
Abstract
Mesenchymal stem cells (MSC) represent a readily accessible source of cells with potent immune modulatory activity. MSC can suppress ongoing inflammatory responses by suppressing T cell function, while fewer studies have examined the impact of MSC on dendritic cell (DC) function. The dog spontaneous disease model represents an important animal model with which to evaluate the safety and effectiveness of cellular therapy with MSC. This study evaluated the effects of canine MSC on the activation and maturation of canine monocyte-derived DC, as well as mechanisms underlying these effects. Adipose-derived canine MSC were cocultured with canine DC, and the MSC effects on DC maturation and activation were assessed by flow cytometry, cytokine ELISA, and confocal microscopy. We found that canine MSC significantly suppressed lipopolysaccharide (LPS)-stimulated upregulation of DC activation markers such as major histocompatibility class II (MHCII), CD86, and CD40. Furthermore, pretreatment of MSC with interferon gamma (IFNγ) augmented this suppressive activity. IFNγ-activated MSC also significantly reduced LPS-elicited DC secretion of tumor necrosis factor alpha without reducing secretion of interleukin-10. The suppressive effect of IFNγ-treated MSC on LPS-induced DC activation was mediated by soluble factors secreted by both MSC and DC. Pathways of DC functional suppression included programmed death ligand-1 expression and secretion of nitrous oxide, prostaglandin E2, and adenosine by activated MSC. Coculture of DC with IFNγ-treated MSC maintained DC in an immature state and prolonged DC antigen uptake during LPS maturation stimulus. Taken together, canine MSC are capable of potently suppressing DC function in a potentially inflammatory microenvironment through several separate immunological pathways and confirm the potential for immune therapy with MSC in canine immune-mediated disease models.
Introduction
M
DCs are the most potent antigen presenting cells playing a pivotal role in both the initiation and determining the nature of adaptive immunity [19]. Thus it is important, from a therapeutic perspective, to gain insight into mechanisms responsible for the influence of MSC on DC function such as antigen presentation, maturation, and cytokine secretion. Several groups have shown previously that MSC interfere with the maturation of DC and the generation of Th1 immune responses, as well as reduction of IL-12 [4,14,17,20]. In mouse and human models, studies that demonstrate impairment of DC maturation, toll-like receptor (TLR) activation, migration, and differentiation of hematopoietic DC progenitors have revealed a variety of ways DCs are attenuated by MSC [13,14,21 –28]. Further studies are likely to reveal additional mechanisms by which MSC disrupt DC transition from immature antigen acquiring cells to mature cells optimized for antigen presentation.
Since results from studies using rodent versus human models have yielded, in some cases, noteworthy differences, veterinary studies involving companion animals are likely to serve as a more effective translational intermediary between preclinical and clinical studies in humans. For example, spontaneous diseases appearing in veterinary patients are more representative of the human condition compared to those artificially induced in the laboratory rodent model. Supplementation of knowledge derived from mouse models through investigative veterinary research will likely expedite the goal of reducing failure rates in human clinical trials.
Since domesticated companion animals are generally outbred, have more variation in their diet, and have a more diverse environmental exposure than laboratory rodents, the external influences potentially contributing to progressive degenerative disease are likely more similar to their human companions [29]. Indeed, the canine spontaneous disease model has been advanced for the value it brings in testing new therapeutics for human diseases, including cancer, degenerative, inherited, and inflammatory disorders [30 –39]. The knowledge and resources obtained from veterinary medicine is often overlooked as potentially more relevant translational approaches to human disease, as well as their value of expediting development of therapeutics, drugs, and devices [29].
In the stem cell field, MSC have been evaluated for treatment of canine osteoarthritis, inflammatory meningitis, spinal cord injury, and inflammatory bowel disease [29,40 –44]. Thus, understanding mechanisms of MSC function in domesticated canines and their application to treatment of spontaneous diseases offers significant potential benefit to advancing our understanding of human stem cell therapeutics. Previous studies have investigated mechanisms of MSC suppression of canine T cell function [45,46], but the consequences of interaction of canine MSC with DC have neither been previously investigated nor have mechanisms of activation/suppression.
We hypothesize that canine adipose-derived MSC (hereafter referred to as “MSC” in this work) suppress peripheral blood mononuclear cell (PBMC)-derived DC function and maturation in much the same way as has been described in either the mouse or human models and that the mechanisms we observe in the canine model may share components observed in both models, as well as reveal novel components unique to the canine model. Therefore, in the present study, our aim is to investigate the interaction of canine adipose-derived MSC with canine PBMC-derived DCs (hereafter referred to as “DC” in this study), assessing their effect on DC activation, maturation, and function. DC can successfully be differentiated from canine PBMC using well-established methods [47 –49]. Our data indicated that canine MSC potently suppress DC functionality in vitro, using multiple distinct and nonoverlapping immunological mechanisms.
Materials and Methods
Institutional animal care and use approvals
All animal protocols for the procurement of blood from dogs at the Colorado State University (CSU) Veterinary Teaching Hospital were approved by the CSU Institutional Animal Care and Use Committee (IACUC). Adipose tissues used for generation of canine Ad-MSC were procured from purpose-bred animals used in teaching studies, after humane euthanasia, in accordance with CSU IACUC approvals.
Generation of adipose tissue-derived MSC
MSC were derived from the stromal perivascular fraction of adipose tissue derived from inguinal fat of specific pathogen-free purpose-bred Walker hounds. Subcutaneous adipose tissue was minced, divided into 1 g aliquots in 1 mL of freezing medium (11% DMSO, 14% complete medium Dulbecco's modified Eagle's medium (DMEM) [see below], and 75% fetal bovine serum [FBS]) and stored in liquid nitrogen before future use. Adipose tissue biopsies were thawed and enzymatically digested with collagenase type IA (Sigma-Aldrich, Saint Louis, MO) at 37°C for 30 min and centrifuged at 670g in complete DMEM (Gibco/Thermo Fisher, Grand Island, NY) containing 15% FBS (VWR-Seradigm, Inc., Aurora, CO), 2 mM
Preparation of canine monocyte-derived DC
Preparation of monocyte-derived DC was performed as previously described [48,49]. Briefly, peripheral blood from dogs was collected into tubes containing EDTA as an anticoagulant. Blood was diluted with an equal volume of sterile phosphate-buffered saline (PBS) and layered over Lymphocyte Separation Medium (LSM®) (MP Biomedicals, Inc., Santa Ana, CA) and centrifuged for 30 min at 370g without break engagement. PBMCs separated from red blood cells in the LSM/PBS interphase were carefully removed and washed 2 × more in PBS and suspended in complete DMEM. The PBMCs were plated in 96-, 48-, or 24-well plates at between 2.5–5 × 106 cells/mL and allowed to adhere to the wells for 3 h at 37°C. The wells were subsequently agitated gently and decanted with a pipette to remove nonadherent PBMCs. Adherent cells were between 70% and 85% CD14+ (Fig. 1A). Freshly prepared complete DMEM containing 50 ng/mL recombinant human granulocyte macrophage colony-stimulating factor (hGM-CSF) and 10 ng/mL recombinant mouse interleukin-4 was added to the remaining (adherent) monocytes to stimulate differentiation to immature DC (iDC). The differentiation medium was changed 48–72 h later, and full differentiation to CD11chi cells was apparent between 5 and 7 days among the high forward and side scattered events (larger cells) (Fig. 1B). Flow cytometric analysis revealed that CD11chi cells were greater than 95% MHCII+/CD86+/CD40+(Fig. 1). Maturation of iDC was achieved by treatment of immature CD11c+ cells with 50 ng/mL Escherichia coli lipopolysaccharide (LPS) 055:B5 (InvivoGen, San Diego, California) for varying time points (24–36 h). PBMC-derived DC were assayed for activation/maturation as determined by upregulation of surface expression of MHCII, CD86, and CD40 by flow cytometric analysis.
