
Abstract
Sepsis is a complex process, including a first wave of damage partially due to the body's response to pathogens, followed by a phase of immune cell dysfunction. The efficacy of a pharmacological approach facing a rapidly evolving system implies a perfect timing of administration—this difficulty could explain the recent failure of clinical trials. Mesenchymal stromal cells (MSCs) are usually defined as immunosuppressive and their beneficial effects in preclinical models of acute sepsis have been shown to rely partly on such ability. If nonregulated, this phenotype could be harmful in the immunosuppressed context arising hours after sepsis onset. However, MSCs being environment sensitive, we hypothesized that they could reverse their immunosuppressive properties when confronted with suffering immune cells. Our objective was to evaluate the effect of human MSCs on activated human lymphocytes in an in vitro endotoxemia model. Peripheral blood mononuclear cells (PBMCs) underwent a 24-h lipopolysaccharide (LPS) intoxication and were stimulated with phytohemagglutinin (PHA) in contact with MSCs. MSCs induced a differential effect on lymphocytes depending on PBMC intoxication with LPS. Unintoxicated lymphocytes were highly proliferative with PHA and were inhibited by MSCs, whereas LPS-intoxicated lymphocytes showed a low proliferation rate, but were supported by MSCs, even when monocytes were depleted. These data, highlighting MSC plasticity in their immunomodulatory activity, pave the way for further studies investigating the mechanisms of mutual interactions between MSCs and immune cells in sepsis. Thus, MSCs might be able to fight against both early sepsis-induced hyperinflammatory response and later time points of immune dysfunction.
Introduction
M
Despite this strong rationale, dozens of clinical trials aiming at inhibiting inflammation and immunity effectors have failed these last few years. Clinicians are now considering that these failures may be due to an already prominent immunodepressive context. Consistent with this conceptual shift, immune-activating therapeutics are now being explored not only in the hope to prevent secondary infections but also to participate in inflammation resolution.
The immune system activation triggers humoral, cellular, and neural mechanisms to activate anti-inflammatory responses to limit local and systemic tissue injury. However, this compensatory mechanism contributes to an increased incidence of nosocomial infections and late death [2].
This immune suppression, affecting both innate and adaptive immune systems, is due to early apoptosis and hypofunctionality of different actors of immunity. In particular, lymphocyte dysfunction, also called T cell exhaustion, includes a progressive decrease in their ability to produce cytokines, a loss of proliferative capacity, and a decrease in cytotoxicity. By contrast, regulatory T cells are more resistant to sepsis-induced apoptosis [3]. T lymphocyte number and function are deeply impacted during sepsis and have been proposed as predictive biomarkers of survival. Enhancing T cell survival and function by cytokines such as IL-7 or blockers of the PD-1/PD-L1 axis has been shown to benefit survival in preclinical models of sepsis [4].
Mesenchymal stromal cells (MSCs) have recently been shown to favor survival and to reduce organ dysfunction in preclinical models of sepsis through a variety of mechanisms [5,6]. Their use in the very first hours of induced sepsis apparently led to dampening the overactive immune system. Indeed, MSCs are able to downregulate proinflammatory markers (IL6, TNFα, and IFNγ) and increase anti-inflammatory markers (TGF-β, IL1Rα, and IL10). MSCs can also help in endothelial regeneration, improve bacterial clearance and macrophage phagocytosis, reduce neutrophil apoptosis as well as their tissue infiltration, and secrete antimicrobial peptides (LL37, C-terminal part of cathelicidin) [7]. However, such an early treatment is difficult to achieve in clinical practice, and MSC potential should be evaluated now in more relevant timings.
Does the immunosuppressive potential of MSCs worsen sepsis-associated immunoparalysis? However, the evolution of knowledge on MSCs has shown their high sensitivity to their environment and the ability to modulate their actions paves the way to new therapeutic potential, especially in situations requiring immune activation. Their participation in maintaining the memory T cell niche [8] as well as their acquisition of a secretory profile, including various potential mediators of immune reactivation under several conditions [9], led us to consider their possible use in late sepsis and the possibility to prime them to further enhance this effect.
We therefore undertook in vitro cocultures of MSCs with lymphocytes to assess whether the dogma of MSC immunosuppressive effect could be challenged when confronted with intoxicated lymphocytes toward an immune supporting effect.
We show that MSCs can reverse from a suppressive to supportive effect in the presence of endotoxin-intoxicated lymphocytes. We further show that this immune activating effect may be liable to MSC prestimulation.
Materials and Methods
MSC isolation and culture
Human MSCs were obtained from bone residues of patients who underwent a total hip replacement surgery (HIA Percy, 92140 Clamart, France). They were isolated by plastic adhesion, then amplified up to 80–90% confluence in a medium containing MEMα (Biological Industries) supplemented with 10% fetal calf serum (FCS) (HyClone), then frozen in MEMα + 10% human albumin (LFB Biomedicaments) + 10% dimethylsulfoxide (Sigma), and finally preserved in liquid nitrogen. For experimental purposes, MSCs were thawed and amplified before coculture experiments. We used MSCs from eight donors after 1–3 amplification passages. Samples for isolating the MSCs and the PBMCs were obtained with informed consent and the study received institutional regulatory approval.
MSC priming
MSCs were seeded in a 96-well culture plate (TPP) at 625 or 104 cells per well (MSC:PBMC ratio of 1:160 and 1:10, respectively) in 100 μL of RPMI (Gibco) + 10% FCS and placed at 37°C, 5% CO2. After 2 h of adhesion, the medium was changed according to four different conditions: unprimed MSCs with RPMI + 10% FCS; Pam-primed MSCs with RPMI + 10% FCS + pamidronate (Hospira) at a 5 μM concentration for 16 h; lipopolysaccharide (LPS)-primed MSCs with RPMI + 10% FCS + LPS (Escherichia coli O55: B5; Sigma) at a 10 ng/mL concentration for 1 h; and Pam+LPS-primed MSCs (Fig. 1A). At the end of the stimulation, all wells were washed twice with PBS before coculture.

Experimental design.
Peripheral blood mononuclear cell isolation, intoxication, stimulation, labeling, and coculture with MSCs
Whole blood samples were obtained from healthy volunteers in a blood donation center (CTSA; 92140 Clamart, France) on citrated tubes (BD Vacutainer). Please note that based on our observations, we believe that heparin is not an appropriate anticoagulant because it inhibits the effect of LPS intoxication on lymphocyte proliferation, as previously suggested [10]. The blood sample was numbered with Sysmex XE2100D (Sysmex), divided, and used immediately to make coculture (D0 condition); or placed in dry tubes (BD Vacutainer) with constant stirring for 24 h at 37°C (LPS– condition); or placed in dry tubes with constant stirring for 24 h at 37°C in the presence of 1 μg/mL LPS intoxication to mimic endotoxemia (LPS+ condition). After these 24 h, peripheral blood mononuclear cells (PBMCs) were isolated by separation on 1.077 Ficoll (Pan Biotech) and then suspended in RPMI + 10% FCS + beta-mercaptoethanol 0.1 mM (Sigma). PBMCs (106/mL) were labeled with CFSE 5 μM (Life Technologies) and centrifuged before seeding in coculture medium (Fig. 1A). PBMCs from 10 different donors were used.
PBMCs were cocultured with already adherent MSCs in a 96-well plate 30 min after the end of priming (105 PBMCs per well) in 200 μL of coculture medium containing phytohemagglutinin (PHA) (Sigma) at a 10 μg/mL concentration or a cocktail of CD3/CD28 antibodies at 0.05 μg/mL each (Sanquin). Coculture was kept for 4 days at 37°C (Fig. 1A). PBMCs from 10 different donors were used for PHA stimulation and five additional donors for PHA and CD3/CD28 comparison.
