
Abstract
According to the Committee for Advanced Therapies, amniotic stem cells were classified as an advanced therapy medicinal product. This work aims to standardize the isolation of amniotic stem cells and the selection of the optimal time of transplantation and cell application methods in burn patients according to the guidelines of the Good Manufacturing Practice. The placenta used in the study was sourced during a Cesarean section. The remnants of the amnion preparation were placed in a sterile container and transferred to a class B environment, where the primary cultures began. The highest average number of cells was obtained by tissue homogenization and culture growth on the AmnioGrow medium. The isolation of the pure monoculture should be performed using the antibodies against CD105. On the basis of an analysis of population doubling, the aging of a population, the cells' viability, and the severity of injury, the cells should be used between passages 3 and 6. Significant differences were found in the number and viability of cells that were transferred as a full sheet, depending on the transfer method. To sum up, amniotic cells are a promising source in the treatment of burns and can be used as a hospital exemption.
Introduction
A
This work aims to standardize the isolation of stem cells and amniotic cells, and the selection of the optimal time of transplantation and application methods of cells in patients with burns and chronic wounds, all while adhering to the guidelines of GMP. The various methods of isolation, and various culture media, were analyzed. The quality control points were selected and analyzed. The optimal transit for transplantation and tool for the cell transfer were defined. This is a complete work that allows for the implementation of amniotic stem cells into clinical practice.
Materials and Methods
Required contents
The consent of the National Centre for Tissue and Cell Banking is required for the isolation, processing, and storage of the amniotic cell. Furthermore, the consent of the Chief Pharmaceutical Inspector for the manufacturing of medicinal products of advanced therapies is required. The consent of the Bioethics Committee for application cells was therefore obtained.
Qualification, registration, and material preparation
The studied placenta was sourced during a Cesarean section in surgery block conditions from 21 donors (mean age 31, min 18, max 44 years old). Eight pieces were obtained from every amniotic membrane. The transport of tissue material must be validated and its parameters must be archived. Laboratory tests in relation to the living donor include HIV 1 and 2 (anti-HIV 1 and 2), hepatitis B (HBsAg and anti-HBc), hepatitis C (anti-HCV), and syphilis-specific test. An individual donation number, compatible with the ISBT128 system, has been given to each donor. The software (Bank Tkanek; ABG S.A.) and the computer, on which the operations are carried out, are associated with the production of cell transplants and must be previously validated. After being released from the quarantine (−80°C refrigerator Platinum 500; Angelantoni Industrie), the tissue material was admitted for preparation. A transport container with the material was moved into room class C, where the material preparation is held in sterile conditions. The preparation is conducted in the Cell and Tissue Culture Laboratory in vitro, together with the Tissue Bank in room class C, which is intended for tissue processing using the laminar airflow cabinet (Heraeus HSKS 18). The remnants of the amnion preparation are placed in a sterile container (Maco Biotech freezing Storage Pots 40–80 mL; Macopharma) and transferred to class B, where the primary cultures begin to grow. A Protocol of the culturing process is established for each newly found culture. The Protocol of the accomplished transplant is created after a finished transplantation. Each culture requires a Line Protocol. We should keep an accommodation plan in class B in mind because of the possibility of material cross-contamination from cells donated from different sources. Furthermore, a risk analysis must be performed, taking into account the risk of cross-contamination and the scientific basis in relation to the patient's safety. A documentation of the risk management process should be commensurate with the level of risk. Quality control assumes a biological analysis after the isolation and after each transit, and after transplantation. Fruition by the particular culture (line) of minimal criteria for stem cells should be applied to additional quality parameters. In accordance with the planned day of the experiment, a culture medium was swayed from the vessel and cells were separated by adding 1.5 mL of TrypLE™ Select (1 × ) solution, no Phenol Red (Life Technologies).
Isolation of amniotic cells
The standardization of the primary amniotic cell culture aimed to select the most efficient quantitative method while maintaining the desired parameters of the viability and amount of the apoptotic cells. The pieces of the amnion were divided into 4 × 4 cm parts to preserve the initial number of cells. To achieve the optimal cell isolation method, a selection of the following enzymes was used: Dispase II, Trypsin, and Collagenase I. Additionally, in comparison, mechanical isolation by tissue homogenization was carried out using the mesh of 70 μm (BD Falcon). It was then that the isolated pools of cells underwent a preliminary test–the optimal isolation method was selected on the basis of research into the amount of obtained cells, their vitality, and the percentage of apoptotic cells by using the set The Tali® Apoptosis Kit on image cytometer Tali® Image-Based Cytometer (Life Technologies). The analysis was performed by the producer's protocol. We also examined the long-term effects of the isolation method (21st day of culture) on growth kinetics and doubling of the population cells.