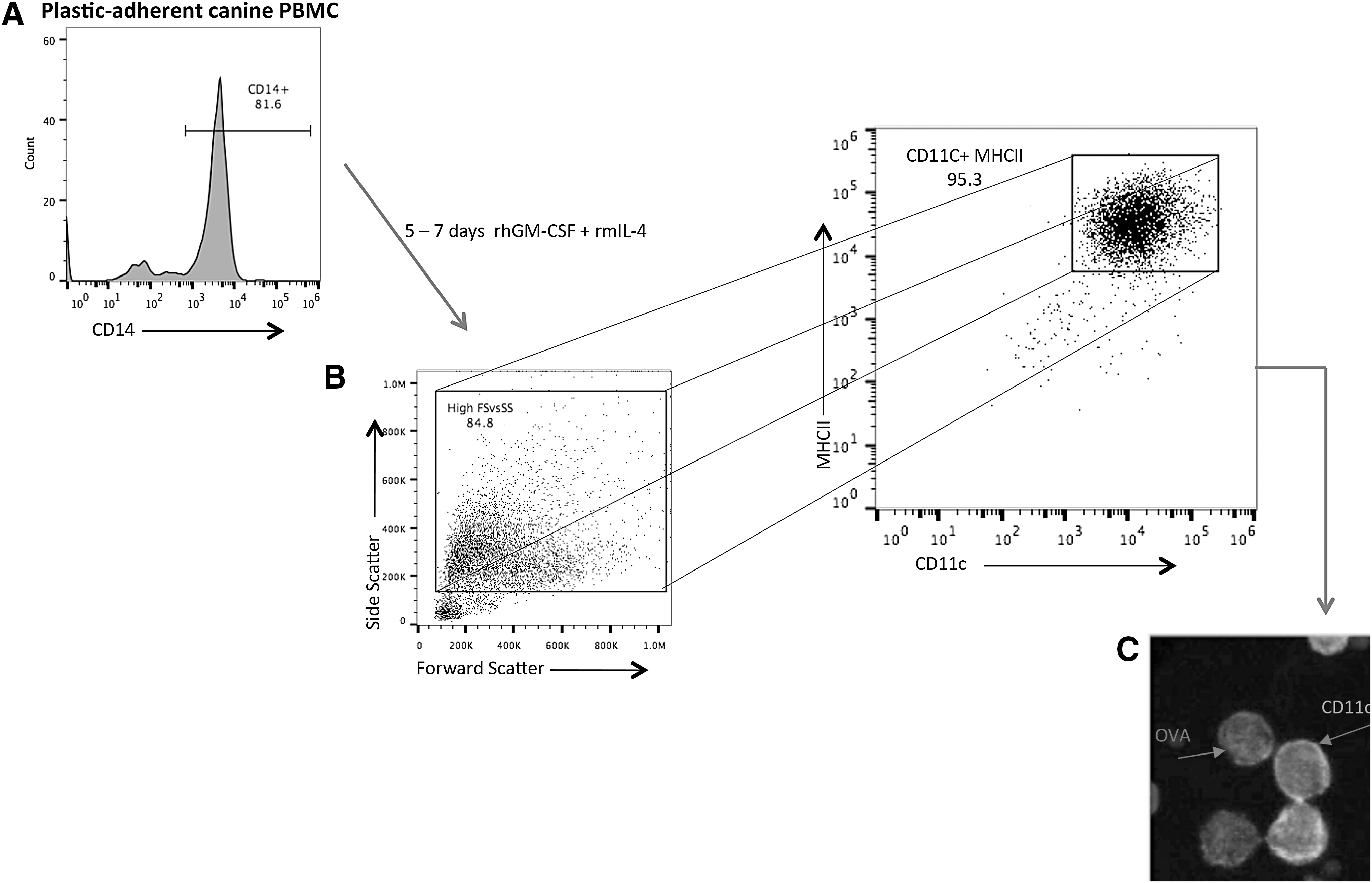
Preparation of monocyte-derived dendritic cells (DC).
MSC and canine DC cocultures
MSC were treated for 18 h with or without 100 ng/mL recombinant canine interferon gamma (IFNγ) (R&D Systems, Minneapolis, MN) and harvested and counted. MSC were mixed into wells containing canine DC approximately 30 min before stimulation with LPS. The ratios of MSC to DC in cocultures were 1:10, 1:100, or 1:1,000 depending on experimental protocol. After 24–36 h of culturing with or without LPS, loosely adherent DC were harvested for flow cytometric analysis from the tightly adherent MSC by gentle agitation of the wells through pipetting. The influence of MSC on DC activation/maturation was assessed as described above for DC maturation determination. DC were identified as those events that were CD11chi and expressing MHCII.
Biochemicals and blocking antibodies
Chemical inhibitors used in inhibition assays were aminoguanidine (AG), a nitric oxide synthase (NOS) inhibitor; SB-431342, a transforming growth factor beta (TGFβ) inhibitor; indomethacin, a cyclooxygenase (COX) 1/2 inhibitor; ZM 241385 and 8-(3-cholorostyryl)-caffeine (CSC)-A2A and (Adenosine A2A receptor antagonists) (all from Tocris Bioscience, Minneapolis, MN); and 1-methyltryptophan (MT), a competitive inhibitor for indoleamine 2,3-dioxygenase (IDO) (Sigma Aldrich, St. Louis, MO). Anti-canine-programmed death ligand-1 (PD-L1) and programmed death protein-1 (PD-1) monoclonal antibodies were from Merck Research Products, Kenilworth, NJ.
