
Abstract
White adipose tissue is a source of mesenchymal stromal/stem cells (MSCs) that are actively studied for their possible therapeutic use in bone tissue repair/remodeling. To better appreciate the osteogenic potential of these cells, we compared some properties of MSCs from human subcutaneous adipose tissue [subcutaneous-adipose stromal cells (S-ASCs)] and dental pulp stem cell (DPSCs) of third-impacted molars, the latter representing a well-established MSC source. Both undifferentiated cell types showed similar fibroblast-like morphology and mesenchymal marker expression. However, undifferentiated S-ASCs displayed a faster doubling time coupled to greater proliferation and colony-forming ability than DPSCs. Also, the osteogenic differentiation of S-ASCs was greater than that of DPSCs, as evaluated by the higher levels of expression of early osteogenic markers Runt-related transcription factor-2 (RUNX2) and alkaline phosphatase at days 3–14 and of extracellular matrix mineralization at days 14–21. Moreover, S-ASCs showed a better colonization of the titanium scaffold. In addition, we investigated whether S-ASC osteogenic commitment was enhanced by adenosine A1 receptor (A1R) stimulation, as previously shown for DPSCs. Although A1R expression was constant during DPSC differentiation, it increased in S-ASC at day 21 from osteogenesis induction. Accordingly, A1R stimulation by the agonist 2-chloro-N6-cyclopentyl-adenosine, added to the cultures at each medium change, stimulated proliferation only in differentiating DPSC and enhanced the osteogenic differentiation earlier in DPSCs than in S-ASCs. These effects were counteracted by cell pretreatment with a selective A1R antagonist. Thus, our findings suggest that S-ASCs could be advantageously used in regenerative orthopedics/dentistry, and locally released or exogenously added purines may play a role in bone repair/remodeling, even though this aspect should be more thoroughly evaluated.
Introduction
M
MSCs are today actively studied for their capability of self-renewal and multi-lineage differentiation [5] that seem to offer advantages in regenerative medicine in comparison not only to traditional therapies but also to the potential use of embryonic or induced pluripotent stem cells. Indeed, MSCs do not raise ethical issues; they show no evident antigenic capacity when used for allogeneic transplantation and have a very low tumorigenic potential, even though some reports indicate that a risk exists [6,7].
MSCs were first isolated from bone marrow [8], and they are currently considered as the reference cell type with which those deriving from other sources such as dental tissues (ie, pulp, apical papilla, periodontal ligament) or embryonic annexes (ie, amniotic fluid, umbilical cord) are compared [2]. We and other researchers have demonstrated that cells isolated from the tissues or fluids mentioned earlier are multi-potent [9 –13], as well as preclinical and clinical studies have pointed out the possibility to use these cells in tissue engineering, especially for bone repair/remodeling [14 –19].
However, the actual availability of MSCs from the currently analyzed sources is doubtful. Indeed, bone marrow shows a low rate of MSCs, which further decreases with age [20]. The same is true for dental tissues. Moreover, dentists are more oriented toward conservative or restorative dental practices today [18]. Again, harvesting bone marrow from patients or amniotic fluid from pregnant women requires invasive procedures associated with pain and morbidity that discourage their routine employment; on the other hand, MSCs from umbilical cords, which are usually discarded after childbirth, seem to be useful until the fifth passage in vitro [21]. Therefore, the isolation of MSCs from more accessible sources that can be expanded to large cell numbers, while carrying a good multi-lineage differentiation potential, remains the ideal goal.
A good perspective is offered by the white adipose tissue (WAT), which is widely distributed throughout the body. WAT is constituted by two main depots, namely subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT) adipose tissue; the former represents >80% of the total body fat, and the latter is associated with internal organs. In SAT and VAT cells, different biological properties are present, including a population of adipose stromal cells (ASCs) [22]. However, those isolated from VAT are fewer and exhibit lower proliferation and differentiation capacity than ASCs from SAT, mostly because they are adipogenic precursors [23]. In contrast, SAT, which can be obtained from multiple body sites, offers the possibility of harvesting large ASC numbers from a single and less technically demanding procedure as compared with other tissues [18,24]. Moreover, cells retain their multi-lineage differentiation potential [25].
Based on this evidence, we started our study on MSCs isolated from human SAT [subcutaneous-adipose stromal cells (S-ASCs)] to examine whether they could represent an effective source of stem cells for mineralized tissue engineering in comparison to human dental pulp stem cells (DPSCs), which we previously studied [12]. The comparison included the evaluation of some distinctive features for MSCs in relation to their expansion and differentiation ability. Moreover, we investigated whether exogenous compounds were able to enhance S-ASC differentiation toward an osteogenic phenotype.
Among the possible candidates, we chose to examine the effects of purines, as in previous years several reports showed that extracellular purines, usually released from all cell types, promote the differentiation of MSCs toward the osteogenic lineage. Since, in particular, (i) the stimulation of metabotropic receptors for the purine nucleoside adenosine are mainly involved in controlling osteogenesis [26 –28]; and (ii) previously we pointed out that the stimulation of the adenosine A1 receptors (A1R) subtype increased the osteogenic differentiation of DPSCs, [12], in the second part of this article, we examined the effects of 2-chloro-N6-cyclopentyladenosine (CCPA), a selective A1R agonist, on the osteogenic differentiation ability of S-ASCs, previously ascertaining the expression of this receptor in these cells along their differentiation process.