Analysis of cell growth kinetics in culture
To analyze the culture doubling time (DT), the cells were detached from the bottom of the vessel [TrypLE Select Enzyme (1 × ); Life Technologies] at certain intervals of time. The DT was calculated with the following formula:
t 2—the time of further measurement (further interval time)
t 1—the previous time of measurement (the previously analyzed time interval)
n 2—the number of collected cells during further measurement (further interval time)
n 1—the number of collected cells during the previous measurement (previous interval time)
An analysis of the population doubling factor
To analyze the population doubling time factor (PD), the cells were detached from the bottom of the vessel [TrypLE Select Enzyme (1 × ); Life Technologies] and, after the planned culture termination (21 days), the cells were compared to the number of seeded cells. The DT was calculated with the following formula:
N h—the number of cells on termination of culture
N 0—the number of seeded cells
Culture conditions
Culture conditions for the primary amniotic cell culture were tested. The culture medium was changed every 72 h using the laminar air flow cabin in accordance with GMP. The impact of the used culture medium on the amniotic and apoptotic cell's viability and the obtained number of cells were tested using The Tali Apoptosis Kit. The vessels with cells were kept in the incubator (Heracell 240; Thermo Scientific) under these conditions: temperature of 37°C, air containing 5% of CO2, and constant air humidity (95%). The culture was conducted in six-well plates of 9.6 cm2 in 3 mL medium. The cell culture flasks of 75 cm2 surface were used to obtain a sufficient amount of cells for the execution of column-based separation and for surface markers' analysis. The amniotic primitive cultures were conducted using the Ham's F12 Nutrient Mixture, AmnioGrow Plus Mediums, KGM-CD, and DMEM-ATMP mediums. The amniotic stem cell culture, which was isolated by the column-based method from the amniotic primitive cell population, was conducted on the MSC Basal Medium with standardized supplement Mesenchymal Stem Cell Stimulatory Supplements.
Stem cell isolation from the amnion cell culture
After choosing the optimal method of isolation and the primitive amniotic cell culture, the stem cells were selected by using the column-based method. The isolation was made from the primitive culture of the opportunity of the first passage. A proper surface antigen, on the basis of which the selection of the cells was effectuated, was chosen thanks to the amniotic cell culture using a flow cytometry method and by the nonvoluntary kit to the mesenchymal stem cells markers analysis (BD Stemflow™ hMSC Analysis Kit). The stem cells were isolated from heterogenic mixture of amniotic cells by using a positive selection after-surface marker CD105 (CD105 MicroBeads, human; Miltenyi Biotec). The manual cell separator (MACS) column was put into the MACS separator (Miltenyi Biotec) ensuring a magnetic field, which is essential to hold the labeled cells.
Quality control
Fulfilling the minimal criteria for stem cells
The cells were assessed after the column-based method (first passage) and before the graft.
Adherence to plastic
The adherence to plastic assessment was effectuated by microscopic observation using the fully-motorized inverted microscope, Olympus IX81 (Olympus).
Differentiation in three cell lines of mesoderm
The above-mentioned analysis was performed with the Human Mesenchymal Stem Cell Functional Identification Kit (R&D Systems). Supplements and growth factors facilitated the differentiation of human mesenchymal stem cells into adipocytes, chondrocytes, and osteocytes. A panel of verifying antibodies consisting of goat anti-mouse FABP4, goat against human aggrecan, and mouse against human osteocalcin was made. The cells were seeded and cultured by the protocol. During adipogenesis, the cells were seeded in a concentration of 3.7 × 105 and cultured by using the alpha modification of minimum essential medium (αMEM) with supplements, which differentiate into adipocytes. During chondrogenesis, the cells were seeded at a concentration of 7.4 × 104 and cultured using the αMEM medium with supplements, which differentiated into osteocytes. During osteogenesis, the cells were seeded at a concentration of2.5 × 105 and cultured using the Dulbecco's modified Eagle's medium (DMEM) with supplements, which differentiated into adipocytes. The medium was changed every 72 h. After 14 days of the culture, the cells were consolidated with 3.7% formaldehyde (Avantor Performance Materials Poland S.A.) solution in phosphate-buffered saline (PBS; Biosera). The cells were consolidated for 30 min. The permeabilization process was effectuated for 20 min with the incubation of 0.5% Triton X-100 (Sigma-Aldrich) solution in PBS. The washing buffer was a 3% solution of bovine serum albumin in PBS. One microliter antibody was put into a one-well plate. The time of incubation with the antibody was 45 min. The incubation was held in the dark. Surface antigens: CD34 CD45 were tested by primary antibody conjugated with fluorochrome Alexa Fluor® 350 dye (Bioss, Inc.). Fixation and permeabilization were performed, as described before. The nuclei were stained by a 30 min incubation with SelectFX® Nuclear Labeling Kit (DAPI, SYTOX® Green, 7-AAD, TO-PRO®-3 Iodide), for fixed cells (Thermo Scientific), according to the manufacturer's protocol. The intensity of the histochemical reaction was assessed by the semi-quantitative method, “Score.” The staining was estimated by the following criteria of intensity: 0—invisible staining, 1—visible staining, 2—weak (but clearly noticeable) staining, 3—medium intense staining, and 4—very strong staining. A 100 randomly chosen cells were assessed in each preparation. The results were calculated on the basis of the following formula: Staining intensity = staining degree (d) × number of cells (n) [12]. A microscopic observation was performed by using the fully motorized inverted microscope Olympus IX8, and photos were taken by the Cell M program (Olympus).
Cell markers analysis, which are specific for mesenchymal stem cells
To analyze the surface markers, a flow cytometer (FacsAria I; Becton Dickinson) was used. The cells were analyzed by the BD Stem flow hMSC Analysis Kit (BD Biosciences).