Monocyte depletion
After 24 h at 37°C, with or without the presence of 1 μg/mL LPS, PBMCs from five donors were isolated by separation on 1.077 Ficoll (Pan Biotech) and numbered with Sysmex XE2100D (Sysmex); 10 × 106 cells were used for CD15+ cell depletion by using the magnetic-activated cell sorting cell isolation kit (MACS® CD15 MicroBead Reagent Miltenyi Biotec) according to the manufacturer's instructions. Then, the depleted fraction was used for CD14+ cell depletion by using the magnetic-activated cell sorting cell isolation kit (MACS CD14 MicroBead Reagent Miltenyi Biotec). CD15+ cells were pooled again with depleted PBMCs. This two-step depletion method was used to keep granulocytes that express CD14 when exposed to LPS. At the end of depletion, complete suspensions and depleted suspensions (without monocytes) were numbered using CD45/CD14 (Beckman coulter) and counting beads (Life technologies) and labeling on flow cytometer (Beckman Coulter Navios).
Flow cytometer analysis
After 4 days of coculture, 5 μL of counting beads (Life Technologies) was added to the wells. Cells were incubated with PBS +2% w/v human albumin (LFB Biomedicaments) +0.5% w/v human immunoglobulin (LFB Biomedicaments) for 20 min at 4°C, then labeled with anti-CD3 antibody for 20 min at 4°C, washed with PBS, and incubated in PBS containing DAPI (Sigma) at a 1 mg/mL concentration. When CD3/CD28 stimulation was used instead of PHA, CD3 labeling was replaced by anti-CD4 and anti-CD8 (Beckman Coulter and Ozyme, respectively). The flow cytometer (Beckman Coulter Navios) analysis gated living T cells with side and forward scatter, CD3 or CD4 and CD8-positive binding, and DAPI exclusion. Cytometric assessment of the number of counting beads allowed the calculation of the total number of cells per well. The range of CFSE MFI provided the number of cells per division (Fig. 1C). Four parameters were measured or calculated to represent data (Fig. 1B) [11]:
The final viable T cell number (measured) representing the total number of CD3+/DAPI− cells.
The parent cell number (calculated) corresponding to the minimal number of T cells accounting for the final cell number (ie, the theoretical number of original cells that survived before any proliferation occurred). This calculated number represents T cell survival.
The precursor frequency (calculated) is the proportion of parent cells that had at least one division; it represents the proportion of cells able to proliferate among the original cells that survived.
The proliferation index (calculated) is the ratio between the final viable T cells, which had at least one division (numerator), and the precursor cell number (denominator); it represents the proliferation capacity of reactive cells.
Protein array quantification of cytokines
At the end of the cocultures, the medium of each well was removed and frozen at −40°C until analyses. Supernatant cytokines (GM-CSF, IFNγ, IL-2, IL-7, IL-13, IL-15, IL-17a, and IL-33; Premixed Mag Luminex Performance Assay high sensitivity [Bio-Techne]) were seeded on a Curiox DA-Bead Droparray plate using a minimum sample volume of 5 μL (Clinisciences), as previously described [12]. Microparticles were read using Bio-Plex®200 (Bio-Rad).
Statistical analysis
Data are presented as median (interquartile range [3d quartile–1st quartile]) or bootstrapped 95% confidence intervals. Statistical comparisons between groups on single quantitative variables were run as follows:
Resampling tests were used for group versus group comparisons, pairing on each level of MSC: PBMC–donor interaction. When multiple comparisons were used inside a single experiment, P-values were corrected using the Holm method.
The significance threshold to accept a difference for a test or group of tests was 0.05, and tests were bilateral.
Tests and graphical representations were done under RStudio (v.0.98.1080) running R (3.1.1). The used packages were stats, coin, lmPerm, and multcomp for tests and ggplot2 for graphics.
Results
MSCs inhibit proliferation of unintoxicated T cells
We first assessed the validity of our functional test. We showed (Fig. 2) that MSCs strongly inhibited proliferation of T lymphocytes used immediately after isolation without any LPS intoxication: the presence of MSCs reduced the final viable T cell number in comparison with the condition without MSCs (45420 [6570] vs. 161700 [32100], P < 0.001). This was associated with a decrease in the proliferation index (5.74 [2.13] vs. 15.04 [0.90], P < 0.001) and the precursor frequency (50.2% [4.0] vs. 72.6% [2.9], P < 0.001) along with a moderate decrease in the survival factor (number of parents 12110 [4850] vs. 14770 [4310], P < 0.05).
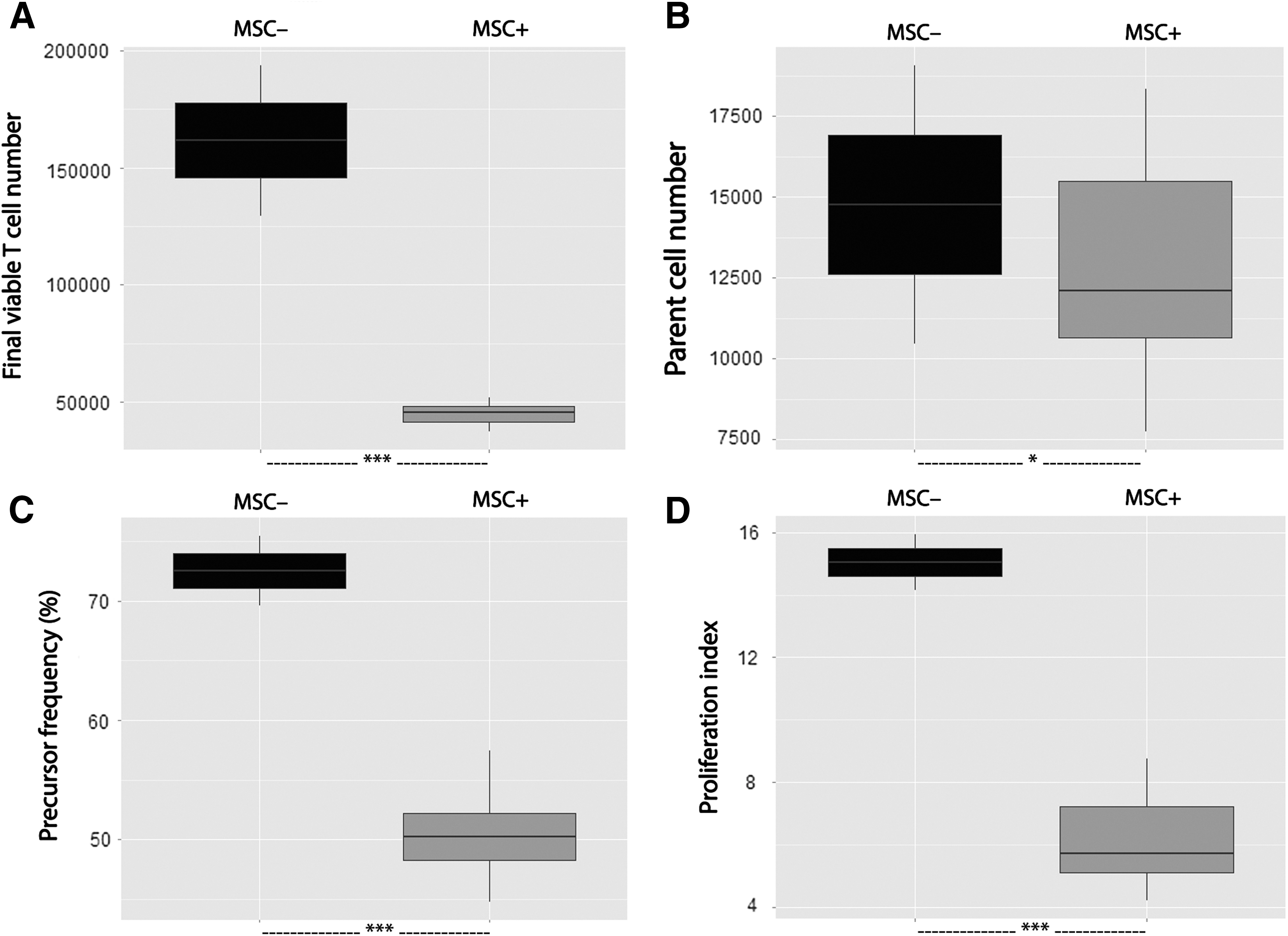
Healthy T cell proliferation was inhibited by MSCs. MSCs from eight different donors were seeded in a 96-well culture plate (104 MSCs/well). CFSE-labeled PBMCs isolated from whole blood samples of two healthy donors were cultured alone or cocultured with MSCs in a PHA-containing medium (105 PBMCs/well, ie, an MSC:PBMC ratio of 1:10). After a 4-day-long culture, the final viable T cell number
LPS intoxication alters T cell survival and proliferative capacity
We then evaluated the effect of LPS on PBMCs without MSCs (Fig. 3). The 24-h LPS preintoxication of PBMCs reduced the number of live T cells at the end of the PHA-mediated proliferation test compared with the condition without LPS (39830 [12100] vs. 163900 [70300], P < 0.001). This reduction was associated with a decrease of T cell survival (number of parents 21380 [6260] vs. 31780 [11300], P < 0.05) and a decrease in the proportion of precursor cells (precursor frequency 30.3% [15.2] vs. 58.6% [10.5], P < 0.001) and in their ability to proliferate (proliferation index 3.55 [0.96] vs. 6.94 [1.31], P < 0.001).
