
Abstract
Pluripotent stem cells hold the potential to form the basis of novel approaches to treatment of disease in vivo as well as to facilitate the generation of models for human disease, providing powerful avenues to discovery of novel diagnostic biomarkers and/or innovative drug regimens in vitro. However, this will require extensive maintenance, expansion, and manipulation of these cells in culture, which raises a concern regarding the extent to which genetic integrity will be preserved throughout these manipulations. We used a mutation reporter (lacI) transgene approach to conduct direct comparisons of mutation frequencies in cell populations that shared a common origin and genetic identity, but were induced to undergo transitions in cell fate between pluripotent and differentiated states, or vice versa. We confirm that pluripotent cells normally maintain enhanced genetic integrity relative to that in differentiated cells, and we extend this finding to show that dynamic transformations in the relative stringency at which genetic integrity is maintained are associated with transitions between pluripotent and differentiated cellular states. These results provide insight into basic biological distinctions between pluripotent and differentiated cell types that impact genetic integrity in a manner that is directly relevant to the potential clinical use of these cell types.
Introduction
T
While this may at first appear paradoxical, we believe it can be explained by the fact that pluripotent cells such as embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) divide rapidly, lack any significant G1 phase, and possess G2- and M-phase checkpoints that function inefficiently [2], thus predisposing the occurrence of large-scale genomic aberrations. However, this does not necessarily pose a significant threat to a population of pluripotent cells because such large-scale aberrations typically trigger apoptosis to eliminate, retrospectively, cells carrying these genetic defects, and pluripotent cells show upregulated cell death activity and so are well positioned to deal with large-scale genetic defects [5].
In contrast, no such retrospective mechanism exists to mitigate the accumulation of point mutations in these same cells. Therefore, the potential for point mutations to accumulate in pluripotent stem cells is high, especially given the rate of proliferation of these cells, the propensity for mutagenesis during replication of DNA, and the abbreviated G1 phase in the cell cycle in pluripotent cells [6,7]. Although point mutations involve minimal changes to the genome sequence, they can have very significant effects. Point mutations are the most commonly occurring type of mutation and are responsible for a majority of genetic diseases in humans [3]. It therefore follows that in the absence of an effective retrospective means to deal with the accumulation of point mutations, pluripotent cells have evolved mechanisms to avoid the accumulation of such mutations prospectively.
In 1977, T.B.L. Kirkwood [8] proposed the disposable soma theory, which posits that because germ cells and early embryonic cells (and, by modern extension, pluripotent stem cells) normally give rise to either entire subsequent generations of individuals or to large populations of subsequent cells within an individual body, it is evolutionarily advantageous for these cells to employ mechanisms that maintain an enhanced level of genetic integrity, even if this requires the expenditure of additional energy. That there is enhanced maintenance of genetic integrity in germline cells was suggested by early studies [9 –16] and has been confirmed by studies using the lacI mutation reporter transgene system, also known as the Big Blue Mouse, developed by Stratagene (now part of Agilent, Santa Clara, CA) [17 –19].
Thus, we have shown that in both sexes, mouse germ cells maintain frequencies of spontaneous point mutations that are 5–10-fold lower than those found in somatic tissues from the same individuals, and this appears to be based on elevated levels of DNA repair and cell death activities in these cells [20 –24]. Similarly elevated levels of DNA repair pathways that specifically limit the initial occurrence of spontaneous point mutations and ameliorate most potentially mutagenic DNA damage have been reported in pluripotent stem cells [4,5,25 –28].
Previous suggestions of differences in the level at which genetic integrity is maintained in pluripotent and differentiated cell types came first from studies of early embryonic cells, which provide an example of pluripotent cells in vivo. Assessments of mutation frequencies that could be performed on the limited number of cells available from this source indicated lower levels than those typically found in differentiated somatic cell types [10,15,18]. ESCs and iPSCs resemble naturally occurring pluripotent cells in the inner cell mass or epiblast of the preimplantation embryo [29,30], but have the advantage that they can be expanded in vitro while maintaining the pluripotent state, and this has facilitated more extensive analyses of genetic integrity in these cells. Initial concerns regarding mutational burdens—especially with respect to mutations that might predispose tumorigenesis in pluripotent stem cells—were raised because of the potential use of these cells for cell-based therapies [31,32].
Despite reports of the incidence of certain types of mutations, including aneuploidies [33] and copy number variations [34,35], as well as certain epigenetic changes [36] in pluripotent cells, some of which may confer growth advantages in vitro [37], overall the results of these studies indicate that pluripotent cells typically do not develop an excessive mutational load regardless of whether they are derived directly from pluripotent cells in vivo (ESCs) or indirectly from differentiated somatic cells in vitro (iPSCs)[34,38 –42]. In addition, it appears that different methods of reprogramming somatic cell types to form iPSCs do not yield significant differences in the incidence of mutations [35]. However, the extent to which changes in mutational load accompany transitions between pluripotent and differentiated cell states, or vice versa, has been less well investigated.
