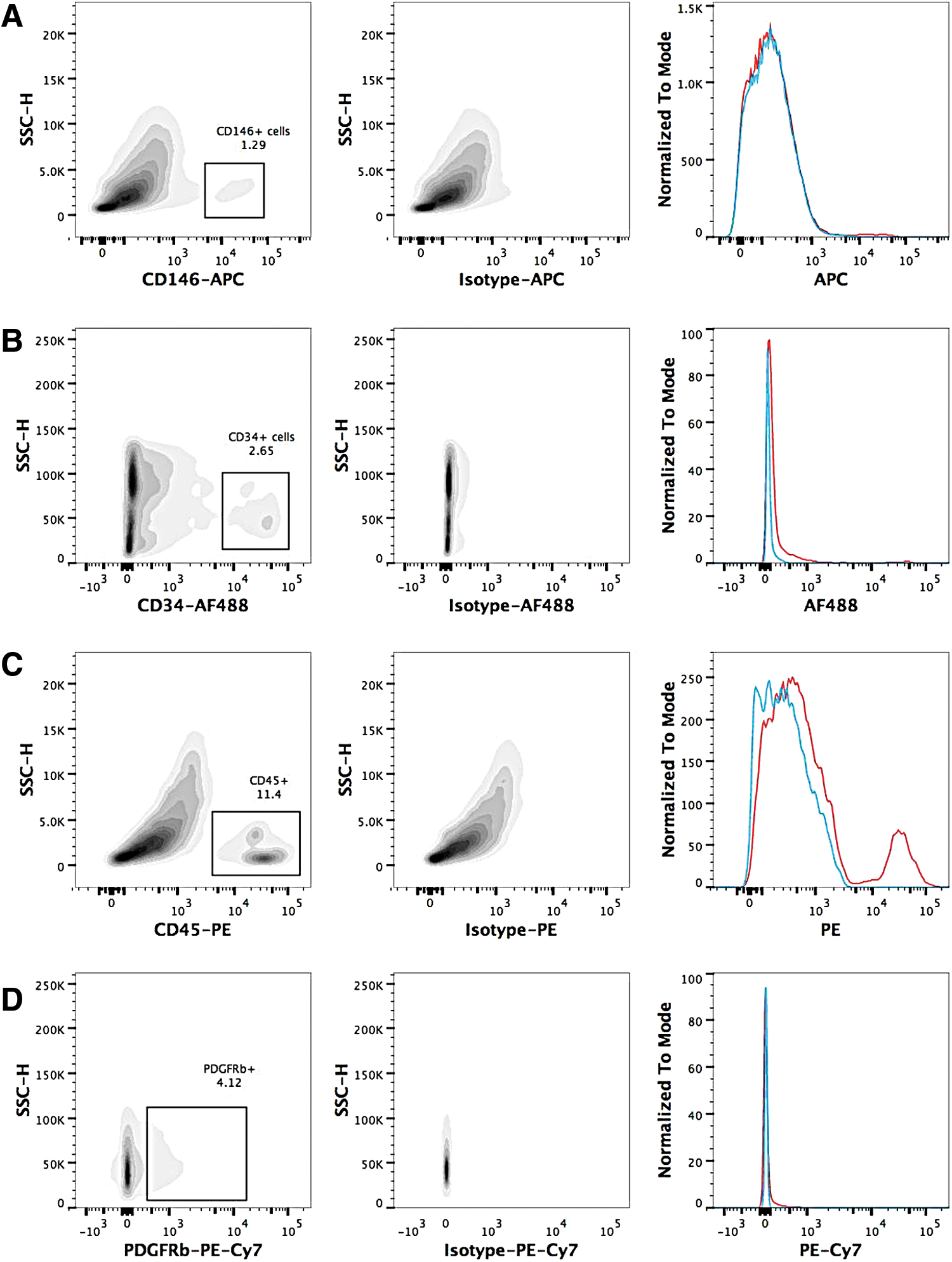
Flow cytometry analysis of single antibody staining of ovine stromal vascular fraction following single-cell selection and exclusion of dead cells. Antibody staining is shown in the left-hand side density plots for CD146
Details of the antigen clone as well as the specific isotypes used are also detailed.