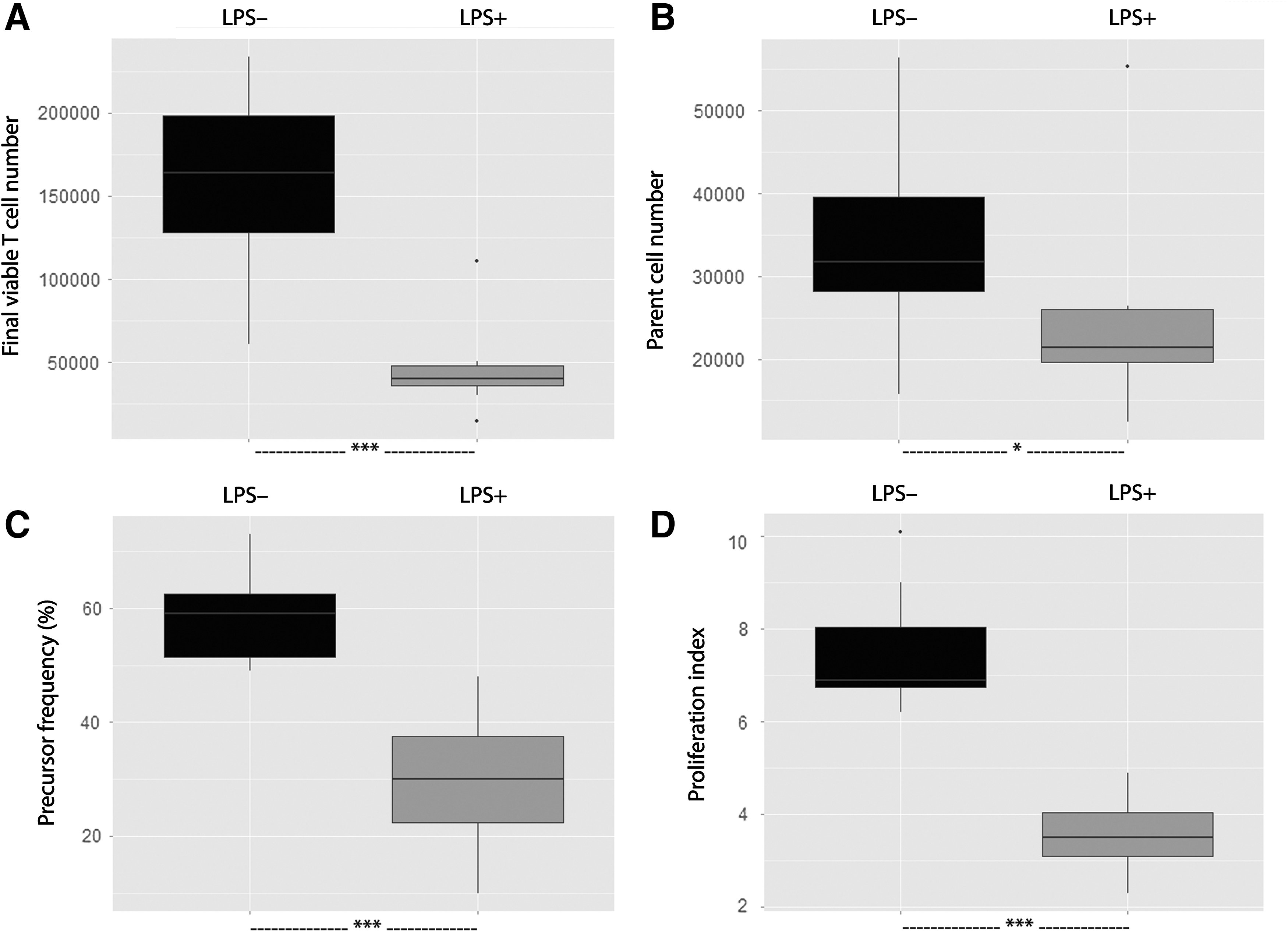
T cell proliferation and survival were altered by LPS intoxication. Whole blood samples were obtained from eight healthy donors and incubated for 24 h at 37°C with or without LPS (1 μg/mL). CFSE-labeled PBMCs isolated from these samples were cultured in a 96-well culture plate (105 PBMCs/well) in a PHA-containing medium. After a 4-day-long culture, the final viable T cell number
MSCs improve survival capacity of LPS-altered T cells
The same protocol was used in the presence of MSCs to evaluate their impact on survival and function of these LPS-altered T cells (Fig. 4). The presence of MSCs in coculture with LPS-intoxicated PBMCs at the highest 1:10 ratio resulted in an increased number of viable T cells on day 4 (77310 [41980] vs. 39830 [12100], P < 0.01), an increase in parent cell number (39850 [8800] vs. 21380 [6260], P < 0.05), and an increase in precursor frequency (38.1% [18.2] vs. 30.3% [15.2], P < 0.05), but no significant increase in T cell proliferation index (3.86 [1.54] vs. 3.55 [0.96], NS). At the lowest 1:160 ratio, no significant difference was noted for these 4 criteria (final viable T cell number 46030 [16550] vs. 39830 [12100], NS; parent cell number 25000 [4710] vs. 21380 [6260], NS; precursor frequency 30.3% [19.2] vs. 30.3% [15.2], NS; and T cell proliferation index 3.26 [0.82] vs. 3.55 [0.96], NS).