Methods
Materials
Disposables for tissue culture were from Falcon (Steroglass, Perugia, Italy). L-Glutamine for culture medium was purchased from EuroClone S.p.A. (Milan, Italy). Minimum essential medium (MEM) Alpha Medium, penicillin/streptomicin (P/S), amphotericin B, ascorbic acid, dexamethasone, 3-isobutyl-1-methylxanthine (IBMX), β-glycerophosphate disodium salt, and all the other chemicals, unless differently indicated, were from Sigma-Aldrich (Milan, Italy).
Cell culture
Pulp samples (n = 10) were obtained from the third molars, scheduled to be removed for orthodontic purposes at our Department from six patients (two women and four men), with a mean age of 18.00 ± 1.34 years. Informed consent for clinical research and for the processing of personal data was obtained from each of them. All teeth samples were de-identified.
Dental pulps were isolated after tooth extraction as previously reported [12,29]; they were digested (1 h; 37°C) in MEM Alpha Medium containing L-Glutamine, P/S 100 × , 500 μg/mL clarithromycin (Abbott, Italy), 0.1 U/mL collagenase, and 0.8 U/mL dispase (Sigma-Aldrich). Cells were separated by filtering through a 70-μm strainer (Falcon, Becton Dickinson, Franklin Lakes, NJ); then resuspended in growth medium consisting of MEM Alpha Medium, 15% fetal bovine serum (FBS; Gibco by Life Technologies Italia, Monza, Italy), 1% P/S, and amphotericin B 1,000 × ; and centrifuged (10 min; 1,200 rpm). The cell pellet was resuspended in the same medium and plated in 25 cm2 flasks. Cultures were incubated at 37°C in 5% CO2, and the medium was changed twice a week.
The human S-ASCs were purchased from the Zen-Bio Company (Research Triangle Park, NC) and cultured by using a growth medium consisting of MEM Alpha Medium, 20% FBS (Gibco), 1% P/S, and 1% Amphotericin B. The mean age of six human subjects (women) was 28 ± 3 years. Cultures were incubated at 37°C and 5% CO2, and the medium was changed twice a week. Experiments were performed only in the first six–eight cell passages.
Detection of surface and intracellular markers by flow cytometry
Samples of 5 × 105 cells were treated and then stained for surface or intracellular antigens, as previously described [30], using the following fluoresceinisothiocyanate (FITC)-, orphycoerythrin (PE)-, or allophycocyanin (APC)-conjugated antibodies: Anti-CD13 (CD13-FITC), anti-CD45 (CD45-FITC), anti-CD29 (CD29-PE), anti-CD105 (CD105-FITC), and anti-CD166 (CD166-FITC) were obtained from Ancell (MN); anti-CD14 (CD14-FITC) was purchased from Miltenyi Biotec (Bergisch Gladbach, Germany); anti-CD90 (CD90-FITC), anti-CD73 (CD73-PE), and anti-CD146 (CD146-PE) were obtained from Becton Dickinson; anti-CD144 (CD144-FITC) was obtained from Acris Antibodies (Herford, Germany); and anti-CD34 (CD34-PE) was purchased from Beckman Coulter (Fullerton, CA).
Cells were incubated with these antibodies for 15 min at room temperature (RT). Then, tubes with cells were washed (3 mL of washing buffer) and centrifuged (4°C, 400 g, 8 min); cells were resuspended with 1 mL 0.5% paraformaldehyde, incubated for 5 min at RT, washed, centrifuged (4°C, 400 g, 8 min), and stored at 4°C in the dark until the acquisition. Finally, the antibody-bound cells were analyzed by using a flow cytometer (FACS Calibur, BD) and CellQuest™ software (BD). Debris was excluded from the analysis by gating on morphological parameters; 20,000 nondebris events in the morphological gate were recorded for each sample. All antibodies were titrated under assay conditions, and optimal photomultiplier gains were established for each channel. Cells incubated with phosphate-buffered saline (PBS) served as a control, and all experiments were performed in triplicate.
The data from flow cytometry were then analyzed by using FlowJo™ software (TreeStar, Ashland, OR) and are presented as mean fluorescence intensity ratio, which was calculated by dividing the MFI of positive events by the MFI of negative events.
Cell proliferation
Counting of live cells
It was performed by using the trypan blue exclusion method. Briefly, cells were harvested after different culture periods (from 0 up to 10 days), incubated with trypan blue, and counted with an hemocytometer (three different fields for each sample evaluated in triplicate). Results are expressed as number of live cells/mL.
Doubling time
Cells at the passage 5 (P5) were seeded at 1 × 104 cells/cm2 and cultured for different periods. They were detached by trysin-EDTA (Gibco) at 48, 72, and 96 h after seeding, and alive cells were counted by the trypan blue exclusion assay. The doubling time (DT) can be computed as follows:
Colony-forming unit-fibroblast
Cells at P5 were plated in six-well plates at different densities (100, 250, and 500 cells/cm2) and cultured in humidified 5% CO2 at 37°C. Culture medium was usually changed every 3 days. After 2 weeks, the dishes were washed twice with PBS, fixed with cold 100% methanol, and stained with 3% Crystal violet (Sigma). Colonies were counted under an optical microscope (Leica Microsystems, Germany) that was equipped with a digital camera (Canon, Tokyo, Japan), and the aggregates formed by 16 to 20 or more cells were scored as a 1 colony-forming unit-fibroblast.
In vitro osteogenic differentiation assay
The osteogenic differentiation was induced by culturing the human S-ASCs or DPSCs with MEM Alpha medium that was supplemented with 10% FBS, 0.05 mM ascorbic acid, 10 mM β-glycerophosphate, and 100 nM dexamethasone for as many as 28 days, with changes of medium every 3 days. To visualize calcium phosphate depositions, cultures were stained at different time points with Alizarin Red S (ARS; Sigma-Aldrich). Calcium depots were either visualized by a phase-contrast microscope (Eclipse TS100; Nikon), equipped with a D200 digital camera (Nikon), or measured by the following spectrophotometric measurements.