The maximal time analysis of the amniotic stem cell culture
Cells were seeded in the amount of 1 × 104 cells per flask (75 cm2-Sarstedt) on a six-well plate to analyze the population doubling with the aim to assess the aging of culture by the Cellular Senescence Assay test (Cell Biolabs). The population doubling, which had come after the achievement of a 90% confluence with each cell passage, was analyzed. Meanwhile, the cells from the six-well plate (Thermo Fisher Scientific) were detached and the aging of the culture was analyzed following the producer's protocol.
Transfer standardization
Various tools were used to transfer to the biggest cell sheet. Several different materials were used in order to obtain the sheet with greatest amount of cells. The use of Dispase II was the reference (2.4 U/mL; Gibco). The cells were seeded in six-well plates at 300,000 per plate. The culture was conducted with the aim of achieving 100% confluence. Each tool used for the transfer was manipulated for 1 min. The cells were examined in regards to their number and viability using the Tali Viability Kit. The cells were then transferred to a clear plate for 48 h before a readhesion assessment was made.
Cell transfer by the thermo-scientific mediums
Three hundred thousand of the amniotic stem cells were seeded for thermo-scientific, available Nunc™ Dishes with UpCell™ Surface (Thermo Scientific). The cells were detached following the producer's protocol.
Statistical analysis
The study was repeated three times (seven amniotic membranes from different donors every time). Statistical analysis was performed with STATISTICA 12 software. Shapiro–Wilk was used for confronting the assumption that the population is normally distributed. A one-way analysis of variance (ANOVA) was used for samples with a homogenous variance. The nonparametric equivalent of ANOVA was the Kruskal–Wallis test. For equal variations, Tukey's post hoc test was used, and for unequal variations, the Games-Howell test was used. The significance level was set to 0.05 (5%).
Results
Amniotic cell isolation
The average number of cells obtained by the tissue homogenization process is the highest (117.8 × 104/mL) and is statistically higher than cells obtained by Collagenase (P < 0.01) and the Trypsin enzyme (P < 0.01). However, it has been proven that a statistically significant difference between the mechanical tissue fragmentation and the cell's etching by Dispase enzyme does not exist, as the average number of obtained cells by using this method did not reach a million to 1 mL and is only 88.7 × 104 (Fig. 1). The statistically significant difference among the cells obtained using Dispase enzyme, Collagenase enzyme (P < 0.01), and Trypsin enzyme (P < 0.01) was observed. In both cases, the number of isolated cells was four times higher when using the Dispase enzyme. The lowest average number of cells was obtained by etching cells using Trypsin enzyme (16.1 × 104/mL). Furthermore, a statistically significant difference between the cell capacity and the population doubling during the growth of the culture depended on the isolation method. The doubling population of cells was significantly smaller than in the case of cells isolated by the Collagenase enzyme against mechanical tissue fragmentation (P < 0.01), etched by Trypsin (P < 0.01) and etched by Dispase (P < 0.01). The highest doubling population rate was noted for the cells etched by Trypsin enzyme, and the smallest—for cells obtained by etching of the Collagenase enzyme (Fig. 1). There was no statistically significant difference in the other groups treated. The time of the doubled culture was also examined in the time intervals. The highest rate was noted on the 12th day (Fig. 1) for cells obtained by Collagenase etching (2.7 of a day) and Trypsin etching (2.7 of a day), while the smallest rate was the case of cells analyzed on the 21st day of the culture obtained by homogenization (for 0.5 of a day) and of cells etched by Dispase (0.5 of a day). A negative rate of DT was observed on the third day after cell isolation by the Trypsin enzyme (−4.2). An analysis of amniotic cell viability showed that a statistically significant difference between the viability of isolated cells by the mechanical method (homogenization) and cell trypisinization (P < 0.04) exists. It was found that an amniotic cell's viability is significantly greater in the case of mechanically isolated cells than in the case of cells isolated by Trypsin etching (Fig. 1). The average viability for homogenization is 70% and in the case of Trypsin etching, it is 58%. In the other cases of isolation, there was no significant difference in the cells' viability. There were no statistically significant differences observed in terms of the amount of apoptotic cells in treated groups. The average amount of apoptotic cells oscillated between 8% and 13% (Fig. 1).