The majority of results indicating that pluripotent stem cells maintain genetic integrity at enhanced levels relative to differentiated cells have been based on comparisons of independently derived populations or lines of pluripotent and differentiated cell types, respectively [4,25 –28]. However, because pluripotent cells can be manipulated to adopt any of a wide variety of cellular fates, it is now possible to compare subpopulations of cells that emerged from a common initial population and were subsequently induced to adopt distinct fates, including either the loss or gain of pluripotency. Thus, ESCs can be maintained indefinitely in a pluripotent state or they can be induced to differentiate to form any of a variety of different somatic cell types [43]. Similarly, differentiated somatic cell types can be induced to undergo reprogramming to form pluripotent iPSCs that can then be either maintained indefinitely as pluripotent cells or induced to differentiate back into either the same somatic cell type from which they were derived or other somatic cell types [44].
Mutation reporter transgenes provide a sensitive approach to assess the frequency of genetic mutations in any cell sample in a manner that facilitates statistically significant comparisons. The most extensively studied mutation reporter system is the lacI, “Big Blue” rodent system originally developed by Stratagene [17,45]. This approach is based on the colorimetric detection of expression of the lacZ reporter gene as an indicator of mutations that have occurred in the lacI (repressor) gene while it was resident in the transgenic cells. Copies of the transgene expressing the mutant phenotype are sequenced to confirm the presence and type of relevant mutation in the lacI gene and to identify clonal mutations, which are counted as a single mutagenic event regardless of how frequently they are detected in the same sample. This approach yields quantitative assessments of mutational loads in distinct populations of cells that can be analyzed for statistical significance.
We generated ESCs and iPSCs from mice carrying the lacI mutation reporter transgene and exploited the ability to induce pluripotent or differentiated cellular states to directly test the hypothesis that the stringency with which genetic integrity is maintained undergoes dynamic changes as a function of entry into or exit from the pluripotent state. Our results provide definitive evidence that this is indeed the case and support our contention that enhanced maintenance of genetic integrity is a fundamental characteristic of pluripotent cells based on coregulation of genetic integrity and pluripotency at the genomic and epigenomic levels in these cells [5,18,29].
Materials and Methods
Animal models
lacI mutation reporter transgenic mice
The lacI mutation reporter transgenic mice (aka: Big Blue Transgenic Mice, originally established by Stratagene [La Jolla, CA], now part of Agilent [Santa Clara, CA]) were developed to screen for spontaneous point mutations in the lacI mutation reporter transgene recovered from any cell or tissue type [45]. We obtained mice homozygous for a lambda shuttle vector containing the lacI gene from Stratagene. Confirmation of the presence of the lacI mutation reporter transgene was performed by genotyping as described in the Supplementary Data (Supplementary Data are available online at
lacI–B6/129/Sv transgenic mice
Mice homozygous for the lacI transgene on a C57BL6/J background were bred to 129/Sv mice to produce blastocysts hemizygous for the lacI transgene on an F1 hybrid C57BL6/J × 129/Sv background. Confirmation of the lacI hemizygous transgene genotype was performed on derived ESCs as described in the Supplementary Data.
Reprogrammable transgenic mice
Carey et al.[46] generated a reprogrammable transgenic mouse model in which the four Yamanaka genes, Oct4, Sox2, Klf4, and cMyc, encoding transcription factors that promote reprogramming are expressed from a single polycistronic genomic locus (reprogrammable expression cassette termed 4F2A) under the direction of a doxycycline (Dox)-inducible promoter. A breeding pair of these mice homozygous for the reprogrammable cassette was purchased from The Jackson Laboratory (Stock#011004; Bar Harbor, ME).
Reprogrammable lacI transgenic mice
To establish the reprogrammable lacI model, we bred mice homozygous for the reprogrammable cassette to mice homozygous for the lacI mutation reporter transgene. The resulting double hemizygous F1 offspring carrying both the reprogrammable cassette and the lacI transgenes were then back-crossed to homozygous reprogrammable transgenic mice. F2 mice homozygous for the reprogrammable cassette and hemizygous for the lacI transgene were identified by genotyping (see Supplementary Data). All procedures with live mice were conducted in accordance with approved guidelines in the animal facility at The University of Texas at San Antonio, and approved in advance by the UTSA Institutional Animal Care and Use Committee.
Cell models
Embryonic stem cells
ESCs were generated from lacI–B6/129/Sv blastocysts using standard methods as described [47] (see Supplementary Data for details). ESCs were cultured on feeder layers of mouse embryonic fibroblasts (MEFs) in ESC medium containing LIF: DMEM, 15% FBS (Atlanta Biologicals, Norcross, GA), 0.1 mM β-mercaptoethanol, 2 mM
Induced pluripotent cells
iPSCs were generated from tail-tip fibroblasts recovered from adult reprogrammable lacI transgenic mice using doxycycline induction of the reprogrammable transgene cassette as described [46] (see Supplementary Data for details). Each line of ES or iPS pluripotent cells was characterized for karyotype, expression of pluripotency markers, and potential to give rise to cell types characteristic of each germ layer in a teratoma assay (see Supplementary Data). Pluripotent stem cells (PSCs) were purified by MACS sorting for SSEA-1 before analysis. Purity of sorted PSCs was independently confirmed by staining for alkaline phosphatase (see Supplementary Data for details).