Briefly, 800 μL of 10% (v/v) acetic acid was added to each well; cells were incubated for 30 min with shaking, then removed by scraping, transferred into a 1.5-mL vial, and vortexed for 30 s. The obtained suspension was overlaid with 500 μL mineral oil (Sigma–Aldrich), heated to 85°C for 10 min, transferred to ice for 5 min, carefully avoiding the opening of the tubes until fully cooled, and centrifuged at 20,000 g for 15 min. The samples were acidified (pH between 4.1 and 4.5) with 200 μL of 10% (v/v) ammonium hydroxide. Aliquots (150 μL) were read in triplicate at 405 nm by a spectrophotometer (Spectramax SM190; Molecular Devices, Sunnyvale, CA).
Evaluation of early osteogenic markers
Alkaline phosphatase activity assay
Tissue-nonspecific alkaline phosphatase (ALP) activity was measured after 1 week of cell culture. Cell monolayers were washed with PBS, lysed with 1 mL of Tris buffer (10 mM, pH 7.5, 0.1% Triton X-100), and centrifuged (2,000 rpm; 1 min). Then, 20 μL of supernatant from each sample was combined with 20 μL of 1 mM p-nitrophenyl phosphate (p-NPP, Sigma; solution at pH 10.3 with MgCl2-diethanolamine buffer) substrate and dispensed into 96-well plates. The samples were incubated in the dark at RT for 30 min. The reaction was stopped with 10 μL of 2 N NaOH. The amount of p-NPP released was measured as absorbance at 405 nm on a microplate spectrophotometer (Spectramax SM190). The protein content of each sample was determined by the BioRad protein assay (Bio-Rad Laboratories, Milan, Italy). The enzyme activity was expressed as nmoles of p-NPP released per mg of protein per 30 min.
RNA isolation and real-time PCR analysis of osteogenic markers
Total RNA was isolated from adipose and DPSCs by using the RNase Mini Kit (Qiagen, Inc., Valencia, CA) following the manufacturer's protocol. In detail, cells were lysed with 350 μL of RLT lysis buffer. After the addition of an equal volume of 70% ethanol, a total of 700 μL of each sample was transferred to the RNase spin column. After several washes with elution buffers, 30 μL of water was added to each column to elute the RNA. Eluted RNA concentration (OD at 260 nm) and purity (OD260/280) were quantified by the use of a NanoDrop (Agilent). Afterward, RNA was reverse transcripted into cDNA by using the Omniscript Reverse Transcriptase Kit (Qiagen). In brief, 7 μL of RT mix solution containing 2 μL of oligo (dt) was added to 2 μg of total RNA solution. The reaction mixture was loaded to the Gene Amp PCR system 9700 (Applied Biosystem, Foster City, CA) undergoing the cycle at 37°C for 60 min.
Real-Time RT-PCR was carried out with the ABI Prism 7900 Sequence Detection System (Applied Biosystems). The expression of ALP and RUNX2 was evaluated at 0, 3, 7, and 14 days; cells were cultured in osteogenic medium. Commercially available TaqMan Gene Expression Assays (RUNX2, Hs00231692_m1, ALP, Hs01029144_m1) and the TaqMan Universal PCR Master Mix (Applied Biosystems) were used according to standard protocols. Samples were normalized (ΔCt) against the housekeeping gene beta-2 microglobulin (B2 M, Hs99999907_m1, Applied Biosystems), and the -ΔΔCt value of ALP and RunX2 relative to undifferentiating medium was calculated by using the ΔΔCt method.
Scanning electron microscopy and electron-dispersive X-ray analysis
Sandblasted and etching (SLA) Ti6Al4 V disks [diameter 12 mm; thickness 3 mm, SLA (grade 5 Titanium Alloy); Strumann, Switzerland] were placed into 24-well plates and on each one 5 × 105 cells were plated in MEM alpha medium that was supplemented with 10% FBS. After 3 days, the culture medium was replaced with the usual medium (described earlier for the induction of the osteogenic differentiation) that was renewed every 3 days till 6 weeks. After this period, samples were fixed in 2% of glutaraldehyde in 0.1 M cacodylate buffer at pH 7 for 16 h at RT. To remove the fixative, the samples were washed three times with ice-cold PBS and then, they were first dehydrated by immersion in solutions of crescent concentration of ethanol/water (20%, 50%, 90%, 100% v/v) and subsequently immersed in hexamethyldisilazane for 15 min.
Afterward, the samples were dried for 1 day and sputter-coated with gold for 40 s before a scanning electron microscopy examination (Philips XL30CP) was conducted at an accelerating voltage of 20 kV. Electron-dispersive X-ray microanalysis (EDAX) of calcium, phosphorus, carbon, and titanium was performed (EDS, Oxford, Inca Energy 250) at a voltage of 20 kV. Spectra were collected at a magnification of 1,000× and a collection time of 1 min.