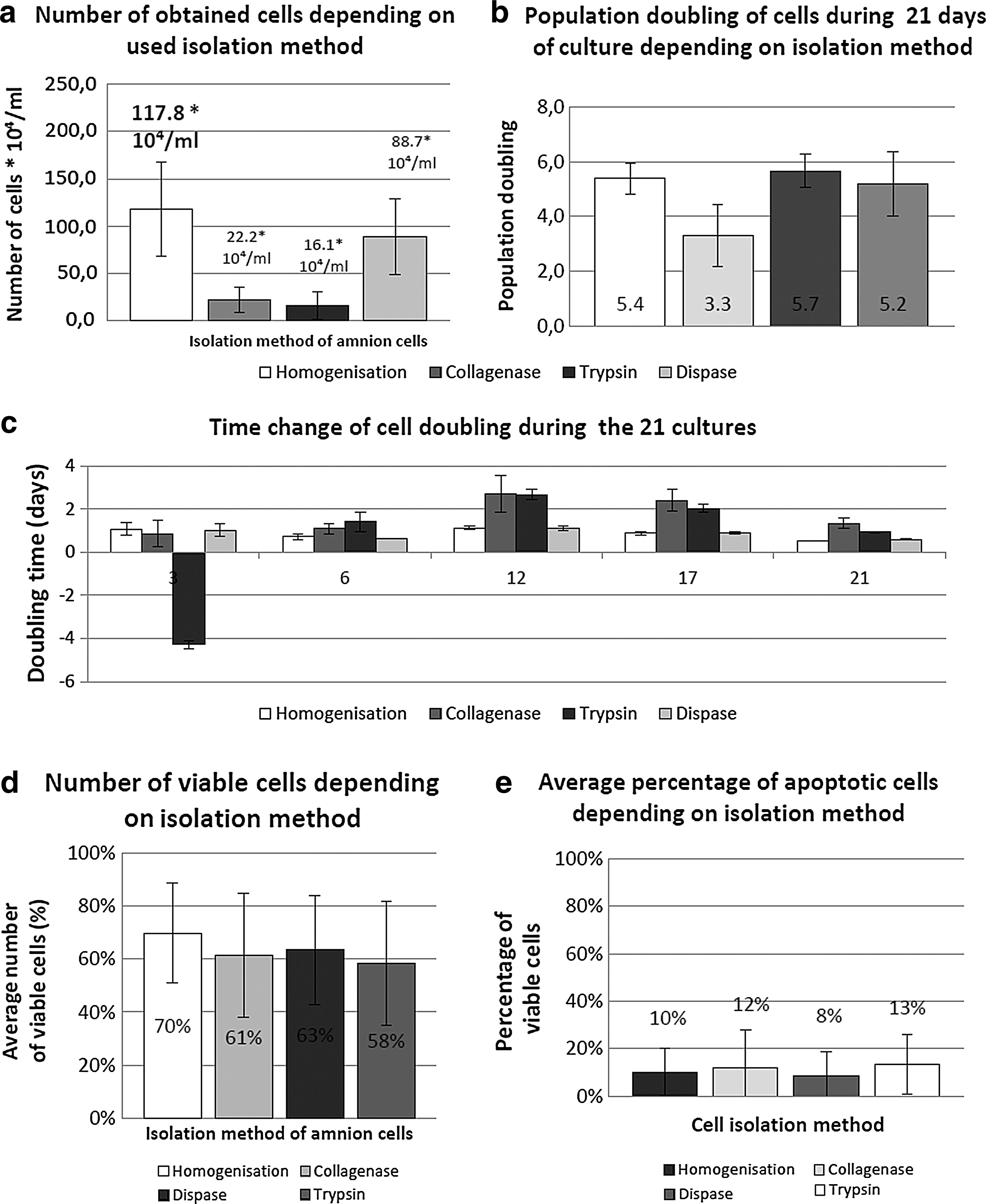
Culture conditions
Depending on the used culture medium, various amniotic cell morphologies were observed. The use of KGM-CD and Ham's F12 medium promoted cell growth morphology, which is similar to the epithelial cells (Fig. 2), while the cultured medium of the AmnioGrow and DMEM stimulated cell growth, which has a fibroblast-like character. One of the conditions for the recognition of isolated cells for the stem cells (quality control) is similar to the fibroblast's morphology, which was confirmed in this study.

It was found that there is a statistical significance difference in the number of cells obtained only using the AmnioGrow and the KGM-CD medium (P ≤ 0.01). The highest average number of cells obtained was the culture on the AmnioGrow medium (82.0 × 104/mL), while the lowest average was on the KGM-CD medium (26.5 × 104/mL). In the cases of cultures on the rest of the mediums, there are no statistically significant differences in the number of cells obtained. It was proven that there is a statistically significant difference between the ability of cells to double the population during the growth of the culture depending on the culture medium used. The differences between the number of doublings in the culture were observed between the AmnioGrow, and KGM-CD medium and was (P < 0.01). The highest doubling factor was noted for cells cultured using AmnioGrow (1.5 time), and the lowest for the cultures that were conducted using KGM-CD medium (−0.8; Fig. 2). A change of the DT of the cell population depends on the day the culture was examined. The highest rates apply to the cultures on the DMEM (1.2 days) and the cells incubated in the KGM-CD medium (1.1 days) noted on the 17th and 12th day (Fig. 2). The lowest rate occurred in the case of cells cultured using the KGM-CD medium (−1.7 day), which means we cannot expect the cell separation for nearly 2 days when using this medium. An analysis of impact of the culture medium used for cell viability showed that the use of AmnioGrow medium provides a statistically much greater viability than the other mediums, such as DMEM (P < 0.01), KGM-CD (P < 0.01), and Ham's F12 (P < 0.01). AmnioGrow ensures the highest average of cell viability, 80% (Fig. 2). The smallest average is obtained when using KGM-CD medium (54%). It has been observed that there are no statistically significant differences in the number of apoptotic cells in the treated groups. The average number of apoptotic cells was 11% when using AmnioGrow and Ham's F12 medium (Fig. 3). The highest average of apoptotic cells was noted in the culture grown in DMEM (12%).

Stem cell isolation from the amniotic cell culture
Research into the presence of cell differentiation markers, conducted using the flow cytometer, indicated that the amniotic cells showed the lowest expression of this marker in comparison with the two others (CD73 and CD90). Therefore, the isolation was performed using the antibodies against the CD105 marker. These are essential to confirm and to show that the examined cells are mesenchymal stem cells. Because of the amniotic cell's tendencies to creating sheets using the column-based method causes a loss about 30% of cells.