Ovine SVF was stained with anti-human CD146 as well as anti-ovine CD34 and CD45 antibodies with DAPI used to exclude dead cells (Fig. 3A). Full minus one controls were used to determine the level of true staining (Fig. 3A, bottom, middle, and right plots). Distinct cell populations were identified as pericytes (CD45−CD146+CD34−) and adventitial cells (CD45−CD146−CD34+) in the bottom left plot in Fig. 3A. Ovine pericytes and adventitial cells collected using FACS were reanalyzed immediately after cell sorting. The pericytes are shown in the upper plots in Fig. 3B and demonstrate that 84% of the reanalyzed events were the same cell size and 100% of these events were CD146+ indicating a pure cell sort. More than 90% of the adventitial cells were in the cell selection gate with 100% of these cells being CD34+. The events that were outside of the cell selection gate are likely to represent debris from nonviable cells. It was impossible to determine the true level of cell death as one cell would lead to multiple pieces of debris in an unpredictable manner.

FACS of oPSCs. The FACS plots in
As the articular cartilage repair model was to use the IFP, the variability in the number of pericytes that could be obtained was investigated using the infrapatellar fat from four separate stifle joints (two animals). The FACS plots used to select the pericytes and adventitial cells are shown in Fig. 3C. While there was variability in the levels of staining, pericytes and adventitial cells were identified and isolated from each of the specimens.
In vitro characterization of oPSCs, including mesenchymal differentiation
Ovine pericytes and adventitial cells in monolayer culture are shown in Fig. 4A and B, respectively. Pericytes from ovine bone marrow were analyzed using FACS and were CD34−CD45−CD146+. The histogram in Fig. 4C for the anti-human CD146 antibody demonstrated a large CD146+ population (red) with clear staining compared to the isotype control (blue). Cultured ovine pericytes from the IFP did not demonstrate CD146 staining on FACS but did with immunocytochemistry compared to controls (Fig. 4D, E).
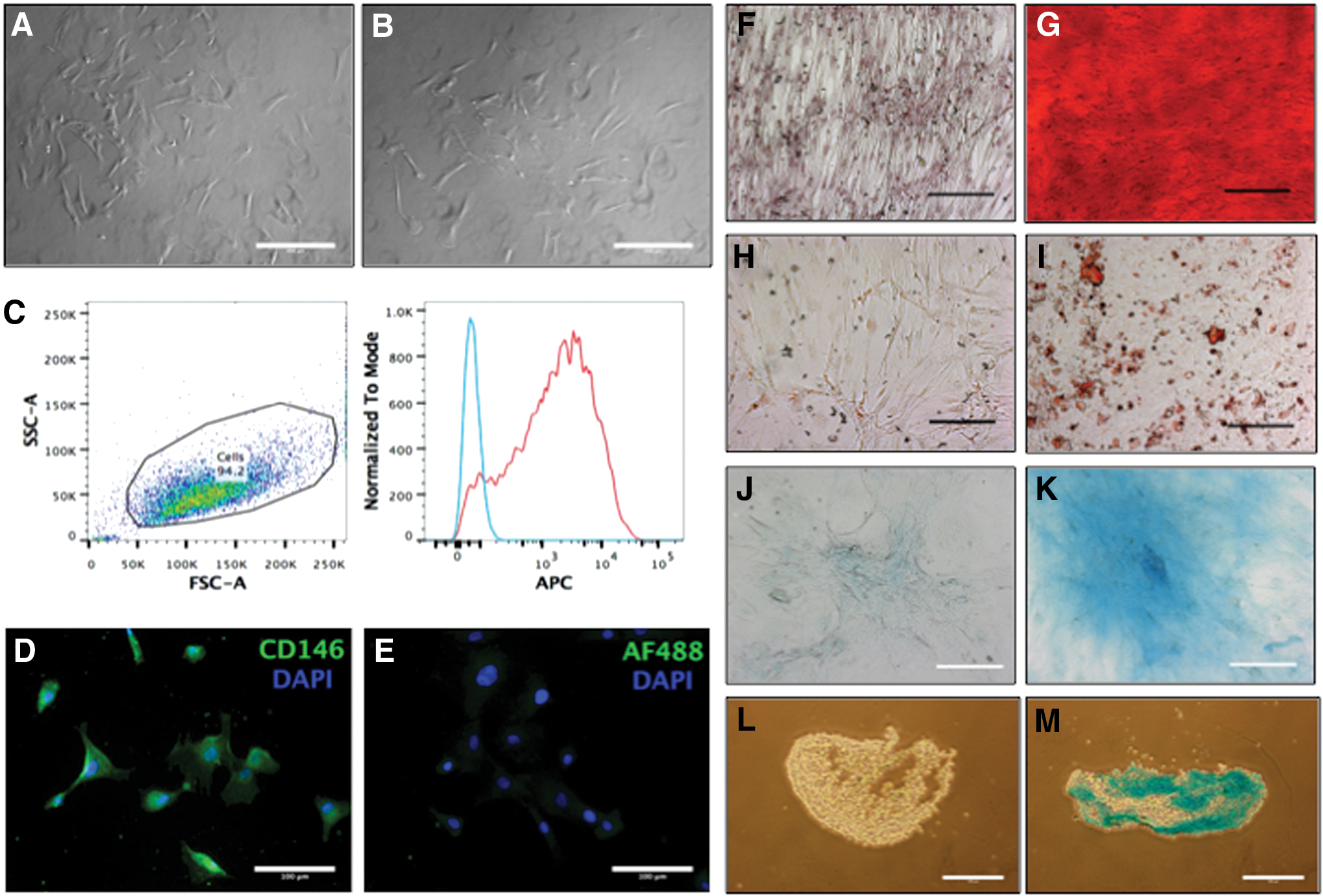
Ovine pericytes
Ovine pericytes and adventitial cells both demonstrated the ability of osteogenic, adipogenic, and chondrogenic differentiation; the differentiation of ovine pericytes is shown in Fig. 4. After 21 days in differentiation media, the pericytes stained positively for Alizarin red (calcium deposition with osteogenic medium, Fig. 4G), Oil Red O (intracellular fat vacuoles, adipogenic medium, Fig. 4I), and Alcian blue (proteoglycans with chondrogenic medium Fig. 4J, L). Each of the stains was positive compared to cells from the growth medium controls (Fig. 4F, H, I, K).
Autologous oPSCs remain in situ following insertion using a hydrogel
Ovine adventitial cells transfected with an eGFP virus demonstrated some positive signal on day 1 post-transfection (Fig. 5A), which appeared significantly increased by day 6 post-transfection. An aliquot of the transfected cells was analyzed using flow cytometry and demonstrated an 88% transfection rate compared to nontransfected cells at day 6.