Expression of A1R
At the indicated time points, total protein extracts were prepared to perform western blot analysis as previously described [12]. Protein concentration was determined by the BioRad protein assay. The antibodies used were a rabbit polyclonal anti-A1 adenosine receptor primary antibody (final dilution 1:1,000; overnight incubation at 4°C; Alomone Labs, Israel) and a donkey anti-rabbit IgG HPR-conjugated secondary antibody (final dilution 1:5,000; 1 h incubation at RT; GE Healthcare Life Sciences, Milan, Italy). To determine the equal sample loading, the blots were stripped and reprobed with an anti-ß-actin antibody (1:100 dilution; Santa Cruz Biotechnologies, DBA Italia, Segrate, Milan, Italy). Immunocomplexes were visualized by using the enhancing chemiluminescence detection system (GE Healthcare Life Sciences) and quantified by densitometric analysis using ImageJ software (U.S. National Institutes of Health, Bethesda, MD,
Statistical analysis
The Graphpad Prism 6 Software was used for statistical analysis. The data are expressed as mean ± standard error of the mean (SEM). One-way analysis of variance was used when three or more groups within one variable were compared. To analyze two groups within each cell type, the unpaired Student's t-test was used; to compare data from the two cell types, the multiple t-test was used; and the statistical significance was determined by using the Holm–Sidak method. Values of P < 0.05 were considered significant.
Results
Morphological, phenotypical, and proliferative characteristics of undifferentiated MSCs from human dental pulp and WAT
We started our study with the comparison of some characteristics in both kinds of undifferentiated MSCs. We first considered their morphology in vitro as well as the expression of some markers selected according to the criteria of ISCT [31]. At passage 5, S-ASCs exhibited a spindle-shaped (fibroblast-like) morphology comparable to that of DPSCs (Fig. 1a). Flow cytometry analysis revealed that both kinds of cells were also phenotypically similar (a representative scheme of marker expression is reported in Fig. 1b). Undifferentiated human S-ASCs and DPSCs were negative for hematopoietic markers (CD13, CD14, CD34, CD45) and for the surface vascular endothelial-cadherin adhesion molecule CD144, whereas they positively stained for the typical MSC markers CD29, CD73, and CD90 and to a lesser extent for CD105, 166, and 146.
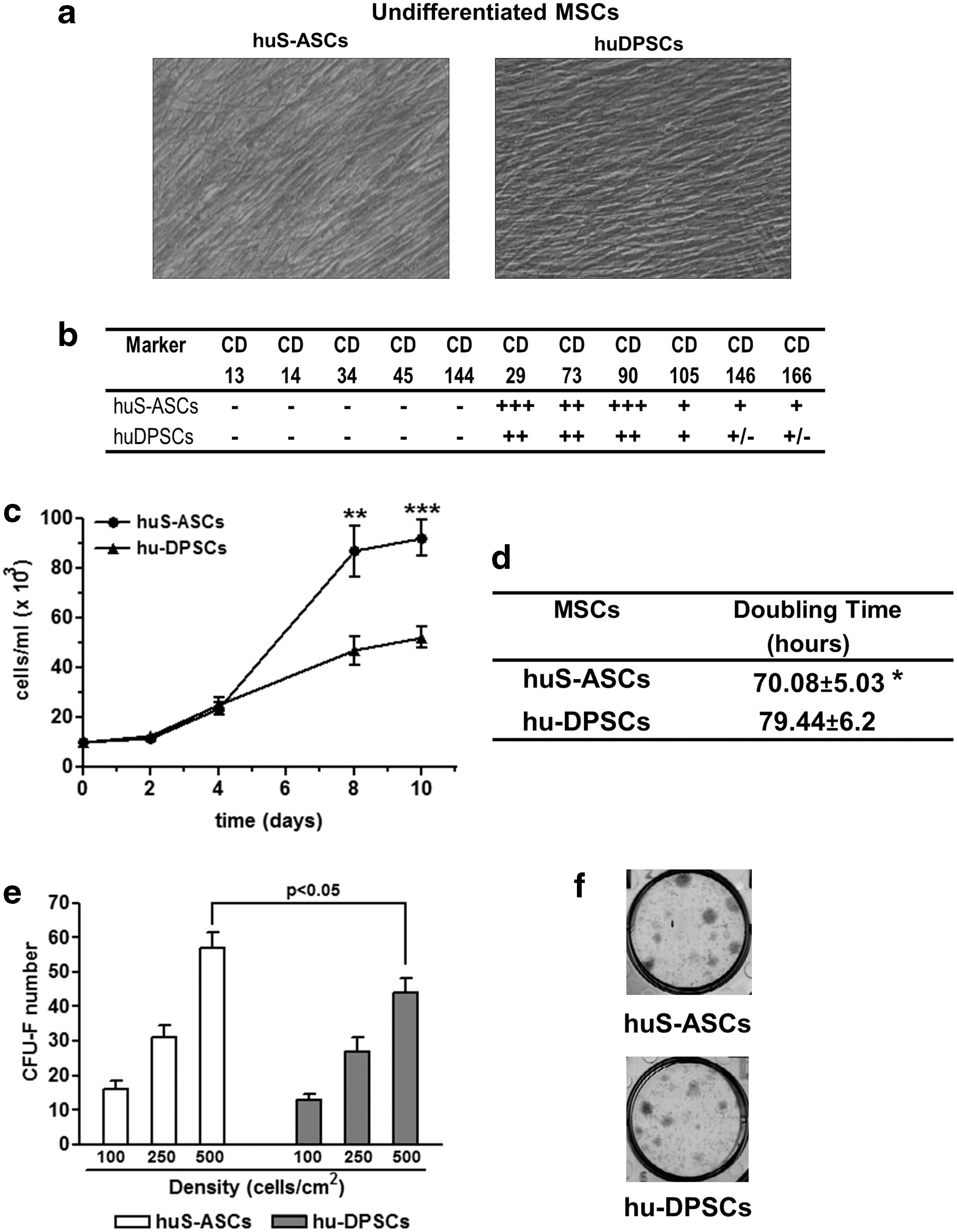
Characterization of undifferentiated S-ASCs and DPSCs.