Quality control
On the basis of an analysis of the previous parameters including population doubling, aging of a population, cell's viability (Fig. 3), and severity of injury the cells should be used between passages 3–6. A flow cytometry representative of the multipotent stem cells markers in freshly isolated hAMSCs occurs between the third and sixth passage. Data from the flow cytometer is enclosed (Supplementary Figs. S1–S3; Supplementary Data available online at
The maximum time analysis of the amniotic stem cell culture
An extensive quality control assuming the minimal assessment criteria for stem cells was performed on the selected passages (Fig. 4).
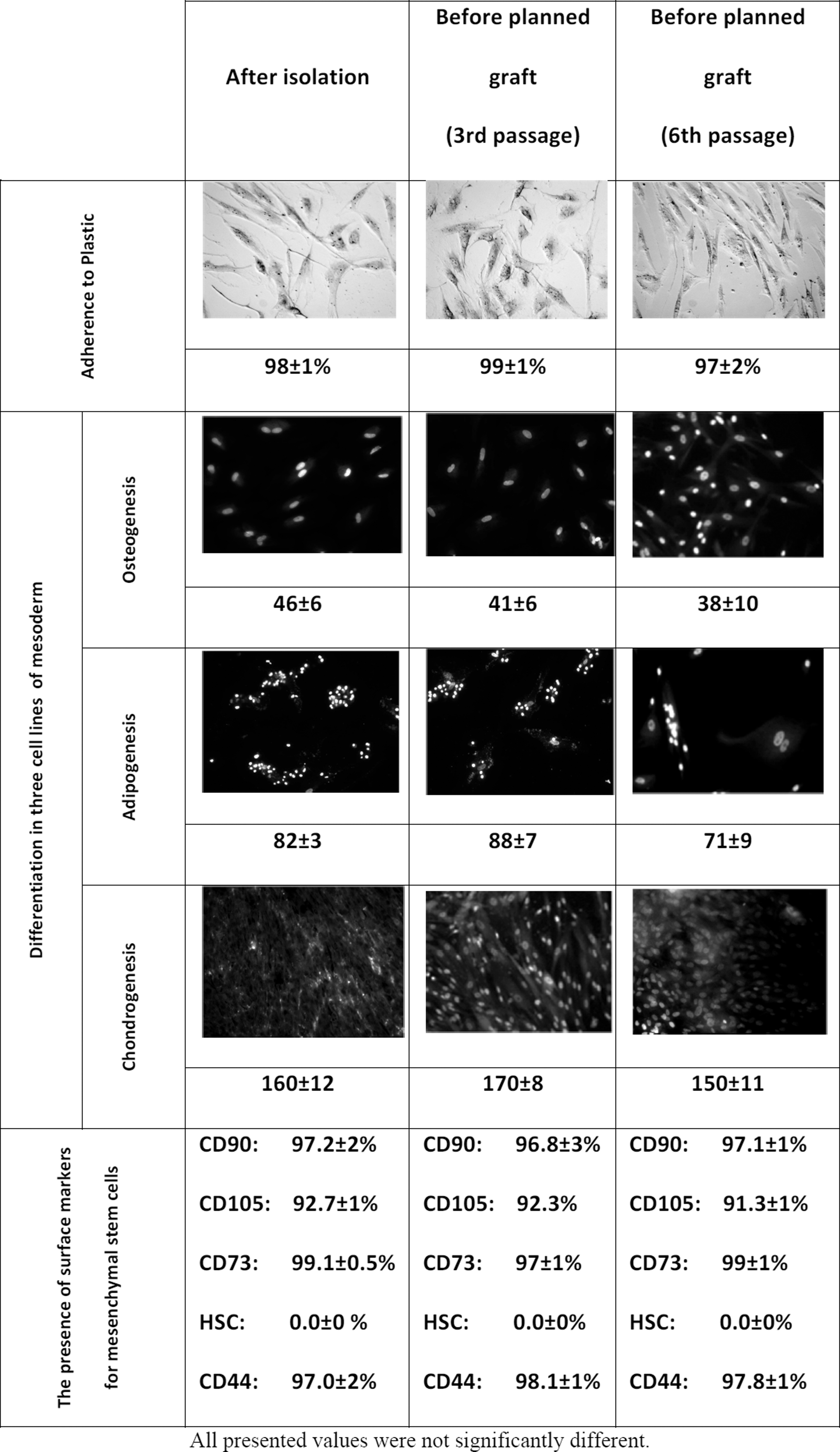
Assessment of the quality control parameters in three time intervals.
Transfer standarization
It has been proven that statistically significant differences exist in the number of cells that were transferred depending on the selected transfer method (P < 0.01). The highest number of cells is transferred when using the enzymatic method (87.8%), causing the sheet's disintegration (Fig. 5). The use of Dispaza II enzyme ensures that a statistically higher number of cells is obtained than while using the HydroGel dressing (P < 0.01), Suprathel dressing (P < 0.01), medical mesh (P < 0.01), and the mesh together with flushing Pasteur pipette 3 mL (P < 0.01), or the dried collagen dressing (P < 0.01). A statistically significant number of cells, transferred by lyophilized collagen, was observed in comparison to those observed when transferring by HydroGel (P < 0.01), Suprathel dressing (P < 0.01), medical mesh (P < 0.01), the method combining the Pasteur pipette flushing and the use of the medical mesh (P < 0.01), allogeneic skin (P < 0.01), and dried collagen dressing (P < 0.01). The smallest number of transferred cells is obtained by the application of dried collagen (P < 0.01) dressing. Number of cells transferred by dried collagen in comparison to the number of cells transferred by the allogeneic skin and amnion, as well as those transferred using liophylized collagen wound dressing, and those obtained with the Pasteur pipette flushing 3 mL (P < 0.01) differs significantly.