DC antigen uptake assay
iDC or mature DC (mDC) were generated as described above, which were admixed with MSC for indicated periods of time and then pulsed with 5 μg/mL ovalbumin (OVA) coupled with allophycocyanin (APC) (Molecular Probes-Thermo Fisher, Waltham, MA) and assayed by flow cytometry to quantitate antigen uptake. In brief, triplicate cultures of 1 × 105 DC were plated either alone or with 1 × 104 IFNγ-treated MSC and pulsed with 50 ug/mL APC-OVA at time = 0, and cultures were harvested and processed for flow cytometry at 1, 3, 6, 12, and 24 h.
Generation of canine skin fibroblasts
Canine skin biopsies of 5 mm diameter were taken from adult dog through punch biopsy. Samples were washed twice with PBS and placed in sterile Petri dishes. The epidermis was removed by scraping with a scalpel blade. In addition, care was taken to prevent any contamination by MSC by carefully removing adherent adipose tissue from the dermal surface by scraping with a scalpel blade. Next, the skin biopsy samples were then cut into small sections measuring 2 mm2. Two to three skin specimen pieces were placed in six-well cell culture dishes and covered with 22 mm glass cover slip. Fibroblast (FB) outgrowth was observed after 15 days of culture in DMEM, 10% FBS, 10 mM HEPES, 1 × Pen/Strep, 1 × nonessential amino acids, and 1 mM sodium pyruvate. Once confluent, skin FB was removed from surrounding edges of biopsy specimen using trypsin EDTA and frozen at P2 for further use. Cell surface phenotype of the FB was determined by flow cytometry. Only P2 and P3 FB was used in this study.
Flow cytometry
Single cell suspensions of canine MSC, DC, or FB were prepared as described above. DC were immunostained with primary antibodies for 20 min at RT in FACs buffer (PBS with 2% FBS and 0.05% sodium azide) following a 5-min incubation with normal dog serum (Jackson ImmunoResearch) to block nonspecific binding. Antibodies used were as follows: To stain CD11c+ cells, mouse anti-canine CD11c was used and, subsequently, tagged by a donkey anti-mouse (conjugated with either APC or Alexa Fluor 488 (Affymetrix-eBioscience, San Diego, CA) depending on specific experiments) followed by FITC-conjugated anti-canine MHCII (Bio-Rad, Hercules, CA), PE-conjugated anti-human (canine cross-reactive) [47] CD86 (Clone IT2.2; eBioscience, San Diego, CA), or Alexa Fluor® 647-conjugated anti-human (canine cross-reactive) CD40 (clone LOB7/6) that recognizes the canine identical immunogen sequence as human CD40 (Bio-Rad, Hercules, CA). The DC and MSC were analyzed using a Beckman Coulter Gallios flow cytometer (Beckman Coulter, Miami, FL), and data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Cytokine analysis
Cell culture supernatants from cultures of either MSC or DC alone or in cocultures of various ratios of MSC and DC were analyzed for canine interleukin-10 (IL-10) and tumor necrosis factor alpha (TNFα) using the DuoSet® canine IL-10 and TNF-α ELISA Kit (R&D systems, Minneapolis, MN) according to the manufacturer's instructions.
Confocal microscopy
MSC and DC were stained in a similar manner as those prepared for flow cytometry above. In addition, DC were stained by uptake of APC-conjugated OVA as described above for the antigen uptake experiments. Confocal images of DC and MSC were obtained using an Olympus (Waltham, MA) IX3 series confocal microscope fitted with a DSU-spinning disk unit for confocal imaging. Images were processed and analyzed using the Olympus cellSens® software.
Statistical analyses
Nonparametric continuous and grouped variables were compared using either a one-tailed or two-tailed (depending on the experimental analysis) Mann–Whitney U-test. P values for statistical variance of biological replicates (seven dogs) were determined using Repeated Measures ANOVA with Tukey's multiple comparison tests. Statistical analyses were performed using GraphPad Prism 6. Significance was set at P < 0.05, *; P < 0.01, **; P < 0.005, ***; and P < 0.0001, ****.
Results
Generation and characterization of canine monocyte-derived DC