We then evaluated further characteristics of the cells under study that were useful for their potential utilization in bone regenerative medicine. The doubling time (DT) was significantly lower for S-ASCs than for DPSCs (Fig. 1d). Accordingly, cell proliferation, evaluated by counting live cells by the classic trypan blue exclusion method, was greater in S-ASCs than in DPSCs, leading to a significantly higher number of cells on the eighth day in vitro (DIV) in the former than in the latter cultures (Fig. 1c). On the 10th DIV, proliferation plateaued in both types of cells, but their number remained significantly different.
When cells were examined for clonogenicity, the increase in colony formation ability was parallel to that of the seeding density in both cell types (Fig. 1e, f). However, S-ASCs formed a greater number of colonies than DPSCs that was statistically significant when cells were seeded at 500 cells/cm2.
Characterization of the osteogenic differentiation of S-ASCs and DPSCs
We then characterized the osteogenic differentiation of both types of cells by measuring the extracellular matrix mineralization and the expression of early osteogenic markers during the differentiation process. In Fig. 2a and b, besides the visualization by microscopy (Fig. 2c, d), we quantified ARS staining by spectrophotometry. S-ASCs in comparison to DPSCs displayed an enhanced extracellular calcium deposition, with a maximal increase on day 21 (P < 0.0001 measured by multiple t-test and Holm–Sidak method).
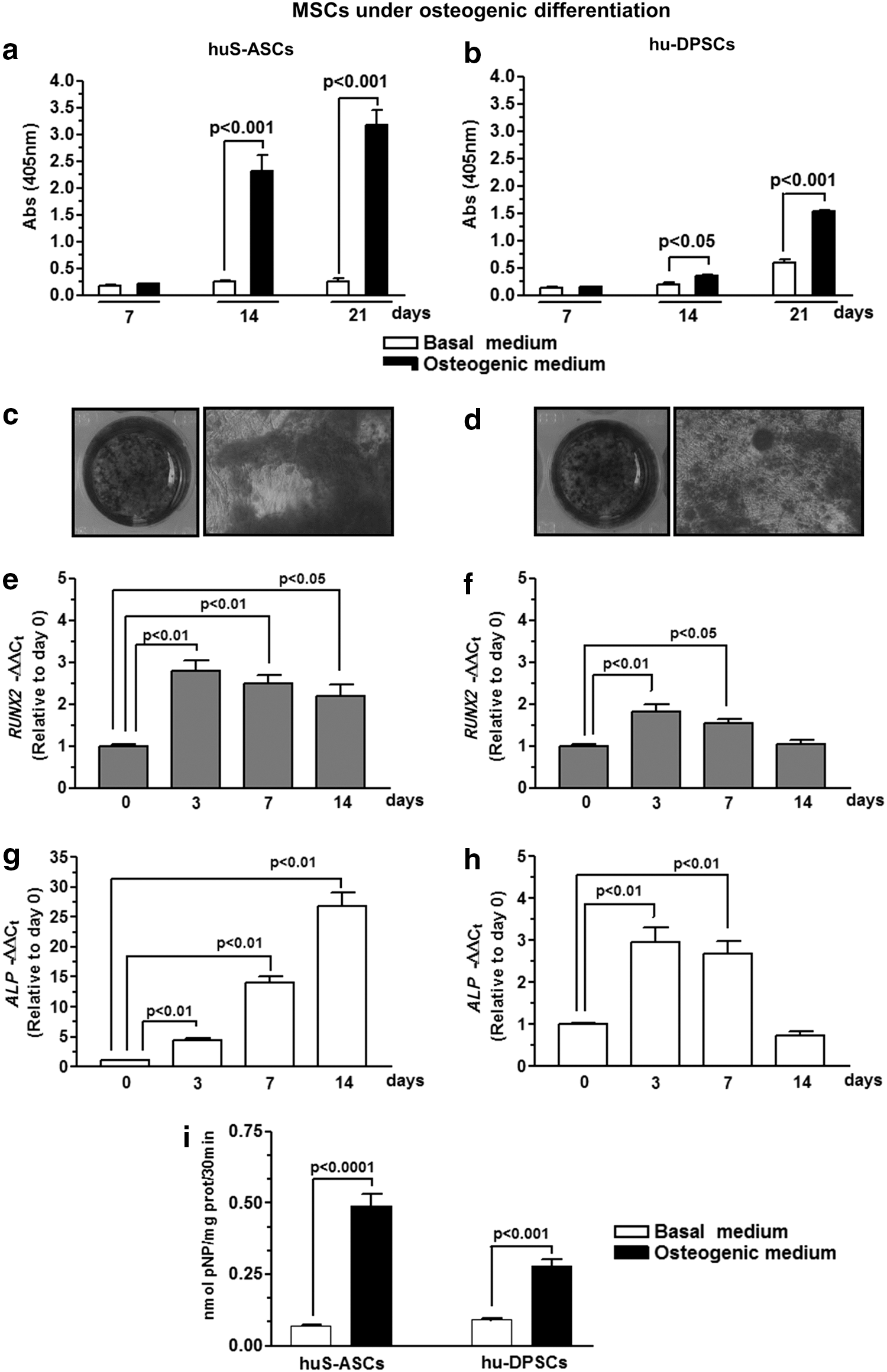
Evaluation of the osteogenic differentiation of S-ASCs and DPSCs. Panels
Data from real-time PCR performed to evaluate the expression of early osteogenic markers along the differentiation process indicated that ALP was massively expressed in S-ASCs over a period of 14 days, during which RUNX2 expression was also significantly increased (Fig. 2e, g). The increase in ALP and RUNX2 expression was smaller in DPSCs (P < 0.0001, multiple t-test) and limited to the first 7 days of differentiation (Fig. 2f, h). In agreement with these findings, the ALP activity, which was significantly enhanced on the seventh DIV in both cell types, was more remarkably increased in S-ASC than in DPSC cultures (Fig. 2i; P < 0.01, multiple t-test). Finally, morphological analysis by scanning electron microscopy (Fig. 3a, b, d, e) showed that after 40 days, S-ASCs, but not DPSCs, covered most part of the standard titanium scaffold, demonstrating a good ability of these cells to adhere and colonize the scaffold surface.