Furthermore, there was a statistically significant difference observed in the transferred cell's viability (P < 0.01). The cell's viability when transferred by the allogeneic skin is higher than when using HydroGel wound dressing (P < 0.01), Suprathel dressing (P < 0.01), medical mesh (P < 0.05), and when using a combined method consisting of a medical mesh and a Pasteur pipette 3 mL (P < 0.01). An application of the amniotic membrane as a membrane used for the transfer also ensures a higher cell viability when transferred using HydroGel dressing (P < 0.01), Suprathel dressing (P < 0.01), medical mesh (P < 0.02), and the method using the medical mesh and Pasteur pipette 3 mL (P < 0.04). Additionally, the use of the amnion to the transferring cells ensures a statistically higher viability than the cell transfer by the dried collagen dressing (P < 0.04). A good viability obtained by the lyophilized collagen dressing ensures a statistically higher viability than when using HydroGel wound dressing (P < 0.01), Suprathel dressing (P < 0.01), medical mesh (P < 0.01), or the combined method (P < 0.02). The application of the lyophilized collagen dressing, like that of the amniotic membrane to cell transfer, ensures a significantly higher viability than the cell's transfer with dried collagen (P < 0.02). The cell's transfer with the Pasteur pipette (3 mL) flushing method also ensures higher viability than the membrane's transfer in the HydroGel (P < 0.01). The highest viabilities observed include those using the lyophilized wound dressing (97% of transferred cell population was alive), allogeneic amnion (96% of cells), and allogeneic skin (95% of cells; Fig. 5). According to research, the best results in transferring the full cell sheet, in relation to the transfer of the enzymatic method, are given with the use of the lyophilized collagen dressing, and amnion transferring, in addition to the allogeneic skin and use of the Pasteur pipette 3 mL. It should be noted that using a pipette may cause a change of spatial orientation of the sheet, while using the skin, amnion and collagen dressing leads to the transfer of a “straightened” cell sheet in the originally orientated formula, harvested from the bottom of the vessel.
The cell transfer was conducted using thermo-scientific mediums. After 48 h, the cells showed the tendency to spot, detach from the medium unprompted and uncontrolled, and roll (Fig. 5). After the transfer, the cells were swimming in the medium in the shape of an involuted sheet (Fig. 5), which rolled up, despite many attempts to straighten it by using sterile tweezers. An intense manipulation of the tweezers caused the tearing of the sheet. Due to the uncontrolled formation of the sheets and swaying cells from the medium during the culture standing, further research was desisted.
Discussion
Various conditions of isolation and cell cultures, and the number of passages, will probably affect the phenotypes and the functioning of the cells [8]. Taking into account the heterogenicity of the stem cell population derived from different sources, the isolation of the specific cell types is difficult and requires the fulfillment of exact requirements [11]. Roubelakis et al. proved that amniotic cells are composed of heterogeneous groups of adherent cells [19]. A selection of specific methods for the isolation of cells before their application in the clinic is crucial, as is the definition of the culture procedures and the maximum time for the culture [13]. Different protocols of amniotic stem cell isolation consisting mainly of mechanical separation from the amniotic membrane from the chorion and latest enzyme digestion have been determined so far [14]. There are three major protocols for the isolation of AFSCs from the human amniotic fluid [20], but very few up-to-date protocols exist concerning the isolation from the amniotic membrane. Methods relying on oxygen tension, two-stage culture, and alternative media formulations have been reported [11,12]. One mechanical method and three enzymatic methods of amniotic cell isolation were analyzed in this article. The highest viability and the number of cells were obtained by mechanical isolation. However, these parameters do not significantly differ in relation to the Dispase enzyme. The application of the Collagenese enzyme results in less multipliable doubling than those occurring in the other isolation methods. The use of the Trypsin enzyme affects the initial number and viability of the isolated cells, which also reflects population doubling at the time that was recorded on the third day after the isolation. However, the amniotic cells isolated in this way show the largest number of doubled population, although they are not significantly greater than the cells isolated mechanically when using the Dispase enzyme. Miao et al. isolated adherent cells from trypsin-digested on-term placentas [21]. We can note from his results that the amniotic cells should be isolated by either using the Dispase II enzyme or through the tissue homogenization process. The references indicate that the use of the Dispase II enzyme affects the amniotic membrane structure, which may have an impact on the growth of the cells cultured from the isolated explants of this method [15]. Soncini et al. suggested a two-stage isolation by mechanical separation and subsequent enzymatic digestion [22]. In the view of obtained results, the author of the work suggests that the cell should be isolated by a one-step procedure—the mechanical fragmentation of the amniotic membrane Four medium cultures were used for culture process standardization. In ‘t Anker et al. cultured cell on M199 (Gibco Laboratories), Tsai et al. on αMEM (Gibco‐BRL), Steigman et al. on DMEM (Lonza), and Roubelakis et al. on DMEM (Sigma-Aldrich) [8,19,23,24]. No standardization or even comparison between the media was performed before this study. We have decided to choose two standard mediums (Ham's F12 and DMEM), one for amniocytes (AmnioGrow Plus) and one promoting epithelial-like cells (KGM-CD). The highest viability was obtained in cultures using the medium intended for the amniotic fluid cell culture–AmnioGrow. The viability of the cultured cells with application to this medium was significantly higher than in the case of cells treated with different mediums. Also, the amount of population doubling and the amount of cells obtained from a culture with the use of this medium is