DC were generated from blood of purpose-bred Beagle dogs or from outbred dogs (mixed breed, Australian Shepherd and Standard Poodle). DC was prepared from plastic adherent monocytes. Adherent cells were between 70% and 85% CD14+ identified by flow cytometry using the Alexa Fluor 647-conjugated (Tük4 clone, IgG2a) canine cross-reactive CD14 antibody (Fig. 1A) (
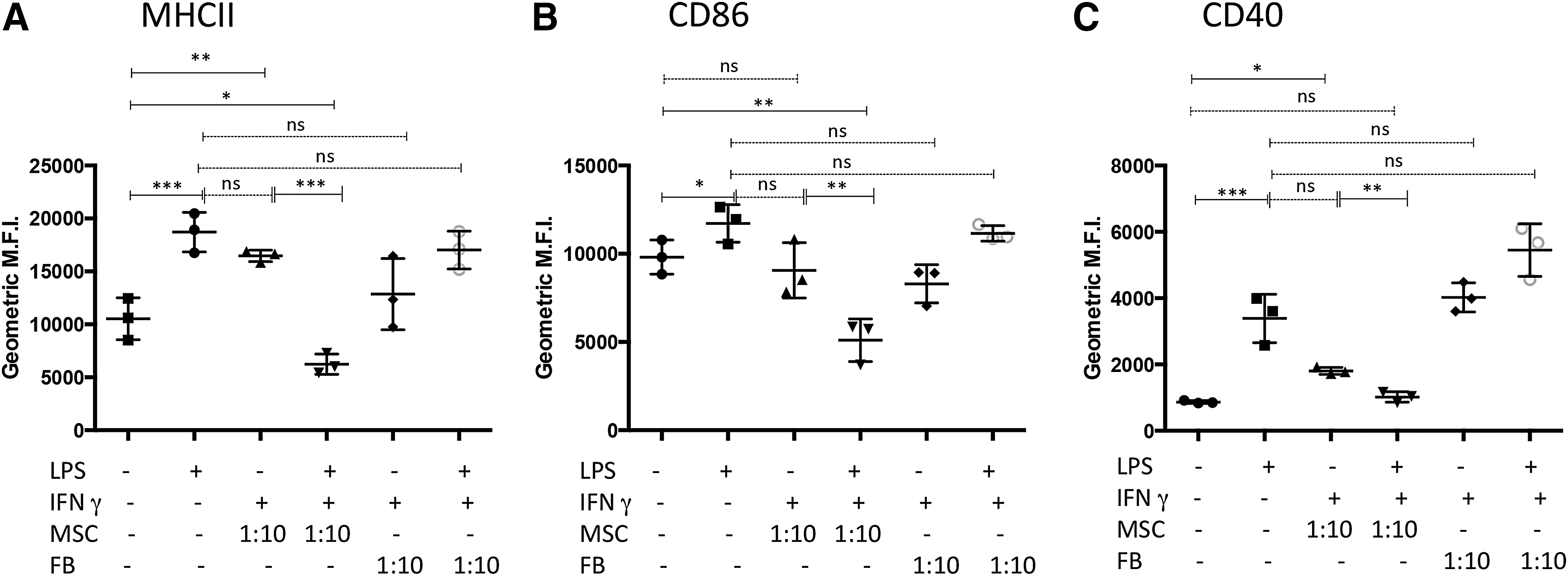
Treatment of MSC with IFNγ enhances suppression of cellular translocation of endosomal MHCII and surface expression of both CD86 and CD40 by LPS-activated DC
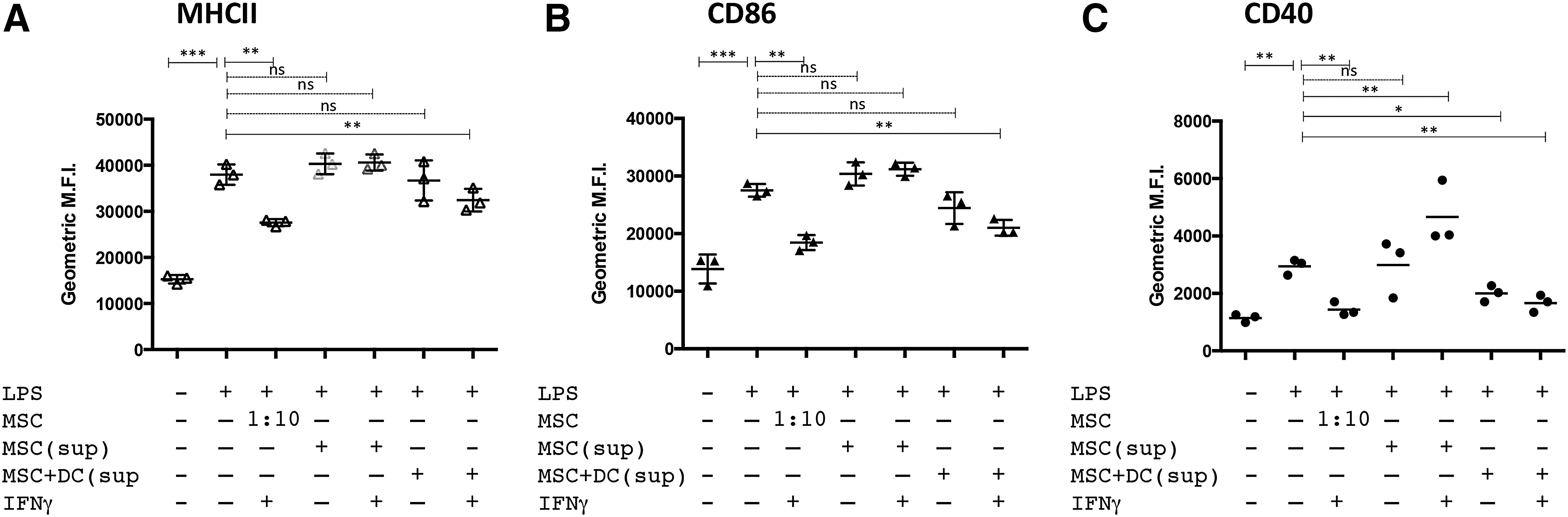
To drive maturation/activation, monocyte-derived DC were treated with 50 ng/mL (E. coli 055:B5) LPS. Surface expression of MHCII, CD86, and CD40 was significantly increased in DC treated for 24 h with LPS. However, when DC were cocultured with MSC and, subsequently, treated with LPS, there was a significant decrease in expression of MHCII and CD86 (Fig. 2, panel A.). The suppression of expression of MCHII, CD86, and CD40 was further accentuated when the MSC were pretreated for 18 h with 100 ng/mL canine IFNγ. This finding suggests that MSC in an inflammatory microenvironment are further stimulated to suppress optimal DC activation, as has been reported previously for MSC suppression of T cell function [12,50,51]. The levels of suppression of LPS-mediated upregulation of DC costimulatory and MHCII molecules by IFNγ-treated MSC could also be titrated based on the ratio of MSC:DC in the cocultures. For example, as shown in Fig. 2B, there was retention of significant suppression of DC maturation when the MSC:DC ratio was reduced to 1:100 (MSC:DC) and even at ratios of 1:1,000 (for CD40 expression). This effect has also been observed with human MSC and DC and suggests that MSC immunomodulating properties require “priming” by inflammatory mediators released from activated immune cells such as IFNγ, IL-1β, and TNFα [52,53]. No significant suppression of either MHCII or CD86 was observed when the ratio of MSC:DC was reduced to 1:1,000 (Fig. 2B), suggesting that CD40 expression was more sensitive to the effects of MSC suppression compared with MCHII or CD86. Consistent patterns of DC activation/suppression were observed in repeated experimentation of seven different dogs (Supplementary Fig. S1).

Activation of mesenchymal stem cells (MSC) with interferon gamma (IFNγ) enhances suppression of DC activation. DC were cocultured with MSC for 24–36 h that were either untreated or pretreated with 100 ng/mL IFNγ for 18 h.
DC TNFα release reduced by coculture with MSC
Since the maturation of canine DC by LPS was significantly inhibited by IFNγ-treated MSC, we assessed the effects of MSC on secretion of the pro-inflammatory cytokine TNFα. The activation of inflammatory Th1-promoting DC can be monitored by the secretion of this pro-inflammatory cytokine. When cocultured in the presence of MSC, LPS-treated DC released significantly less TNFα (Fig. 2; panel Ci). The suppression of TNFα release was further reduced by pretreatment of the MSC with IFNγ (Fig. 2; panel, Ci). In addition, treatment of DC:MSC cocultures with LPS in the presence or absence of IFNγ failed to significantly alter secretion of IL-10 (Fig. 2; panel, Cii). Inhibition of DC TNFα secretion has been demonstrated to inhibit their maturation, migration to lymph nodes, as well as their ability to stimulate allogeneic T cells, by altering the expression of several receptors and coreceptors necessary for antigen capture and processing [54 –56]. It should also be noted that treatment of MSC alone with IFNγ, GM-CSF, IL-4, or LPS did not result in any detectable TNFα secretion (Fig. 2C). Thus, the source of TNFα release observed in these cocultures was likely from DC and not MSC. These results indicate that activated MSC suppressed TNFα production by canine DC, while leaving IL-10 production intact, thus resulting in an overall increase in the amount of IL-10 produced relative to TNFα production and, thereby, driving the DC to an anti-inflammatory phenotype.