GFP transfection of ovine perivascular stem cells increased from day 1
Nontransfected cells were loaded onto a collagen I/III membrane and stained with CMFDA and PI. These cells were then imaged to determine that the loading protocol worked and that there were viable cells attached to the membrane before surgical implantation. Figure 4D and E show the cells attached to the membrane and demonstrate that almost all of the attached cells were viable. Transfected cells were successfully loaded into a hyaluronic acid-based hydrogel at concentrations from 1 to 20 × 104 cells/μL. Figure 5F is an extended focus view and demonstrates that the cells were not over crowded. Figure 5G shows the GFP-emitting virus in a single cell staining the cytoplasm clearly.
Four weeks following surgical implantation, the sheep were sacrificed and the stifle joints excised. The cartilage did not receive any fluorescent staining, and confocal laser scanning microscopy was able to find GFP-emitting cells in the base of the defect treated with the cell-loaded hydrogel (Fig. 5H). A single cell with processes that did not look like a differentiated chondrocyte is shown at higher magnification in Fig. 5I. No GFP-emitting cells were found at the base of the defect treated with the cell-loaded collagen membranes. Cross sections of both defects stained with Masson's trichrome did not demonstrate any repair tissue either with or without the traced PSCs.
Discussion
This work has successfully managed to identify and prospectively identify oPSCs using a similar set of perivascular markers to those used in humans [1,30].
As there was a lack of anti-vine specific antibodies available to identify and isolate oPSCs, the main challenge in developing an autologous model was the identification of antibodies to accurately select PSCs, this included determining if antibodies raised against another species (such as anti-human antibodies) would cross-react appropriately with ovine cells. Immunohistochemistry demonstrated that anti-human CD146 antibodies cross-reacted with ovine tissue and that the antigens had a perivascular location (Fig. 1D–I). The cross-reactivity of the CD146 antibody was validated further when the cultured pericytes from bone marrow were analyzed with flow cytometry (Fig. 3C) and immunocytochemistry (Fig. 3D, E). An ovine-specific antibody was required to identify CD34 accurately [31]. Despite there being antibodies for ovine endothelial cell markers, this research was unable to show that the anti-CD31 antibody worked. The anti-CD31 antibody was described by Pintado et al. [32], who did not describe the use of any negative controls in their work; the fluorescence reported as representing a positive signal could therefore just represent increased voltage or autofluorescence. Zannettino et al. used the same anti-ovine CD31 antibody in their article comparing human and ovine MSCs [33]. All of their results were negative and no positive controls were used to demonstrate that the antibody worked. Their negative results could be true-negatives but without positive controls it could just represent the lack of antibody reactivity.