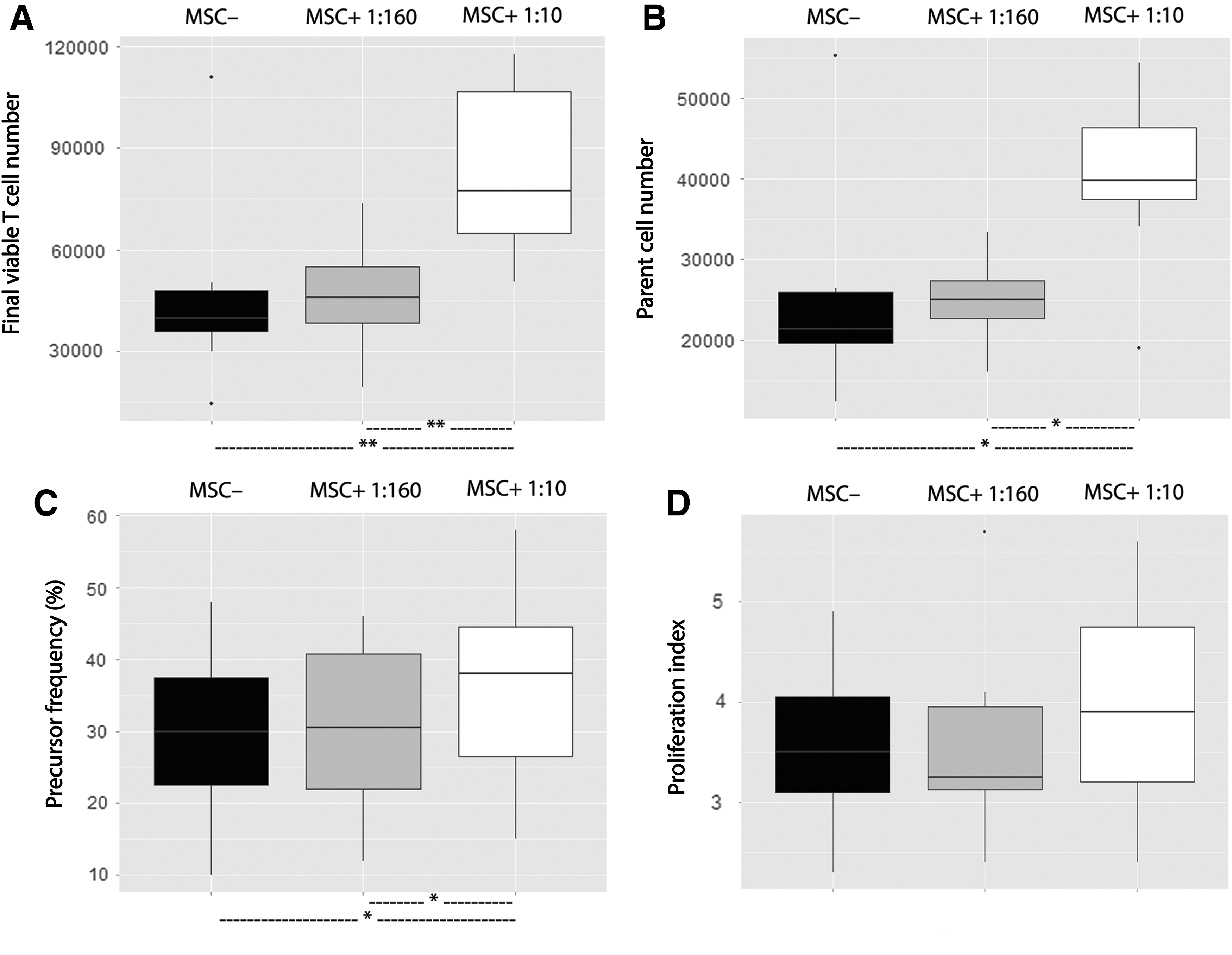
Proliferation and survival of LPS-altered T cells were improved by MSCs at an MSC:PBMC ratio of 1:10, but not 1:160. MSCs from eight donors were seeded either at 625 or 104 MSCs/well. Whole blood samples obtained from eight healthy donors were incubated for 24 h at 37°C with or without LPS (1 μg/mL). CFSE-labeled PBMCs isolated from these samples were cultured alone or cocultured with MSCs in a PHA-containing medium (105 PBMCs/well, ie, an MSC:PBMC ratio of either 1:160 or 1:10). After a 4-day-long culture, the final viable T cell number
The effect of MSCs on T cell functions depends on PBMC LPS intoxication status
We wanted to highlight a differential effect of MSCs on T cells depending on PBMC condition (Fig. 5A–D). When PBMCs were cultured without MSCs, the number of final viable T cells, precursor frequency, and proliferation index significantly decreased when PHA stimulation started 24 h after sampling (LPS–), compared with the condition where PHA stimulation started immediately after sampling (D0), and further decreased when these first 24 h took place with endotoxin (LPS +). The effect of MSCs was assessed in three conditions (D0, LPS−, and LPS+) by the ratio of the final viable T cell number with or without addition of MSCs (a ratio of 1 implying no effect of MSCs). We showed (Fig. 5E) that MSCs had an inhibitory effect on D0 T cells, a less inhibitory effect on LPS − T cells, and a stimulating effect on LPS + T cells (with/without MSC ratio 0.271 [0.087], 0.854 [0.207], and 1.675 [0.985], P < 0.05). This might suggest that MSCs exhibit a differential effect according to the status of T cells: MSCs rather exert an inhibitory activity in contact with highly proliferative cells (D0) than a stimulating activity in the presence of low proliferative cells (LPS+).
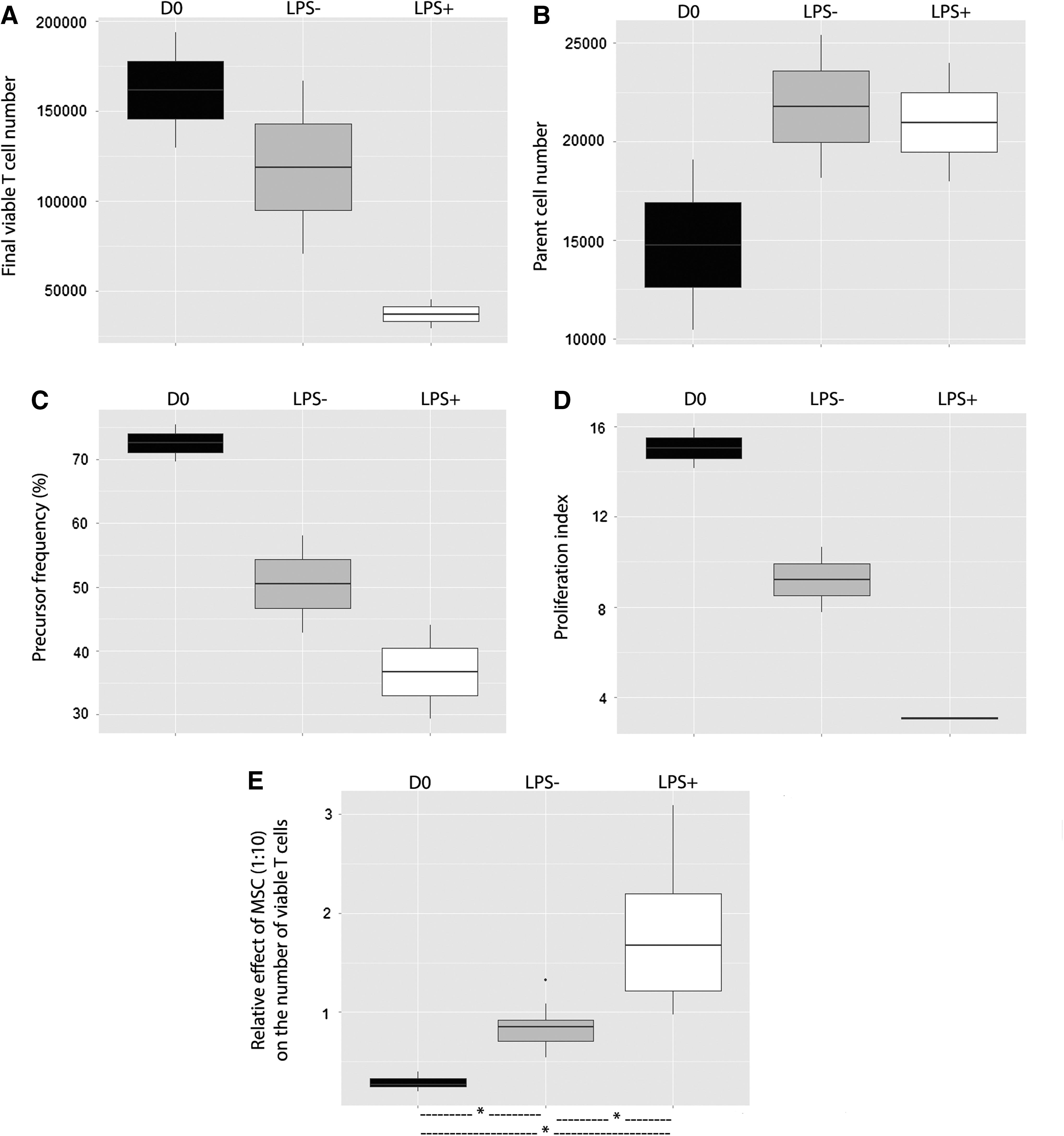
Inhibitory or stimulatory effect of MSCs was, respectively, correlated with high or low proliferative baseline activity of T cells. MSCs from eight donors were seeded at 104 MSCs/well. Whole blood samples obtained from two healthy donors were immediately used (D0) or incubated for 24 h at 37°C without LPS (LPS−) or with LPS (1 μg/mL) (LPS+). CFSE-labeled PBMCs isolated from these samples were cultured alone or cocultured with MSCs in a PHA-containing medium (105 PBMCs/well, ie, an MSC:PBMC ratio of 1:10). After a 4-day-long culture, the final viable T cell number