Analysis by scanning electron microscopy and EDAX assay. Cells were seeded on titanium disks (diameter: 12 mm; thickness: 3 mm); after 3 days, basal growth medium was replaced with osteogenic medium, in which cells were cultured for a further 40 days.
The presence of a fiber network is relevant, presumably due to the deposition of extracellular matrix proteins and minerals (Fig. 3b), as impressive is the EDAX-identified mineralized depots (Fig. 3c). The spectrum related to DPSCs showed only the presence of titanium (Fig. 3f), whereas that of S-ASCs elicited minerals such as calcium, phosphorus, as well as carbon and oxygen (Fig. 3c). Particles analyzed at 4 weeks of cell cultures, indicated by the white circle in the figure, displayed a mean Ca: P ratio of 1.23 (Fig. 3b, c).
Expression of A1R in S-ASCs and DPSCs during their osteogenic commitment
We previously demonstrated that purines interacting with A1R can stimulate the proliferation and the osteogenic differentiation of DPSCs [12]. Here, we wanted to verify whether the A1R activation may influence the same parameters as in S-ASCs. First of all, S-ASCs and DPSCs were grown under osteogenic conditions and were analyzed at different time points for the expression of A1R by western blotting (Fig. 4a, b). Noteworthy, the A1R protein content, which was low in undifferentiated cells (corresponding to the lane at time 0), significantly increased along the S-ASC differentiation process, becoming maximal at 21 DIV after the differentiation induction (Fig. 4a), whereas it remained constant during the osteogenic differentiation of DPSCs and was similar to the A1R expression in undifferentiated cells (lane at time 0) (Fig. 4b).
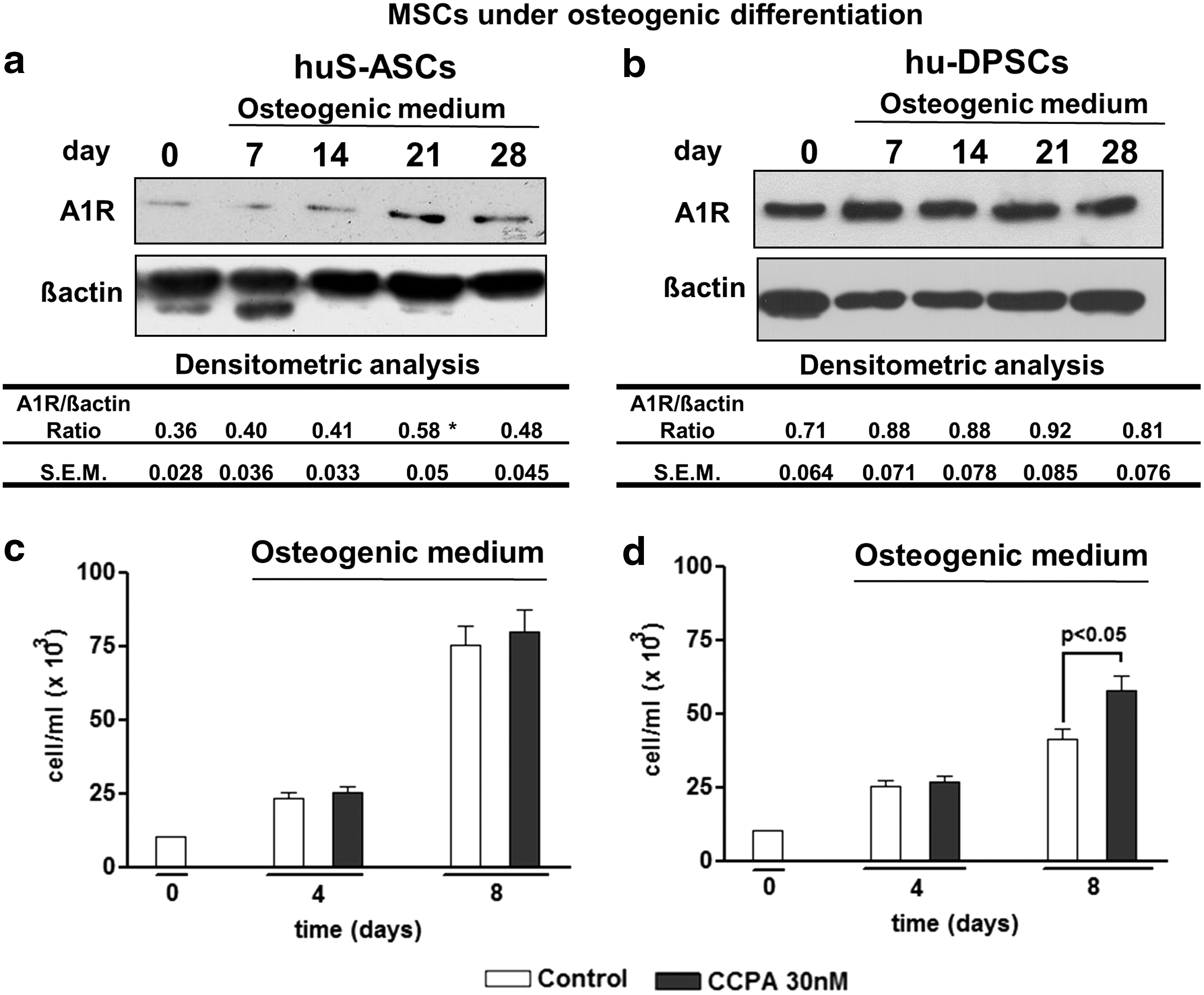
Expression of adenosine A1 receptor (A1R) and effect of its stimulation on S-ASC and DPSC proliferation.