Suppression of DC maturation/activation phenotype by MSC mediated by soluble factors
To determine whether MSC-mediated suppression of DC maturation/activation required direct contact between MSC and DC, DC were treated with LPS in the presence of filtered supernatants from MSC treated with or without IFNγ alone, or supernatants obtained from cocultures of IFNγ-treated MSC and DC. In Fig. 3, it is apparent that supernatants from IFNγ-treated MSC and DC cocultures significantly abrogated the ability of LPS treatment to upregulate the DC surface maturation markers MHCII, CD86, and CD40. No significant DC suppressive effect was observed in DC cultures when using supernatants from untreated or IFNγ-activated MSC alone. These data indicate that coculture of MSC and DC stimulates the release of soluble factors that are capable of suppressing expression of DC maturation. The source of these yet-to-be-identified secreted DC suppressive factors could be either MSC, DC themselves, or both.
Supernatants from MSC:DC cocultures prevent full maturation of LPS-stimulated DC. DC were cocultured with MSC (1:10 ratio, MSC:DC) that were either untreated or treated with IFNγ. In parallel studies, DC were cultured with supernatants obtained from MSC:DC cocultures. The DC were then activated with LPS (50 ng/mL) for 24 hr. The DC were subsequently analyzed for expression of MHCII
Suppression of DC maturation by MSC is mediated through multiple pathways
The mechanisms by which MSC suppress canine DC have not been previously identified. Nor have prior studies carefully considered the possibility that multiple mechanisms may operate concurrently for MSC to suppress DC function. To address this question, inhibitors of specific biochemical pathways known to be operative in MSC suppression were added to canine MSC and DC cocultures. These studies revealed that both shared and distinct immune modulatory pathways were utilized by MSC to suppress individual DC maturation markers (Fig. 4). For example, MSC suppression of LPS-mediated DC MHCII surface translocation was almost completely reversed if the specific NOS inhibitor AG was included in the medium (Fig. 4, Panel Ai). In addition, partial reversal of the suppression of MHCII surface expression was also observed when indomethacin, an inhibitor of the cyclooxygenase (COX) 1/2 inflammatory pathway, was added to LPS-stimulated DC:MSC cocultures (Fig. 4Ai). In addition, the adenosine A2A selective receptor antagonist, CSC-A2A, partially reversed the MSC suppressive effect on MHCII expression (Fig. 4Ai). Finally, blockade of PD-L1 with an anti-canine PD-L1 specific antibody weakly reversed MHCII surface translocation in these cocultures (Fig. 4i). It should also be noted that IFNγ activation strongly upregulated PD-L1 expression on canine MSC in vitro, while PD-1 expression remained the same. For DC, PD-L1 expression was slightly upregulated on DC treated with either IFNγ or LPS while significantly downregulated when in coculture with LPS+MSC. Whereas PD-1 was downregulated on LPS- or IFNγ-treated DC in comparison to iDC and further downregulated when in coculture with LPS+MSC (Supplementary Fig. S2).
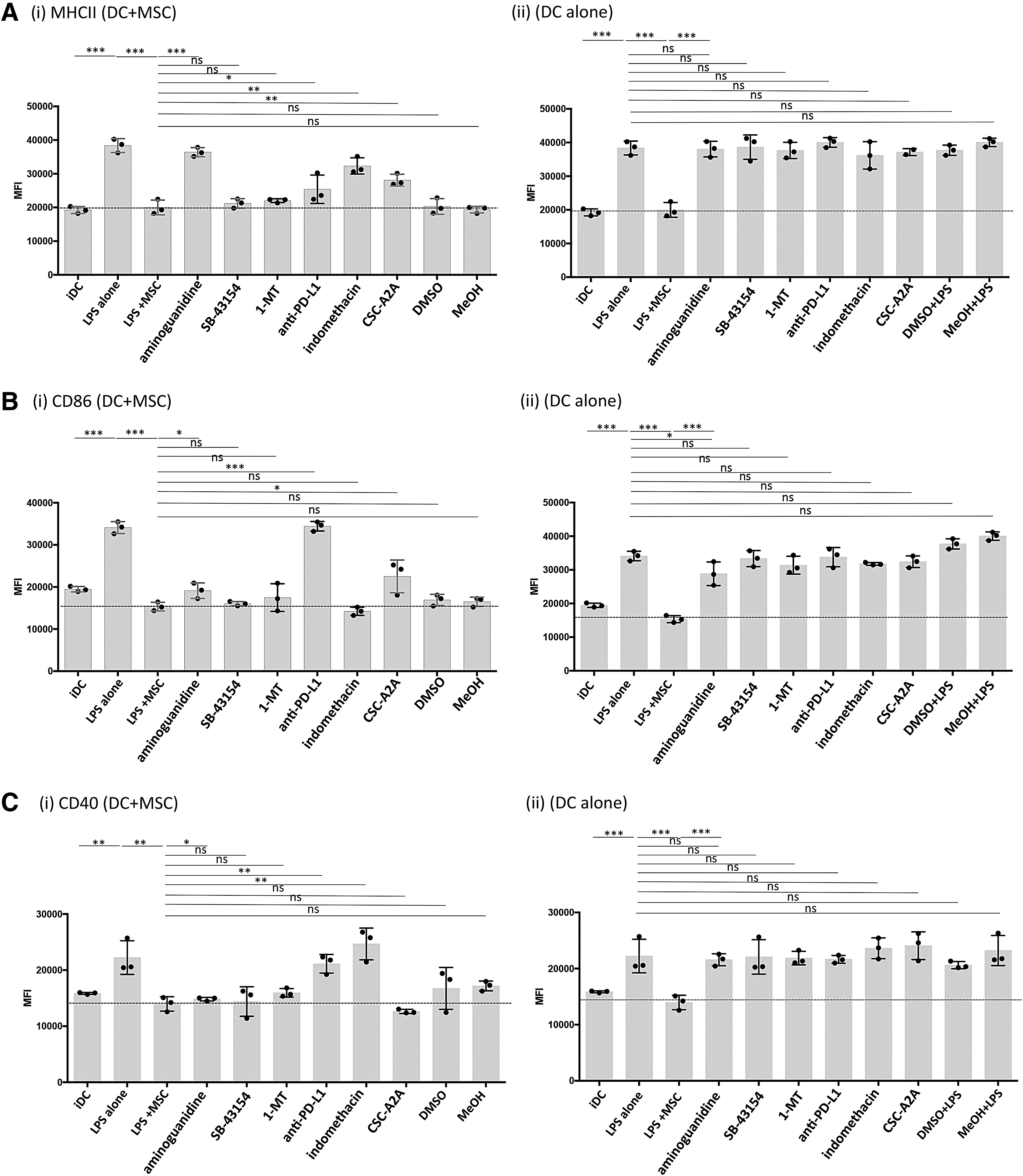
MSC use multiple immune suppressive pathways to abrogate DC activation. IFNγ-treated MSC and DC were cocultured (1:10 ratio) for 24 hr and activated with LPS (50 ng/mL) for 24 hr. The MSC:DC cocultures were also incubated with the following immune pathway inhibitors: aminoguanidine (0.5 nM), indomethacin (100 μM), CSC-A2 inhibitor (0.2 μM), SB-43154 (10 μM), 1-methyltryptophan (MT) (500 μM), or programmed death ligand-1 (PD-L1) blocking antibody (10 μg/mL). Controls included medium with DMSO or methanol vehicle or with an isotype matched antibody for PD-L1 (data not shown).