MSCs increased the expression of IFNγ when PBMCs were intoxicated with LPS
Among the cytokines potentially participating in the effect of MSCs on lymphocyte proliferation, IL-33 and IL-7 were under the limit of detection. IL-2 and IL-15 were present in small amounts (<10 pg/mL) and their expression did not appear to be dependent on the presence of MSCs (Supplementary Fig. S1; Supplementary Data are available online at
MSCs had a supportive effect on T cell proliferation stimulated by CD3/CD28
As PHA is an unspecific way of activating lymphocytes, we performed a more specific T cell stimulation with CD3/CD28 agonist antibodies (Fig. 6). Using the same protocol at the highest MSC:PBMC ratio of 1:10, results were similar: the presence of MSCs in coculture with LPS-intoxicated T cells was associated with an increase in the number of viable T cells on day 4 (15123 [8466] vs. 8882 [4301], P < 0.05), in parent cell number (7455 [5384] vs. 5177 [4739], P < 0.05); and a trend toward an increase in proliferation factors (precursor frequency 26.6% [23.8] vs. 7.1% [8.1], NS; T cell proliferation index (3.74 [3.15] vs. 2.72 [2.72], NS).
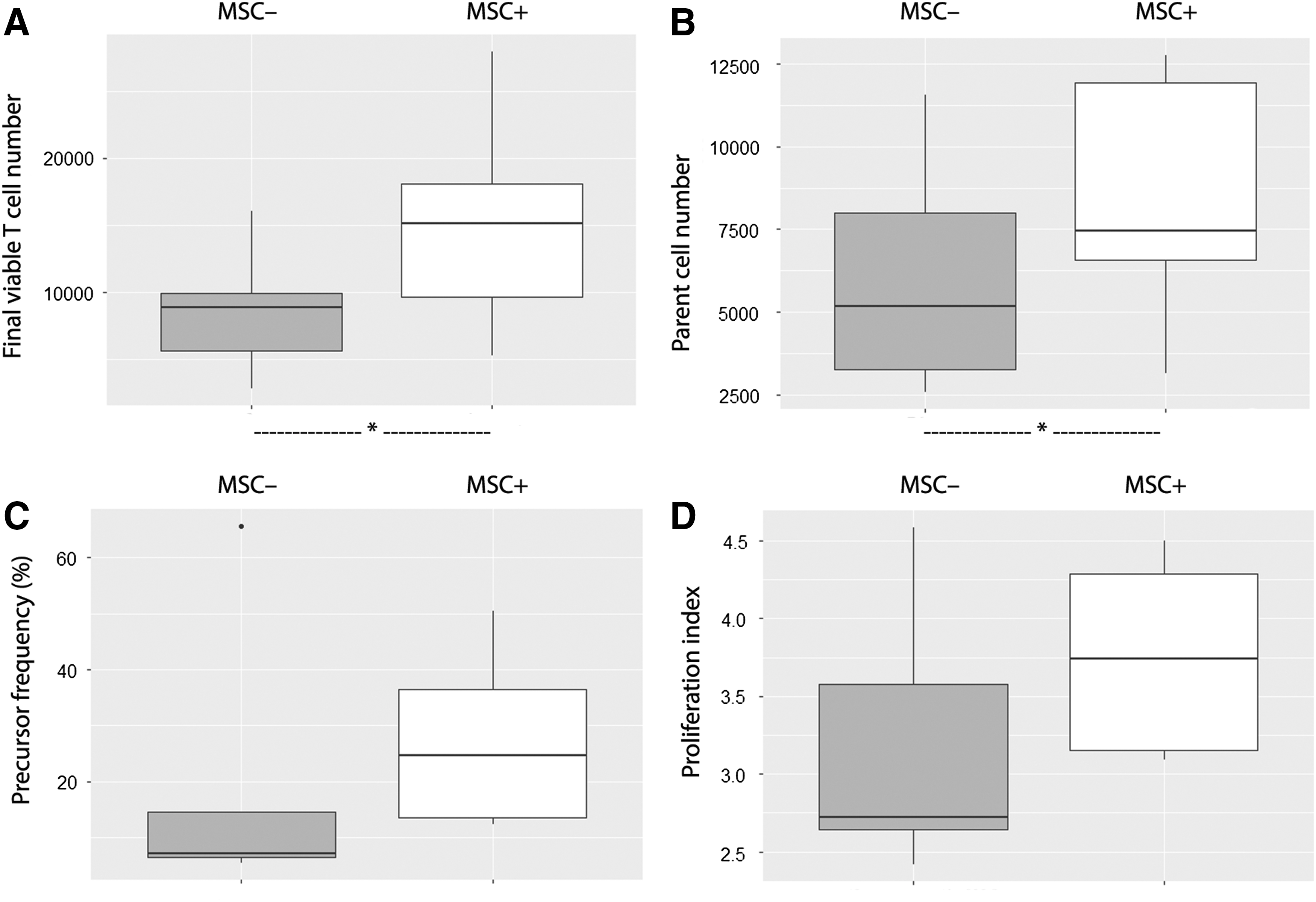
Supportive effect of MSCs on LPS-intoxicated T cells using CD3/CD28 stimulation MSCs from one donor were seeded at 104 MSCs/well. Whole blood samples obtained from five healthy donors were incubated for 24 h at 37°C with LPS (1 μg/mL). CFSE-labeled PBMCs isolated from these samples were cultured alone or cocultured with MSCs in anti-CD3 + anti-CD28-containing medium (105 PBMCs/well, ie, an MSC:PBMC ratio of 1:10). After a 4-day-long culture, the final viable T cell number
CD4 and CD8 cells were equally supported by MSCs
We looked if CD4+ and CD8+ subpopulations were differentially affected by MSCs (Fig. 7). Using CD3/CD28 stimulation, the final viable T cell number seemed lower in these two populations after LPS intoxication (CD4+ cells: 3494 [4213] vs. 5883 [2669], NS; CD8+ cells: 2004 [1743] vs. 14732 [11055], NS) with a significant decrease in the CD8/CD4 ratio (0.57 vs. 2.44, P < 0.05). However, the presence of MSCs increased final viable CD4+ and CD8+ cell numbers (CD4+ cells: 5663 [3969] vs. 3494 [4213], P < 0.05; CD8+ cells: 4780 [3840] vs. 2004 [1743], NS) without significant change in the CD8/CD4 ratio (0.72 vs. 0.57, NS).