S-ASC and DPSC exposure to CCPA, an A1R agonist, differently affected cell proliferation and osteogenic differentiation
Based on our previous experience, we exposed both cell types undergoing osteogenic differentiation to the known A1R agonist, CCPA, that was added to the cultures at each medium change at the concentration of 30 nM. CCPA was able to stimulate DPSC proliferation and osteogenesis [12]. In comparison to untreated (control) cells, CCPA did not modify the S-ASC proliferation (Fig. 4c), whereas it increased the number of DPSCs on the eighth day from their commitment toward osteogenesis (Fig. 4d), as previously observed [12].
In contrast, CCPA significantly enhanced the differentiation process in both kinds of cells, even though with different timing. Indeed, in comparison to untreated S-ASCs undergoing differentiation, CCPA significantly increased the expression of the osteogenic markers RUNX2 and ALP in the period between 14 and 21 days as well as the accumulation of extracellular calcium depots at 21–28 DIV (Fig. 5a–c) after the differentiation induction. In contrast, in DPSCs, CCPA induced an earlier increase of osteogenesis, as demonstrated by the greater expression of RUNX2 and ALP, which was significant at 3–7 days and at 7–14 days, respectively, in comparison to the control, and by the evaluation of the extracellular matrix mineralization, which was enhanced from the 14th day onward (Fig. 5d–f).

Effect of the stimulation f A1R on S-ASC and DPSC osteogenic differentiation. The cell treatment with CCPA was performed in DPSCs and S-ASCs for different periods from the cell induction till the osteogenic differentiation. When present, the A1R antagonist PSB36 was added to the culture medium 1 h before the agonist CCPA. The effects on the expression of osteogenic markers (RUNX2 and ALP) were evaluated by real-time PCR
The CCPA stimulatory effect induced in differentiated S-ASCs or DPSCs was abolished by cell pretreatment with the selective A1R antagonist 1-Butyl-8-(hexahydro-2,5-methanopentalen-3a(1H)-yl)-3,7-dihydro-3-(3-hydroxypropyl)-1H-purine-2,6-dione (PSB36, 10 nM) (Fig. 5), which when administered alone did not affect the differentiation process (data not shown).
Discussion
Regenerative medicine based on the experimental use of MSCs is a science in rapid development, especially in the fields of Orthopedics and Dentistry, where MSCs have a great therapeutic potential in bone repair/remodeling (ie, see Refs. [18,32,33]). A great body of evidence has demonstrated that MSCs, isolated from embryonic as well as adult tissues, are able to differentiate into osteogenic or chondrogenic precursors, interestingly lacking histocompatibility antigens, and can be implanted into allogeneic tissues [33].
However, MSCs show a different ability to proliferate and/or to respond to external or microenvironmental influences depending on their tissue origin [34 –36]. Since this behavior could lead to different in vivo results after transplantation, it seems crucial to investigate the characteristics of selected MSCs to choose the most appropriate MSC type for healing injured bone tissue. Other important features to consider are the ease of removing tissues containing MSCs with abundant cells inside the tissue and the possibility to expand these cells to obtain a great number of MSCs.
With this in mind, our study aimed at investigating the properties of cells isolated from the SAT, which may correspond to the requirements reported earlier. Previous studies have pointed out that those deriving from the subcutaneous component of WAT have the properties of MSCs [37,38]. A number of papers as well have compared the differentiation properties of human S-ASCs with those of cells derived from other human sources such as bone marrow, umbilical cord, or placental and dental tissues (periodontal ligament, deciduous teeth, or maxillary periosteal tissue) other than adult dental pulp [39 –41]. However, the results so far obtained are conflicting, as they point out a greater osteogenic differentiation potential of MSCs from one or another source, with this also depending on the tissue toward which the regeneration was estimated [42 –44].
Eager to contribute toward shedding light on this intricate aspect and based on our previous experience with DPSCs [12,29], here we report findings obtained by comparing some characteristics of S-ASCs with those of MSCs derived from adult dental pulp, which represents another consolidated paradigm for studies in regenerative bone medicine [43]. To our knowledge, only one paper has reported a similar comparison, using, however, cells from rat incisors and inguinal adipose tissue, and the authors showed a relatively limited potential for mineralized tissue engineering of adipose-derived MSCs, in comparison to DPSCs [44]. In contrast, it was demonstrated that S-ASCs from rats are more resistant to cryo-preservation so that they constitute a robust source of MSCs in comparison to bone marrow cells and DPSCs, which are also derived from rats [45].
Our study started with the characterization of some aspects of human adult S-ASCs and DPSCs, the evaluation of which may contribute toward expanding knowledge of the basic biology of these stem cells, and which may constitute the natural premise for the clinical translation of cell-based therapies. Here, we show that the two cell types share similar morphology and expression of mesenchymal markers. For the latter, we refer to a seminal paper by Dominici et al. [31], which has been cited in the majority of articles related to stem cells, even though there is still a certain debate on the list of the most appropriate markers to define the mesenchymal nature/behavior of cells [46].