Suppression of DC CD86 expression by MSC was completely reversed by PD-L1 blockade (Fig. 4Bi). In addition, partial reversal was observed with the inhibitors AG and CSC-A2A. For DC CD40 expression, reversal of MSC suppression was observed with PD-L1 blockade and with indomethacin (Fig. 4Ci). Addition of the IDO inhibitor 1-MT had no effect on reversal of MHCII, CD86, or CD40 expression on DC by MSC. However, transcripts for the predicted canine IDO-1 and IDO-2 genes were detected in canine untreated or IFNγ-treated MSC by reverse transcriptase-PCR analysis using primers for the putative canine IDO-1and IDO-2 loci (data not shown). When DC alone were treated with the inhibitors followed by LPS treatment, there was no effect of MHCII, CD86, or CD40 expression (Fig. 4; panels A ii, B ii, and C ii). Taken together, these data indicate that canine MSC use multiple, nonredundant immune pathways to downregulate DC activation. The pathways utilized include checkpoint molecules (PD-L1 and PD-1), nitric oxide, cyclooxygenase, and the adenosine accumulation pathways.
Pathways used by MSC to suppress DC TNFα production
Blockade of canine PD-L1 in LPS-treated MSC:DC cocultures restored TNFα secretion by canine DC to greater levels than treatment of the DC with LPS alone (Fig. 5A). When DC:MSC cocultures were incubated in the presence of the NOS inhibitor AG or the adenosine receptor inhibitor CSC-A2A, restoration of TNFα production (either partially or fully) was observed (Fig. 5A). Interestingly, treatment of LPS-stimulated DC alone with the TGFβ inhibitor, SB-431342, resulted in an augmentation of TNFα secretion over the single LPS treatment implicating a modulating effect of DC activation mediated by TGFβ (Fig. 5B). However, cocultures of SB-431342-treated DC with MSC abrogated this hyper TNFα secretion indicating a dominant effect over the stimulatory effect of TGFβ inhibition. These data suggest that most of the MSC suppressive mechanisms that thwart expression of MHCII, CD86, and CD40 are also linked to suppression of TNFα secretion by DC.
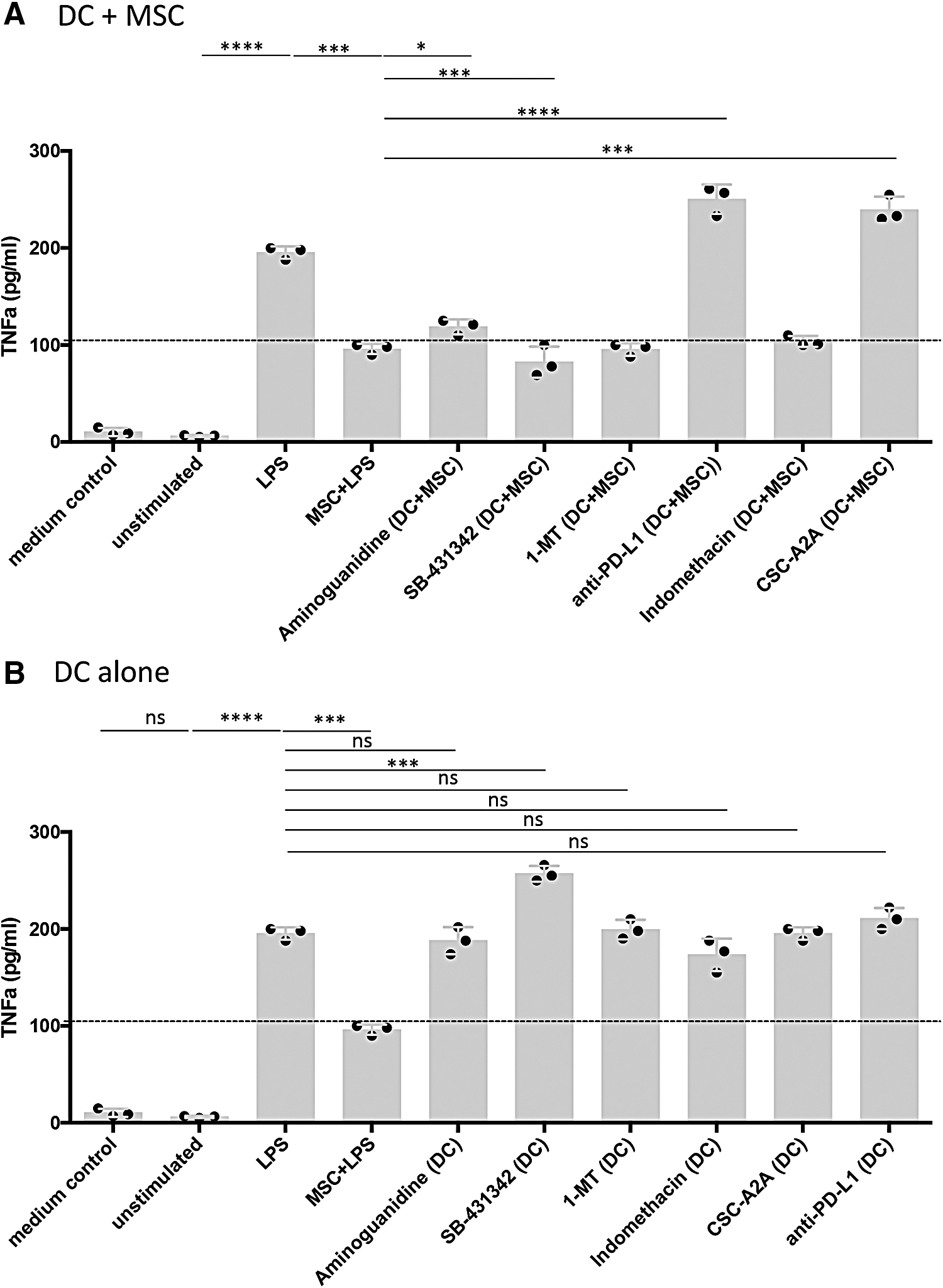
Identification of MSC immune modulatory pathways that regulate DC TNFα production.