MSC effect was not different in CD4 ± and CD8 ± subpopulations. MSCs from one donor were seeded at 104 MSCs/well. Whole blood samples obtained from five healthy donors were incubated for 24 h at 37°C with or without LPS (1 μg/mL). CFSE-labeled PBMCs isolated from these samples were cultured alone or cocultured with MSCs in anti-CD3 + anti-CD28-containing medium (105 PBMCs/well, ie, an MSC:PBMC ratio of 1:10). After a 4-day-long culture, the final viable T cell number was calculated. Histograms represent medians with 95% confidence interval. *P < 0.05.
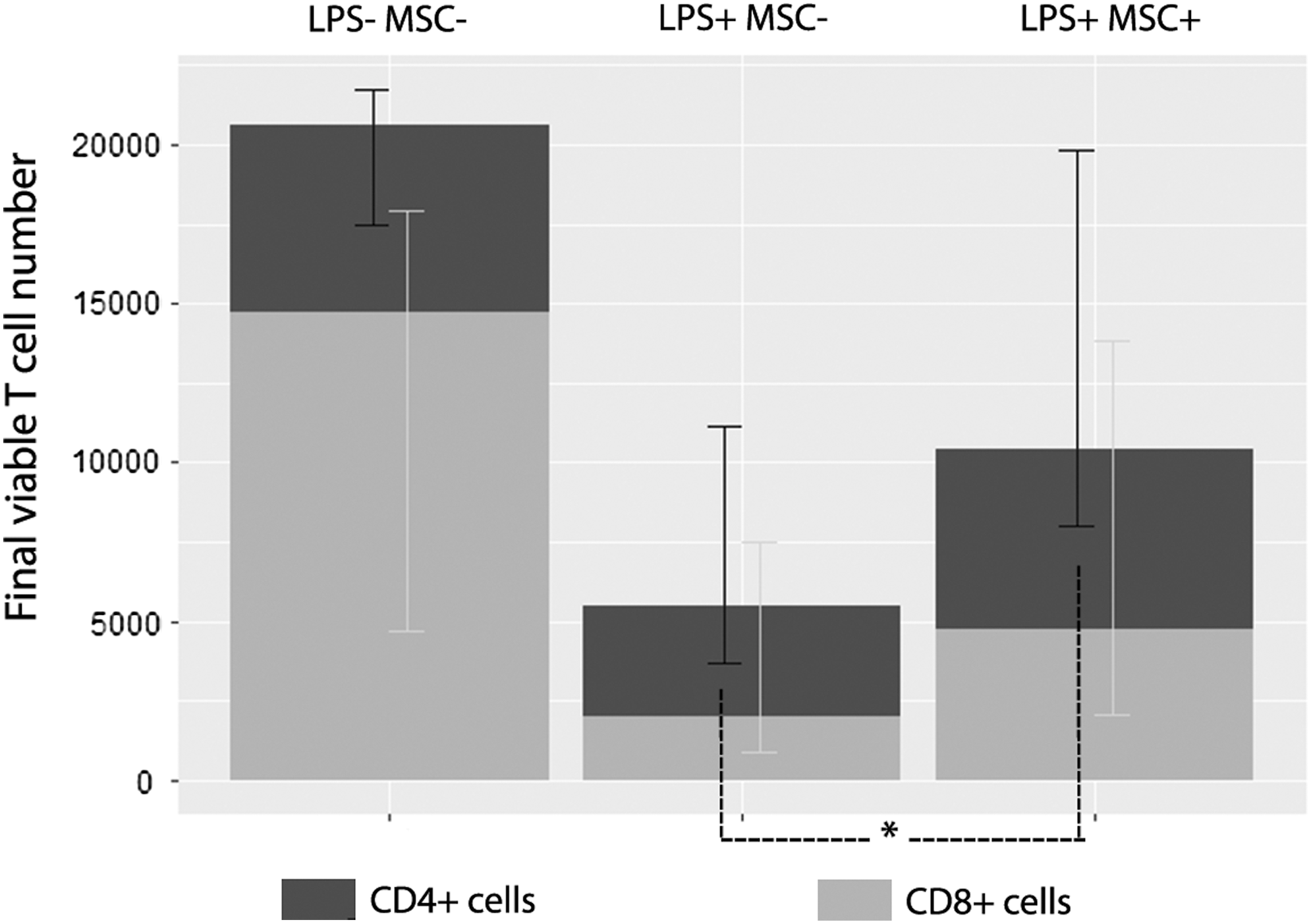
MSCs were still able to support intoxicated lymphocytes in the absence of monocytes
To assess the extent of the effect of monocytes on MSC supportive effect, we depleted CD14+ cells of intoxicated PBMCs before PHA stimulation (Fig. 8). This depletion was associated with a trend toward a decrease in the four parameters: final viable T cell number (7820 [5376] vs. 18762 [18750], NS), parent cell number (4766 [2528] vs. 8250 [8075], NS), precursor frequency (24.3% [8.0] vs. 42.1% [9.0], P < 0.05), and proliferation index (3.39 [0.32] vs. 4.26 [3.39], NS). Notably, adding back depleted CD14+ T cells did not result in a decrease of final viable T cells (data not shown), excluding the implication of magnetic-activated cell sorting cell isolation in this effect. In comparison with previous results in undepleted conditions, monocyte depletion was associated with the same effect of MSCs on intoxicated PBMCs, that is, an increase in final viable T cell number (17072 [10999] vs. 7870 [5376], P < 0.05), parent cell number (8249 [6446] vs. 4766 [2528], P < 0.05); and a trend toward an increase in precursor frequency (33.1% [6.17] vs. 24.3% [7.95], NS); but no significant change in proliferation index (3.52 [0.19] vs. 3.39 [0.32], NS).
Monocyte depletion did not alter the MSC supportive effect on intoxicated lymphocytes. MSCs from one donor were seeded at 104 MSCs/well. Whole blood samples obtained from five healthy donors were incubated for 24 h at 37°C with LPS (1 μg/mL). PBMCs isolated from these samples were depleted or not in CD14+ cells, CFSE labeled, and cultured alone or cocultured with MSCs in a PHA-containing medium (105 PBMCs/well, ie, an MSC:PBMC ratio of 1:10). After a 4-day-long culture, the final viable T cell number
LPS ± pamidronate priming potentiates the MSC supportive effect on T cells
We tried to improve the beneficial impact of MSCs on LPS-altered T cells with preconditioning methods. At a high 1:10 MSC:PBMC ratio, we showed (Fig. 9) that combined priming with pamidronate and LPS potentiated the positive effect of MSCs on final viable T cell number (91700 [34360] vs. 77310 [41980], P < 0.05). Conversely, at a low 1:160 MSC:PBMC ratio, no MSC priming method (LPS, pamidronate, LPS + pamidronate) showed any significant effect on PBMC support by MSCs.