However, despite some similarities, when we better analyzed other features that were indicative of cell expansion ability and osteogenic differentiation, we found that S-ASCs exhibited a greater proliferation and osteogenic differentiation than DPSCs. Both these properties may be of value for the use of these cells in bone regeneration medicine. Indeed, the lower duplication time and the higher proliferation shown by S-ASCs in comparison to DPSCs would allow us to obtain a greater number of the former cells in a shorter time during their expansion.
Apart from this, the greater differentiation potential of S-ASCs than DPSCs toward an osteogenic phenotype, also evident when cells were cultured on a standard titanium surface, could be very useful for bone repair that was performed with the aid of standardized scaffolds. Thus, our results confirm what is found at present in the literature, which reports the ability of S-ASCs to colonize biotic and abiotic materials, also in in vivo studies [47 –51]. Moreover, the same results would make the use of S-ASCs very suitable in Dentistry, since MSCs obtained from dental tissues are few, being small in size compared with the original source. This represents a limit, mainly in relation to the entity of bone reconstruction. Conversely, it is possible to select a large number of stromal cells from SAT with minimal discomfort for patients, given the abundance of this tissue in the human body.
Therefore, we hope that our findings may further stimulate the research on the bone regenerative properties of S-ASCs. Given the high variability reported in different studies, in our opinion it would be important that an international Committee establish precise guidelines that researchers should follow, thus rendering the data analysis more homogenous and therefore the targeted use of suitable cells in regenerative medicine.
Of course, in vitro data should be corroborated by further analysis of cell behavior in vivo, where cells are induced to differentiate not by means of a conditioned medium but by local factors, which are released from the cells themselves. Therefore, it is also crucial to investigate the nature of external signals inducing the activation of these cells and the best way(s) to increase the number and the efficiency of their differentiation. Numerous papers investigated the influence of specific growth factors and/or selected scaffolds on the osteogenic differentiation of MSCs [47 –53].
Here, we focused on purines, ubiquitous molecules that are present inside cells, where they perform many vital functions (ie, as fundamental components of nucleic acids or as a source of energy supply). However, it is also recognized that these molecules, in particular adenine-based nucleotides, are released from every type of cell in the pericellular fluid, where they are rapidly transformed by specific ecto-enzymes into the corresponding nucleoside adenosine [26 –28]. Both ATP and adenosine behave as signaling molecules, which are able to interact with specific receptors [26 –28]. In relation to bone homeostasis, a recent review emphasized the useful role played by adenosine and related analogs acting on metabotropic receptors (namely, A1, A2A, A2B, and A3) that are specific for adenosine [28].
Here, we investigated the effects consequent to the activation of A1R subtype, based on our experience on the protective effects exerted by the stimulation of these receptors on neural cells [54] and also on the favorable results obtained with the use of an A1R agonist in DPSCs osteogenic differentiation [12]. Our results confirmed that A1R is expressed at a constant level throughout the osteogenic differentiation of these cells. Likely, this feature is important to elicit an enhanced DPSC proliferation that is induced by CCPA. In contrast, probably due to the late increased expression of A1R in differentiating S-ASCs, and also due to the higher proliferative rate of these cells in comparison to DPSCs during the differentiation phase, the stimulation of these receptors by CCPA was ineffective in S-ASC duplication.
Apart from this, the A1R activation was able to enhance the S-ASC osteogenic process, but only belatedly. Thus, the different expression of A1R between the two cell types influenced the achievable effects by the stimulation of the same receptor, indicating that the tissue of origin can condition the potential results. Indeed, in MSCs derived from other tissues, the A1R-induced effects on cell proliferation and/or osteogenic differentiation were not so evident or prevailing [55,56]. As previously reported [12], the discrepancy might likely be due to differences in the species (animals or humans) or in the health state or age of subjects used to obtain MSCs (ie, aged women with osteoarthrosis versus young healthy subjects, as in our case).
Lastly, in the field of purines, it is also important to underline that other receptors for these substances belonging to the family of P1 receptors for adenosine (A2A, A2B, and A3 receptor subtypes) may control cell differentiation, in particular osteogenesis [26 –28,55,56]. Thus, further experiments are needed to evaluate the involvement of other adenosine receptors in the same cells. On the other hand, it has been reported that A1R activation in osteoclast precursors leads to stimulation of the receptor activator NFkB Ligand (RANK), which, in turn, causes the activation of NFkB, a requirement for osteoclastogenesis [57].
Therefore, in vivo, the effects consequent to the stimulation of a single (purine) receptor are the sum of the effects resulting from the simultaneous activation of that receptor subtype, in our case the A1R, on different cells such as osteoblasts and osteoclasts, which ultimately concurs to bone plasticity and homeostasis.
In conclusion, with our study, we provide new insights on the differentiation of S-ASCs that could represent a useful model to better appreciate biological mechanisms underlying bone homeostasis/repair, as well as a manageable tool to be used in bone recovery/remodeling, thanks to the remarkable cell proliferation and propensity to differentiate in osteoblasts, even on a classical titanium scaffold.
The role played by purines in the osteogenic differentiation of S-ASCs deserves to be further evaluated, as these compounds, released from all kinds of cells, especially during tissue injury, may differently contribute to bone repair. Only more extensive studies performed not only on isolated cells but also on more integrated experimental models (ie, co-cultures of different cell types, isolated organs, animals) can elicit the complex effects consequent to the stimulation of selected receptors by exogenous purinergic compounds and could show the real therapeutic potential of these compounds in bone regeneration.
Footnotes
Acknowledgments
This study was partially supported by a grant to Stem TeCh Group from CARICHIETI Foundation and also by funds to R.C. and P.D.I. from the Italian Ministry of Education, University and Research (MIUR).
Author Disclosure Statement
No competing financial interests exist.