The lack of an endothelial marker in the antibody panel used to sort oPSCs meant that endothelial cells would also have been collected. This was a particular problem for adventitial cells; our previous data demonstrated that anti-CD34 antibodies stained a proportion of endothelial cells as well (Hindle et al., unpublished data). Despite PDGFR-β not being used commonly as a marker to FACS sorted human PSCs, this research suggests that it could be a useful marker for the isolation of oPSCs. All of the CD146+ cells in one sort were PDGFR-β+. The ovine antibody was able to identify a larger number of oPSCs than the anti-human CD146 antibody. These cells would need to be isolated and compared to their human counterparts before using them further. If the anti-ovine PDGFR-β antibody were used, it would mean that the entire antibody panel would have been raised specifically to react with ovine cells.
oPSCs were isolated from bone marrow, subcutaneous fat and the IFP. These data demonstrated that these were all feasible cell sources for future experiments. Ovine pericytes and adventitial cells demonstrated mesenchymal potential with osteogenic, chondrogenic, and adipogenic differentiation. The cultured pericytes from bone marrow expressed CD146 and the ones from the IFP did not, this was the same as the pericytes from the human IFPs [4].
In this article, we have concentrated on the use of CD146 and CD34 as markers to identify PSCs as this is directly comparable to the methods used to isolate human PSCs that have demonstrated improved osteogenic and chondrogenic potential compared to unsorted cells and MSCs [3,4]. CD146 has also been indicated as a marker for a subpopulation of chondroprogenitor cells in osteoarthritis [34], and CD146+ cells have demonstrated increased therapeutic potential compared to CD146− cells in a model of murine arthritis [35]. Other authors have investigated the use of different markers, including Grem1+ and LepR+ cells [36,37]. There is currently a lack of comparative data on these subsets of cell populations.
Finally, and importantly, our data demonstrated that the ovine model for PSCs can be used for autologous articular cartilage repair. Ovine pericytes and adventitial cells were isolated from the IFP; no problems were encountered with cell isolation or proliferation.
Insertion methods mimicking both of the commercially available MACI and ChondroCelect products were used. CLSM confirmed attachment in the MACI group with good cell viability (Fig. 5D, E). No difference was noted with surgical preparation time or insertion. As the membrane was dry and stiff before applying the cells with the ChondroCelect method, the membrane was easier to cut to shape. The other insertion technique was using a hydrogel that crosslinked in situ, previously been described by Liu et al. for osteochondral defects [38].
Hydrogels based on hyaluronic acid and collagen I have been used in rabbits [38] and sheep [15] to deliver bone marrow-derived MSCs. The hydrogel was easy to use and was able to insert a greater number of cells than the collagen membrane. The main challenge using the hydrogel was judging the viscosity so that the hydrogel remained in the defect when inserted but was not too viscous that it could not be inserted. The 8-mm circular defects used had a surface area of 50 mm2, as the density of the cells on the collagen membrane was ∼1 × 106 cells/cm2; this meant that ∼5 × 105 cells would be inserted when the collagen membranes were used. The sheep's cartilage was ∼1 mm thick to the calcified cartilage, giving a defect volume of 50 mm3. The cells in the hydrogel were loaded at 20,000 cells/μL. This means about 1 × 106cells were inserted using the hydrogel. There was even potential to load more cells if research found this to be beneficial, this extra capacity was not demonstrated with the collagen membrane as the cells on an MACI membrane are in a monolayer and already occupy all of the available space [39].
Implanted cells were successfully tracked ex vivo using the GFP virus and CLSM. GFP-emitting cells were identified at the base of the articular cartilage defect 4 weeks after insertion of the hydrogel (Fig. 5H, I), despite no repair tissue being demonstrated on histology (Fig. 5J). The high-magnification image of the single GFP-emitting cell did not have a rounded appearance of a chondrocyte. This might be due to stem cells acting more as trophic mediators [40] rather than engraftment and differentiation or just that 4 weeks is an early time point for seeing articular cartilage repair.
In conclusion, our data have demonstrated that there is now an autologous large animal model for furthering research into PSCs. The ovine model is more likely to be socially acceptable than the current canine model. While we have demonstrated the model's use in articular cartilage repair, this model could be used for tissue engineering and regeneration across multiple organs.
Footnotes
Acknowledgments
This work was made possible by grants from the University of Edinburgh and Tenovus Scotland. B.P. is supported in part by the British Heart Foundation Centre for Vascular Regeneration (RM/13/2/30158).
Author Disclosure Statement
No competing financial interests exist.