the highest, although it is not significantly higher than in the culture using the DMEM, which is normally used for cell culture [16,25,26]. In view of the obtained results, it is suggested that the DMEM should be changed in favor of the AmnioGrow medium. In the published works, the authors achieved a plateau after 21st day of culture [17,27]. In this article, the lowest average of the DT was obtained on the 21st day of cell culture. In ‘t Anker et al. it reached a plateau of growth after 11 days. It should be noted, however, that specimens of amniotic membrane and amniotic fluid were included in the study [23]. Initially, a mixture of amniotic cells, which had the epithelio-fibroblast-like morphology was observed, and disappeared soon after. Furthermore, elongated, fusi form, and fibroblast-shaped cells were all observed in the culture. The other authors suggest that the epithelial cells were rarely detected after the third passage, which would suggest the desirability of the graft after the third passage. Those differences may result from the various methods and cultures used. References suggest the presence of the amniotic cell hybrid phenotype as a sign of pluripotency and a suggestion that the cells derived from the amnion are not completely differentiated, from to the epithelial cells nor from the mesenchymal cells [8,17]. However, the mesenchymal cells (hAM-MSCs) show an adherence to plastic and a fibroblast-like morphology, while the amniotic epithelial cells show the phenotype of the cuboidal epithelium [14]. Eluting cells, which have epithelium's phenotype in aid of adherent cells and the morphology similar to fibroblasts, are one of the existing proofs of the presence of amniotic cells in examined stem cell cultures. They are also a reason for which amniotic cells should be used for further research or for clinical aims after the column-based separation. However, Parolini et al. stated that in culture, both human amniotic epithelial cells and amniotic mesenchymal stromal cells can differentiate toward “classic” mesodermal lineages [28], so they may be valuable for further clinical use. Notwithstanding, Parolini et al. [28] and Miki and Strom support the necessity of discarding epithelial cells [29]. The presence of epithelial makers (CD49f and CK18) that disappear when the culture is in progress [8,17] is proof of the existence of epithelial cells. The subpopulations of amniotic cells can be maintained in undifferentiated conditions [16,18]. The ability to differentiate into three lines of mesoderm was also confirmed in this study, both for the cells after column-based isolation and after the third and sixth passage. According to the references, the hAM-MSCs may differentiate into “classical” cells of mesoderm lines (osteogenesis, chondrogenesis, and adipogenesis), and into all three types of layers: the ectoderm, mesoderm, and endoderm [13,17,28].
Numerous works suggest that the amniotic cells show a low level expression of major histocompatibility complex antigens [29]. Their expression of major histocompatibility complex antigens (MHC class I) is even lower than the mesenchymal stem cells of the bone marrow [30]. The results obtained by the author of the work indicate a very low expression of human leukocyte antigen-D-related (HLA-DR) after the first passage. After the second passage, the result is close to zero, which demonstrates a lack of expression. However, the difference in the primary culture was observed. This may result in the impurity of other cells or, as a consequence, of the immune response of unknown etiology involving the placenta. This hypothesis can be conjectured on the basis of research into the HLA-DR expression on the surface of keratinocytes derived from the biopsy of 52 various skin diseases [31]. It is believed that the lack of the HLA-DR (MHC class II) and a very low level of HLA-ABC antigen (MHC class I), along with the so-called “immune privilege” of the fetus, results in a low immunogenicity of the amnion cell [30]. Furthermore, the mesenchymal stem cells derive from the amnion (hAM-MSCs) and hamper the allogeneic response. The main immunomodulation mechanism is secreted by the MSC-soluble factors, such as the transforming growth factor-beta, hepatocytes growth factor, and prostaglandins E2. Furthermore, cell-to-cell contact is also a possible factor having an impact on immunological modulation [10]. To sum up, the mesenchymal stem cell of the amnion may be used as a source of cells for allograft, which plays a significant role in regenerative medicine [30].
The references indicate that the hAM-MSCs show the fibroblast's morphology, presence of surface markers on the human bone marrow-derived mesenchymal stem cells, and isolated cells both from the amnion and other parts of the fully mature epithelium, such as CD29, CD44, CD105, CD73, CD90, and vimentin [17]. In this work, the presence of the stage-specific embryonic antigen 3 (SSEA-3) was demonstrated. In connection with the obtained results, the author of the work claims that the tested cells are the stem cells, as the definition for progenitor cells states that they are the cells that determine the ability of separation and further differentiation within one germ layer [32]. On the other hand, the tested cells meet the criteria ascertained by the International Society for Cellular Therapy in 2006, necessary to identify the mesenchymal stem cells and even to determine the embryonic antigen expression [9]. This statement was confirmed by the lack of CD34 and CD45 antigens. The lack of those antigens was also stated by Paracchini et al. [17]. To sum up, the studies, carried out on the basis of used literature, indicate that cells that are more primary than mesenchymal cells exist [33,34]. The presence of the cell antigens was confirmed after the third and the sixth passage. On the basis of the results, viability, apoptosis, aging of culture, estimated number of cells, microbiological tests in conjunction with the level of HLA-DR, and SSEA-3 demonstrated that amniotic stem cells are the most dangerous between the third and sixth passage. Therefore, we should not use the cells from the third passage due to a clinically significant number of cells and a small probability of the epithelial cells' impurity. The culture fulfills the minimal criteria for the mesenchymal stem cells to the sixth passage, without any signs of culture aging and the viability on the level of ∼90%.