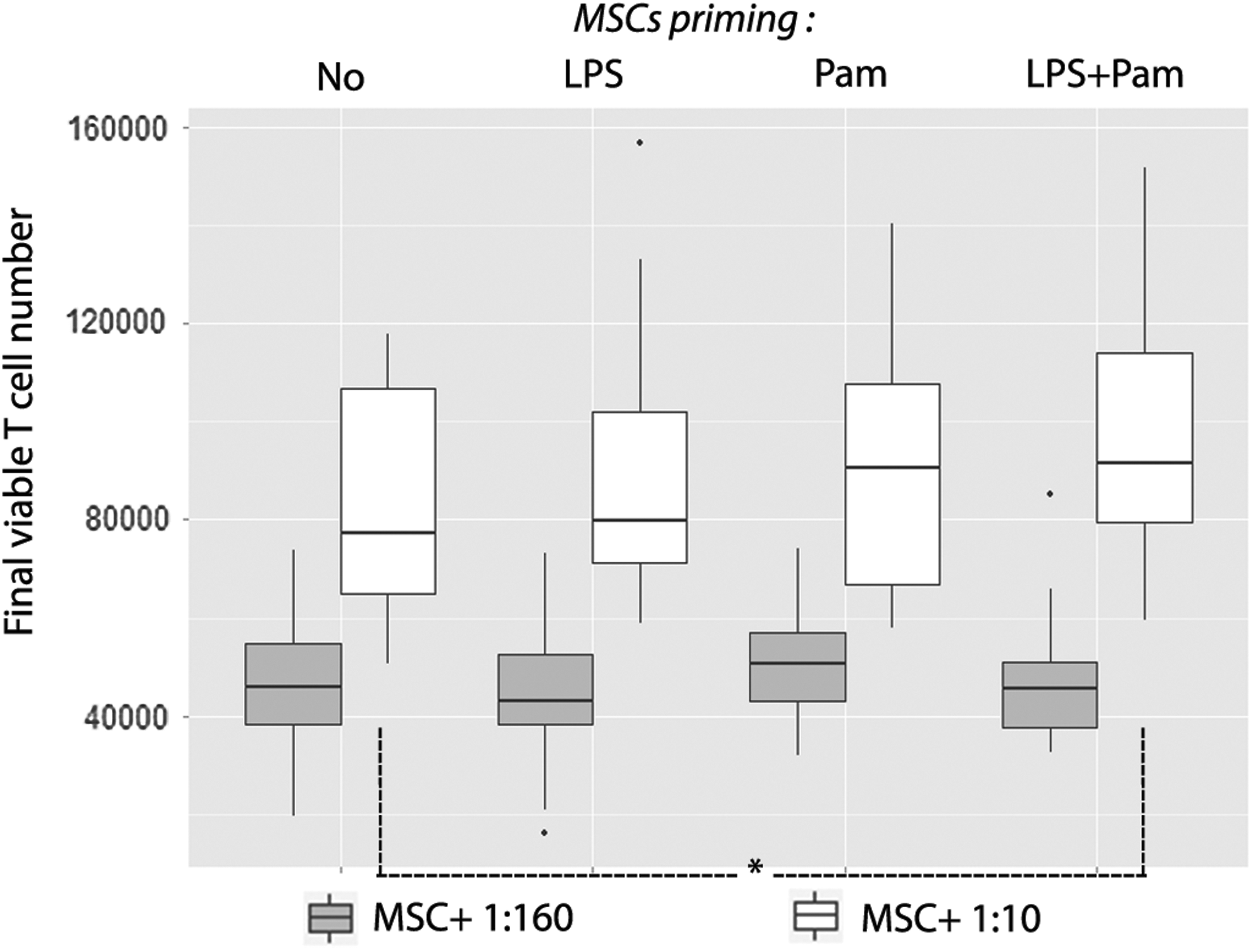
MSC supportive effect on T cells was moderately potentiated by combined priming of LPS and pamidronate. MSCs from eight donors were either unprimed or primed with LPS (10 ng/mL) and/or pamidronate (5 μM), then seeded either at 625 or 104 MSCs/well. Whole blood samples obtained from eight healthy donors were incubated for 24 h at 37°C with or without LPS (1 μg/mL). CFSE-labeled PBMCs isolated from these samples were cultured alone or cocultured with MSCs in a PHA-containing medium (105 PBMCs/well, ie, an MSC:PBMC ratio of 1:10). After a 4-day-long culture, the final viable T cell number was calculated. Box plots represent median, interquartile range, and range. *P < 0.05.
Discussion
MSCs have recently been proposed as a possible therapeutic in sepsis [7]. Preclinical studies have shown that MSC treatment induced a benefit in survival associated with better bacterial clearance. In these studies, MSC administration is almost simultaneous to sepsis induction and therefore corresponds more to a preventive than a curative effect [5]. In sepsis, acute overactivation of immune effectors is deleterious not only to the pathogen but also to the patient tissues and immune system, especially for mature immune cells [3] as well as hematopoietic progenitors [13]. Therefore, the observed improvement of bacterial clearance by MSCs could be due to prevention of immune system self-degradation rather than the true enhancement of an already damaged function. No experiment has so far responded to the question of the possible increase in immune suppression with later administration of MSCs. MSC sensitivity to their environment and the opportunity to improve their efficiency by priming before their administration could lead to reversal of their phenotype when faced with dysfunctional immune cells. In our study, we tried to examine if MSC responsiveness to their environment could lead to immune support when confronted with dysfunctional immune cells and if this behavior could also be induced or enhanced by culture priming. Whole blood intoxication with bacterial endotoxin induced the expected increase in lymphocyte death and hyporesponsiveness to a polyclonal mitogen. The main finding is that for a given PBMC donor, human MSCs showed opposite effects on both parameters depending on PBMC state: inhibitory effects toward healthy PBMCs and supportive effects toward LPS-intoxicated PBMCs. Moreover, the immune supportive phenotype on LPS-intoxicated PBMCs could further be slightly enhanced by preculturing MSCs with a combination of pamidronate and low-dose/short-time LPS, whereas each factor alone did not reach a statistically significant effect.
To mimic the effect of sepsis on human PBMCs, we intoxicated whole blood samples for 24 h with LPS. LPS is derived from gram negative bacteria, which are the most common pathogens responsible for septic shock. LPS can activate TLR4 receptors; this complex is one of the main mechanisms that initiate the immune response leading to sepsis [2]. Moreover, Poujol et al. [14] showed a dose-dependent effect of LPS on T cell proliferation. In this article and in our preliminary experiments, 1 μg/mL altered proliferative functions of T cells when PBMCs were exposed to LPS. We therefore decided to use this dose to obtain an important T cell alteration that was a crucial parameter to evaluate the MSC effect [14]. As expected, LPS intoxication in our experiments led to a significant decrease in lymphocyte proliferation and survival capabilities. These results are consistent with previous in vivo studies describing both numeric and functional alterations in leukocyte subpopulations, including lymphocytes in acute and subacute phases of sepsis [3]. Furthermore, Heidecke et al. found in septic patients a correlation between mortality and lymphocyte functional alterations, with a progressive decrease in proliferation of CD3+CD28+ population induced by sepsis and in their IL-2 and TNF secretion capacity [15]. It can further be pointed out that preclinical data link the long-lasting functional defects of T lymphocytes to the sensitivity to secondary infections [16]. This makes lymphocyte survival and responsiveness clinically relevant issues.
Our functional test is based on coculture with PHA-activated PBMCs (whose lymphocytes are the major population) and MSCs. We used two different MSC:PBMC ratios (1:10 and 1:160). The 1:10 ratio is most commonly found in the literature in mixed lymphocyte reaction experiments. However, we wanted to test the ratio of 1: 160, closer to the conditions of therapeutic use of MSCs in humans. Considering normal values of PBMCs in healthy individuals, a dose of about 2 × 106 MSCs/kg for a 70 kg patient corresponds to an MSC:PBMC ratio ranging from 1:70 to 1:350.
Confronted with freshly isolated healthy leukocytes (D0 condition), MSCs strongly inhibited the proliferation of activated lymphocytes. These results are consistent with those obtained from a large number of teams [17 –19]: Di Nicola et al. showed in 2002 in a mixed-lymphocyte reaction and in a PHA- or IL2-based simulation test that human bone marrow-derived MSCs strongly inhibit proliferation (evaluated by tritiated thymidine incorporation) of autologous or allogeneic ex vivo CD4+ and CD8+ T cells [17]. Noteworthy, coculture experiments between unstimulated PBMCs and MSCs (from all donors) never showed any lymphocyte proliferation activation by MSCs, which were always allogeneic (data not shown), consistent with the well-known low immunogenicity of MSCs [20].