MSC maintain DC in immature state with increased antigen uptake properties
DC maturation following TLR activation is associated with transition from an avid antigen uptake state to a state characterized by antigen presentation with little additional antigen acquisition [19]. This change in function can be measured as a decrease in the kinetics of antigen uptake in DC following treatment with LPS (Fig. 6). This dynamic transformation is characteristic of the maturation program of activated DC. To determine whether MSC alter DC maturation with respect to antigen uptake, we antigen-pulsed untreated or LPS-treated DC and compared the kinetics of APC-conjugated OVA uptake in DC cultured alone or cocultured with IFNγ-treated MSC. OVA uptake was monitored over a period of 24 h. OVA-pulsed non-LPS treated DC showed a continual and sustained pattern of increased APC fluorescence. LPS-treated DC showed only minimal overall OVA uptake and maintained only a marginal increase in APC fluorescence over the time period. Coculture of either untreated or LPS-stimulated DC with MSC resulted in a sustained increase in endocytosis of OVA comparable or greater than DC pulsed without LPS treatment in the absence of MSC (Fig. 6). These results are consistent with MSC suppression of DC maturation and retention of the DC in an immature, antigen avid, highly endocytic state. MSC alone pulsed with APC-OVA failed to indicate any increase in fluorescence over background.
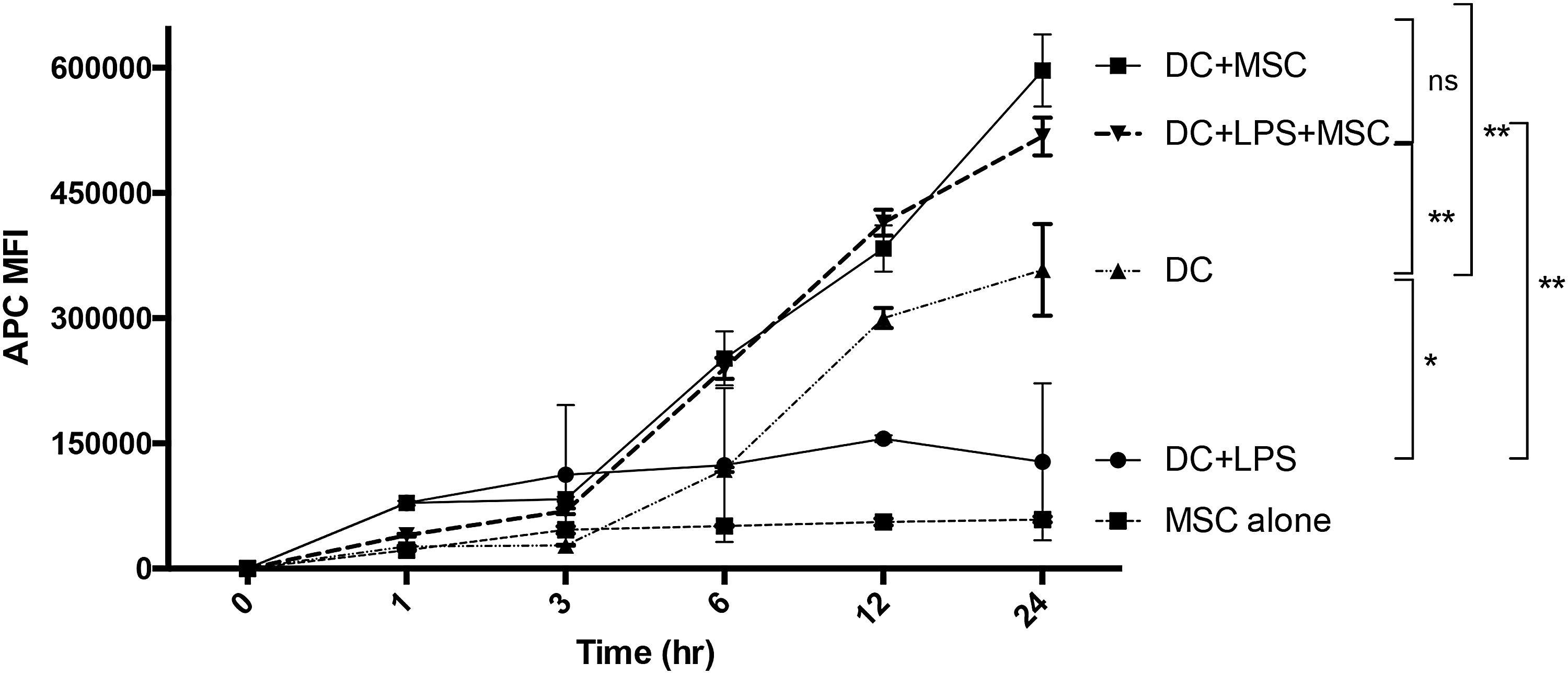
Effect of MSC on DC maturation and antigen endocytosis. Cultures of 1 × 104 DC in 24-well plates were cultured with or without 1 × 103 IFNγ-treated MSC and pulsed with APC-conjugated OVA (final concentration 5 μg/mL) at time = 0, and then DC were harvested and processed for flow cytometry at the indicated time points. As a control, MSC alone were pulsed and harvested identically. Vertical lines represent the range of APC MFI for each time point in triplicate experiments. Result is representative of three DC sets from three separate dogs. P values for statistical variance at the 24 hr time point were determined using Repeated Measures ANOVA with Tukey's multiple comparison tests. *P < 0.05, **P < 0.01, and ***P < 0.005.
MSC suppression of DC activation/maturation is a unique function of MSC not observed with FB
The observed suppression of canine DC activation/maturation by IFNγ-treated MSC may merely be a consequence of coculturing DC with relatively rapidly growing cells, rather than due to unique immune-suppressive functions of MSC. To address this question, P2 canine skin FB was substituted for MSC in the DC coculture system. Canine skin FB was found to be phenotypically distinct from canine MSC, as revealed by flow cytometry and expression of MHCII, CD24, and CD90 (Supplementary Figs. S3 and S4). For example, FB did not express any detectible MHCII following IFNγ activation, while MSC strongly upregulated MHCII expression. In addition, in comparison to MSC, canine FB expressed higher levels of CD24 and generally lower levels of CD90, whereas MSC expressed CD90 at higher levels (Supplementary Fig. S4).
Coculturing DC with canine skin FB failed to suppress any of the DC activation markers to the degree of MSC (Fig. 7). Coculture of DC with skin FB did not significantly alter LPS-stimulated DC upregulation of MHCII or CD86 or CD40 compared to those cultured with MSC. Taken together, these data suggest that immune modulation of DC function by MSC is restricted to the specific functionality of MSC cells and not merely a consequence of limited nutrients due to coculturing with rapidly dividing cells per se.
Skin fibroblast (FB) does not alter DC activation. DC were cocultured for 24 hr with either MSC or primary skin FB at 1:10 ratios (MSC:DC or FB:DC). DC were then collected, and the relative surface expression of
Discussion
The studies described herein demonstrate that canine MSC derived from adipose tissue have the ability to suppress LPS-mediated activation/maturation of canine DC. The impact in vivo of such squelched DC activation almost certainly would result in an attenuated ability to appropriately prime T cell responses. This effect would also be exacerbated if the MSC were first activated with IFNγ, suggesting that the suppressive effect would be optimized in an inflammatory environment typical of autoimmune or pro-inflammatory conditions. In addition, the dosage of MSC required to attenuate DC activation in this canine model is quite low with suppressive effects observed at MSC:DC ratios of between 1:100 and 1:1,000. Such a low-dose efficiency of suppression should limit the potential allogeneic cell burden necessary for therapeutic efficacy and reduce the amount of MSC needed for successful clinical usage.