It is believed that only 5% of the stem cells are injected into the wound's placenta and those cells are able to survive the initial phase. The level of settlement by locally provided stem cells may be low, so that they significantly regenerate and play in full functioning tissue [35]. These problems can be solved by the use of a natural or synthetic scaffold [36]. A perfect mechanism has not been devised yet [37]. Park et al. suggest providing the stem cells with fibrin glue [38]. However, in the experience of the Centre for Burn Treatment, this method is far from ideal and was therefore replaced by the use of a platelet-rich leukocyte concentrate [16]. It is often suggested to use biogels to ensure the maximum integration to the wound without the risk of infection [35]. It has been demonstrated not so long ago that the encapsulation of embryonic stem cells in hydrogel may maintain them in an undifferentiated condition by the sixth month in case there is lack of mechanical or enzymatic passages and no common culture with nourishing cells exists. The hydrogel scaffolds are characterized by hydrophilic properties. When the cell's viability is maintained, stem cell encapsulation happens. [36]. The references show that they create a three-dimensional microenvironment for the stem cells that strictly follows the in vivo conditions, leading to a stimulation of cell proliferation and their differentiation. However, most of the HydroGel dressings are formed by UV cross-linking or require a multi-step modification obtained by chemical reactions and clearing methods, which causes security problems, increases costs, and requires complex manufacturing methods [37]. The unique ability of amniotic cells to create sheets was observed both for the heterogeneous mixture and for stem cells. This may cause the existence of strong intercellular connections; however, this hypothesis requires additional research. The above-mentioned property was noticed during the culture flushing of the PBS before the enzymatic digestion had been used. A strong flushing of the PBS is sufficient for detaching cells from the culture in the form of flakes. This fact is beneficial for four reasons: financial (no need to use the enzyme), viability, number of cells (the negative impact of the use of the enzyme on the amniotic cell culture was demonstrated), and the possibility of transfer of a full sheet of cells to the wound. This appearance was used for developing the amniotic cell transfer method to the wound. Nine transfer methods of full sheet methods were tested. The methods were tested against the enzymatic method. There was no difference among the number of cells transferred by the enzymatic method, lyophilized collagen dressing, allogeneic skin, and amnion. The highest viability of transferred cells ensures the use of lyophilized collagen wound dressing, skin, and allogeneic amnion. It is an innovative solution—a standard clinical application is carried out by using the collagen-fibrin gel or PRLG concentrate (which contains cells) [39]. An alternative application is seeding cells onto biological and synthetic materials. Selim et al. [40] seeded cells onto polylactic-co-glycolic acid, Aho et al. [41] onto bio-absorbable mesh. Solving the technical and methodologic issues are major tasks ahead of us for the clinical application of stem cells [40]. Moreover, before routine implementation in the clinic, additional quality control tests must be considered, such as the assessment of the karyotype after the isolation and before the graft. Our pending studies concern, among others, long-term cryopreservation in different carriers (standard suspension in criomedia, hydrogel, collagen, allogeneic skin, and amnion) and its effect on cell genome stability and multipotency. We will take a closer look on the impact of the mother's age on the quality of the isolated cells. The large-scale production of these MSC for use in clinical studies or research projects has to be validated. Future studies will also pertain to the creation of a ready-to-use, combined ATMP—an acellular dermal matrix with seeded amniotic stem cells and keratinocytes, developed following GMP for the coverage of deep burns (IIb–III°).
Conclusions
The work on the manufacturing of amniotic cells must be carried out in accordance with the regulations of the Minister of Health regarding the Good Manufacturing Practice and the EudraLex—Volume 4 of GMP. This is because the human amniotic membrane-derived mesenchymal stem cells (hAMMSCs) have unique features, such as their eduction from the early embryo-development, a low level of the human leucocyte antigens expression, and a significant potential of differentiation. As they are distinct from embryonic cells, the capacity of the amniotic cells to produce teratomas was not ascertained [13]. The amniotic cells are a promising source in the treatment of burns and chronic wounds, and in the modeling of burn scars. In the Centre for Burn Treatment, the amniotic stem cell graft with hospital exemption was performed on a burned patient. Thirty-five percent of the burns were grafted. Thanks to this treatment, the patient was discharged on the 12th day after the treatment with a completely healed wound. This article shows the complete process of culture standardization, obtaining appropriate consent to the application of cells to a patient, which represents a strong basis for the implementation of amniotic stem cells as a medicinal product of advanced therapy in Tissue Banks across Europe.
Footnotes
Acknowledgments
First, I would like to express my sincere gratitude to my advisor, Prof. Kawecki for his continuous support of my PhD study. Besides my advisor, I would like to thank Dr. Glik for her insightful comments. My sincere thanks go also to Dr. Nowak and Dr. Klama-Baryła who provided me an opportunity to join their team as intern and gave me access to their laboratory and research facilities. I thank my fellow labmates for all the motivating discussions. Last but not the least, I would like to thank my family, my husband, my parents, and my brothers, for supporting me spiritually in writing this article and in my life in general.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.