Contrasting with their effect on freshly isolated lymphocytes, MSCs at 1:10 ratio did not inhibit the proliferation of LPS-altered lymphocytes: the proliferation index was not reduced, and a slight increase of PHA-responsive cells (precursor frequency) was observed in the presence of MSCs. In addition, MSCs promoted T cell survival against the LPS-activated lymphocytes (reflected by the increase in parent cell number). The resulting parameter, final viable T cell number, was thus significantly increased by MSCs in the LPS-intoxicated group; MSCs had the same effect on these four parameters when lymphocytes were activated by CD3/CD28, which better mimics the physiological activation of T cells. In the literature, several experiments had already shown that MSCs had antiapoptotic properties toward various cells such as cardiomyocytes, hepatocytes, or endothelial cells, in particular through the expression of Bcl-2, survivin, and Akt [21]. In a mouse model of sepsis induced by intraperitoneal infection, Pedrazza et al. have shown that simultaneous treatment with murine MSCs reduces apoptosis of mouse splenocytes isolated 12 h after sepsis induction [22]. Moreover, Normanton et al. have demonstrated a downregulation of T cell apoptosis by human MSCs at the same ratio of 1:10; this effect was not associated with an increase of Treg cell number and was contact and IL-7-paracrine dependent [23], a cytokine being currently the focus of a clinical trial per se NCT02640807. According to our experiments, an MSC-positive effect on final viable T cell number relied mostly on protection against cell death, involving both proliferative and nonproliferative populations. In each experiment, a variable proportion of lymphocytes did not divide (0 division peak). We wondered if the MSCs support only nonreactive cells: we showed that MSCs not only promote the survival of nonreactive cells but they also allowed the survival of cells that were still able to perform up to three divisions (data not shown). Moreover, we have evaluated several factors described in the literature as being secreted by MSCs and that can directly or indirectly modulate lymphocyte survival and proliferation [23 –25]. We also evaluated the expression of cytokines to assess the phenotypic orientation of lymphocytes after coculture [26]. The main result concerning the quantification of various factors at the end of culture showed an increase in the expression of IFNγ, that is, maintenance of the TH1 phenotype in the presence of MSCs when PBMCs have been intoxicated with LPS.
We showed that LPS intoxication induces a decrease of CD4+ and CD8+ lymphocytes with an increased CD4/CD8 ratio. These results are consistent with a previous in vivo study by Monserrat et al., which described (1) significantly lower counts of CD3+, CD3+CD4+, and CD3+CD8+ lymphocyte in patients with septic shock than in control subjects; and (2) in surviving patients, a more rapid normalization of CD3+CD4+ lymphocyte numbers, whereas CD3+CD8+ numbers were still low after 14 days [27]. Two other studies also showed that septic patients have significantly lower absolute number of lymphocytes [28,29]. However, in contrast with our findings, the former showed a decrease in CD4/CD8 ratio in blood samples collected within 48 h after patient admission [28], and the latter showed that CD4+ and CD8+ T cell numbers were equally decreased during a period of 48 h of monitoring starting 2 h after the beginning of vasopressive treatment [29]. The discrepancies observed between these studies could be explained by the timing of evaluation, an early decrease in CD4/CD8 ratio being followed by an increased ratio at a later time point. Concerning the effect of MSCs on T cell subpopulations, an in vitro study by Ramasamy et al. showed that PHA could enhance proliferation of both CD4+ and CD8+ T cells and that MSCs equally inhibit CD4+ and CD8+ subpopulations [30]. These results are in line with our results showing a similar effect of MSCs on CD4+ and CD8+ proliferation after LPS intoxication.
Overall, the most interesting element is the apparent adaptability of MSCs when facing lymphocytes in different states. This point is consistent with a study by Bocelli et al. showing that the T cell proliferative capacity was modulated (inhibited or stimulated) in the presence of MSCs, depending on the PBMC:MSC ratio and T cell donor. Highly proliferative T cells under IL-2, IL-7, or IL-15 activation were inhibited in the presence of MSCs, even at low MSC:PBMC ratios; conversely, low proliferative T cells under the same activation were stimulated by MSCs, even at high MSC:PBMC ratios; this plasticity of MSCs depended on soluble factors [31]. The putative mediators of this behavior need to be addressed, but could include IL-7 (currently under clinical trial NCT02640807) or IL-15 [23,25], although we cannot rule out that MSCs may modulate monocytes or other bystander cell behavior. Collectively, these points argue for an adapted behavior of MSCs to promote homeostasis rather than the stereotypical role of a suppressor effect on immunity. However, we cannot exclude that LPS intoxication could also have modified lymphocytes’ sensitivity to outer signals.
In our model, we have decided to intoxicate all PBMCs and not only the lymphocyte population, first because even if LPS can have a direct effect on the CD3-T cell population [33], this effect could be of lower magnitude than the indirect effect of LPS mediated by monocytes [14]. Because we were looking toward circumstances mimicking the postacute phase of sepsis-induced lymphopenia and exhaustion, we tried to ensure a maximal effect on lymphocytes, regardless of the many possible additive pathways under study. However, performing the same experiments after monocyte depletion was important since these cells have been shown to play an important role in MSC-mediated immunomodulatory effects. Indeed, the suppressive effect of MSCs on T cell proliferation was shown to be monocyte dependent [33,34]. Likewise, a gene expression analysis in splenocytes from septic mice treated with MSCs revealed an upregulation of genes in pathways related to antigen presentation, in comparison with untreated mice [35]. First, we showed that lymphocyte survival and proliferation were significantly altered by the absence of monocytes, in line with previous studies using PHA activation [36]. Strikingly, the supportive effect of MSCs was still effective in these CD14+ cell-depleted fractions, suggesting that MSC action toward T cells is partly monocyte independent.
In addition, we have described that the environment influences MSC behavior; therefore, we tried to improve their supportive activity with priming (or preconditioning). None of the two tested primings (LPS and pamidronate) had any effect by itself, neither by potentiating the effect of MSCs at the ratio of 1:10 nor by allowing the emergence of a significant effect at the ratio of 1:160. Unlike Waterman et al., we showed no effect of MSC LPS priming on T cell proliferation, even by using the same priming doses and durations as this team. This study, which focused on healthy cells, has shown that this priming could polarize MSCs toward a proinflammatory phenotype, promoting lymphocyte activation [9]. Moreover, a study by Musso et al. showed that priming of lymph node-derived MSCs with pamidronate or zoledronate induced gamma-delta T cell activation [37]. The differences between cell types used in these two studies could explain the dissimilar results obtained with pamidronate. However, double priming with both LPS and pamidronate seemed to potentiate the MSC-positive effect on T cells; this increase, although modest compared with the overall benefit provided by MSCs, encourages further research of priming modes to modulate MSC influence on T cells and optimize the effectiveness of MSCs at therapeutic doses in humans.
Although MSCs have traditionally been described as anti-inflammatory cells inhibiting lymphocyte proliferation, it is now recognized that their action is highly environment dependent. These results in an in vitro model of endotoxemia showed that MSCs are able to partly protect T cells from LPS-induced cell death, without inhibiting lymphocyte proliferative capacity. Preconditioning MSCs with LPS and pamidronate moderately potentiates this positive effect. This support of T cells by MSCs seems to exist only when lymphocyte functionality is altered, in the case of LPS intoxication, or to a lesser extent by an ex vivo 24-h incubation. Although requiring in vivo confirmation, these results provide some reassuring answers to concerns about the risk of deleterious effects of MSCs on sepsis-induced immunosuppression. Thus these data, if confirmed and extended to other cell types and contexts, would allow considering MSCs as real immune modulators, a term often misused as a synonym of immune suppressors. This opens new ways in therapy as well as of research: how do MSCs sense neighboring cells’ state? Answering this question is a likely prerequisite before being fully able to prime them for eventual immune activation use.
Footnotes
Acknowledgments
The authors would like to specially thank the collect department of Centre de Transfusion Sanguine des Armées, Clamart, for their contribution in obtaining blood samples of healthy volunteers. The authors also extend their thanks to their colleagues for continuous support throughout this work.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.