Our studies extend the results of previous studies of MSC and DC interactions in mice and humans and demonstrate that canine MSC can also suppress secretion of TNFα by DC, while maintaining relatively constant levels of IL-10 secretion. The net result of these MSC effects would be to shift DC, with regards to T cell priming, toward an immunosuppressive, regulatory, or Th2-priming phenotype. We also show that activation of MSC with IFNγ significantly improves MSC suppression of DC activation and maturation. The inflammatory milieu MSC face when administered in vivo is postulated to play a critical role in regulating their immune modulatory potency [2].
One important and unexpected finding from these studies was the complex network of immune modulatory pathways used by MSC to suppress DC function. For suppression of DC activation, at least four different pathways were utilized, including checkpoint molecules (eg, PD-L1 and PD-1 interaction), the nitric oxide, cyclooxygenase, and the adenosine pathways. Not surprisingly, multiple pathways appear to operate concurrently for MSC suppression of canine DC function. Unlike the case with human MSC, we found that canine MSC do not appear to utilize the IDO pathway for suppression of DC function, nor for suppression of T cell function [69]. Thus, canine MSC, in this regard, appear to functionally resemble mouse MSC more closely than human MSC in their immune modulatory pathways. Alternatively, this observation may reflect fundamental differences in pathways that mediate canine DC and T cell activation as opposed to differences in MSC function.
Recent studies have indicated that checkpoint molecules such as PD-L1 may play an important role in MSC-mediated immune modulation [28,57,58]. PD-L1 (B7-H1/CD274) is the ligand for the T cell inhibitory receptor PD-1 and is expressed on epithelial cells and various immune cells, in particular DC, macrophages, and B cells. Indeed, in our studies with canine MSC, it appears that overall the PD-L1 pathway may be quantitatively more important than the nitric oxide, adenosine, and cyclooxygenase pathways. DC have been shown to express both PD-L1 and PD-1 and can thus serve as a target for PD-L1 expressing MSC [59]. Moreover, IFNγ is a strong stimulus for upregulation of PD-L1 on tumor cells, antigen presenting cells, and MSC. We have reported recently that IFNγ strongly upregulates PD-L1 expression on canine tumor cells, monocytes, and macrophages [60]. In the present study, we found that IFNγ also strongly upregulates PD-L1 expression on canine MSC (Supplementary Fig. S2; panel A), while demonstrating only a moderate upregulation in DC treated with either LPS or IFNγ (Supplementary Fig. S2; panel B). PD-L1 expression is significantly downregulated in LPS-treated DC in coculture with MSC. PD-1 expression is not significantly altered in untreated versus IFNγ-treated MSC, whereas for DC, its expression is significantly reduced in cocultures with MSC (Supplementary Fig. S2A, B). Although the precise mechanism is currently unknown, MSC appear to utilize some of the same immune checkpoint regulatory pathways used by tumor cells to suppress T cells and DC.
The most immune relevant adenosine receptors (A2A) are present only at low levels on immature human DC [61]. However, maturation of DC is accompanied by upregulation of A2A signaling responses due to both increases in expression and coupling of A2A [61,62]. The A2A activation on mDC has been shown to shift cytokine profiles from a mostly pro-inflammatory profile to a largely anti-inflammatory program, with reduced secretion of IL-6, IL-12, and IFNα and concomitant augmentation of IL-10 production [63]. Human MSC in coculture with activated T cells significantly increases the expression of the enzyme ectonucleotide triphosphate diphosphohydrolase-1 (CD39) on MSC that converts ATP and ADP to AMP and adenosine nucleosides. Subsequent production of adenosine is mediated by increased expression of ecto-5′nucleotidase (CD73) on both MSC and T cells. MSC were shown to be the primary source of adenosine in this model [64].
Other adenosine receptors that are Gi-coupled (A1 and A3) are expressed on immature human DC and their activation results in cytoskeletal reorganization and migration along different concentration gradients of adenosine suggesting that adenosine receptor activation has a role in the induction of chemotaxis in iDC [61]. In contrast to humans, innate responses of mouse BM-derived DC were blocked by signaling through A1 receptors, whereas both A2A and A2B were undetectable in those cells [65]. Our results showing strong rescue of MSC-mediated suppression of canine DC maturation through blockade of A2A strongly suggest that adenosine affords a significant contribution in the overall suppressive response that MSC have on canine DC maturation. Thus, adenosine likely has a dual role in mediating DC function by: (i) promotion of recruitment of iDC to sites of injury and inflammation through activation of A1 or A3 receptors and ii) imposition of an anti-inflammatory phenotype driving T cell responses away from Th1 toward Th2 or Treg profiles through activation of A2A.
Taken together, this work demonstrates that canine MSC effectively abrogate the ability of canine DC to optimally express activation/maturation surface markers, while altering cytokine secretion profiles resulting in skewing of DC toward a more immune suppressive phenotype. This DC suppressive effect is amplified by MSC activated with IFNγ, underscoring the ability of MSC to respond within a local inflammatory environment to regulate and impose their immune suppressive properties. In addition, it is currently unknown definitively whether LPS treatment of these cocultures may have a synergistic effect on the immunomodulatory function of canine MSC. However, it has been shown that human MSC express TLR, and engagement thereof has been shown to affect their immunomodulatory properties [66 –68]. Preliminary data indicate that LPS treatment of canine MSC alone do not appear to alter their phenotype in terms of the expression of the limited set of canine markers that we currently have available (eg, PD-1, PD-L1, CD11c, MHCII, CD86, and CD40 [data not shown]). Experiments to better understand the effect of TLR-engagement, with or without IFNγ, on canine MSC are slated for future endeavors. Importantly, our studies also demonstrate that immune modulatory properties are unique to MSC and do not reflect nonspecific effects of nutrient competition or secretion of nonspecific inhibitory factors generically by dividing cells in vitro. Experiments are underway in our laboratory to assess the immune suppressive pathways of canine MSC for suppression of T cell function. These studies will help fill in the knowledge gaps to improve our understanding of how MSC regulate the T cell:DC immune synapse.
Footnotes
Acknowledgments
The authors graciously thank the support to the Center for Immune and Regenerative Medicine (CIRM) at Colorado State University through generous donations from organizations such as the Shipley Foundation and Frankie's Fund, which support clinical studies of stem cell therapy and regenerative medicine in companion animals.
Author Disclosure Statement
No competing financial interests exit.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.