Pluripotent cell lines
The presence of a normal karyotype was confirmed for each ES and iPS pluripotent cell line, except for one of the ESC lines (16A), which showed a 2A:XO karyotype. Although the 16A line showed an abnormal karyotype, it met all other criteria for pluripotency, so was included in our study for purposes of assessing relative mutation frequencies in pluripotent cell lines. Importantly, only differentiated cells derived from the karyotypically normal 26A ESC line were used in subsequent studies. Pluripotency of each line was confirmed by immunocytochemistry for pluripotency markers and generation of teratomas, followed by histological analysis to confirm differentiation into cell types representative of all three germ layers. Methodological details and results of these procedures are provided in the Supplementary Data.
Differentiated cells
Pluripotent ESCs or iPSCs were induced to undergo differentiation in either a random or directed manner. Random differentiation was induced by removal of LIF from the medium in which pluripotent cells were cultured. This resulted in formation of embryoid bodies that were maintained first for a period of 14 days (14 cell doublings) before an initial sampling for analysis of mutation frequencies. The remaining cells were cultured for an additional 28 or 42 days, respectively, yielding additional samples of randomly differentiated cells at two later time points following initiation of differentiation. Randomly differentiated cells were generated under two conditions–with (+RA) or without (−RA) addition of exogenous retinoic acid (RA). Details of these differentiation protocols are provided in the Supplementary Data.
Populations of two specific differentiated cell types—macrophages and fibroblasts—were produced by directed differentiation. Macrophages (and monocytes) were produced from ESCs, while fibroblasts were produced from iPSCs. The macrophages represented a third type of differentiated cell population derived from the ESCs. The generation of fibroblasts from the iPSCs returned these pluripotent cells to the same differentiated cell type from which they were originally derived. Additional details of each directed differentiation protocol are provided in the Supplementary Data.
Doubling time assessments
Doubling times were determined for ESCs and iPSCs and for spontaneously differentiated cells and macrophages and monocytes derived from ESCs, as well as for primary fibroblasts recovered from adult mice and fibroblasts generated in culture by directed differentiation of iPSCs. Cell numbers were counted from one duplicated well every 24 h for 4 days. The cell doubling time was determined from cell counts made during the exponential phase of growth (see Supplementary Data for additional details). Although this method does not reveal variation in the rate of cell doubling during the assessment period, it does provide a quantitative measure of the number of cell divisions and hence DNA replications that each population has undergone during this period. This is relevant because DNA replication represents a period of relatively high occurrence of de novo mutations during the cell cycle [48].
Therefore, calculations of doubling times within each cell population allowed us to normalize our measurements of mutation rates as a function of the number of DNA replication cycles each population had undergone. A remaining distinction between pluripotent and differentiated cell types is the abbreviated G1 phase of the cell cycle in the former relative to the latter [6,7], which diminishes the opportunity for postreplication repair of DNA damage in pluripotent cells and enhances the need for elevated activity of DNA repair pathways to maintain genetic integrity in these cells.
Purification of cell types before analysis
MACS sorting was used to purify each pluripotent or differentiated cell type before analysis for either mutation frequencies or doubling times (see Supplementary Data for additional details).
Analysis of mutation frequencies
Isolation of high-molecular-weight genomic DNA
High-molecular-weight genomic DNA was recovered from the sorted cells using a slightly modified version of the protocol provided with the RecoverEase DNA isolation kit developed by Stratagene as described [29,49] (see Supplementary Data for details).
In vitro packaging of lambda phage DNA
To selectively recover the lacI reporter transgene from high-molecular-weight genomic DNA, the DNA was packaged into infectious lambda phage using the Transpack Packaging Extract kit (Stratagene) following the manufacturer's instructions, as described in detail in the Supplementary Data. Following packaging, SM buffer was added to the phage mixture, followed by storage at 4°C.
Escherichia coli infection and detection of mutant copies of the lacI transgene
Packaged phage was used to infect Escherichia coli SCS-8 cells (Stratagene) grown in NZY media plated as a lawn on 25 × 25-cm assay trays containing a sublayer of NZY agar plus X-gal and N, N-dimethylformamide (DMF) to facilitate the blue-clear colorimetric assay. After incubation at 37°C for approximately 18 h, each tray was screened for mutant plaques, which were identifiable by their circular appearance and blue color. Apparent mutant plaques were confirmed by mixing phage from the plaque with fresh E. coli SCS-8 cells and replating the mixture on fresh X-gal/NZY 60-mm plates, all as described [18,29,49] (see Supplementary Data for additional details).
Sequencing of mutant transgenes
The sequence of the lacI gene recovered from each confirmed mutant phage plaque was determined by PCR amplification and sequencing of the lacI gene—a procedure that was outsourced to the ICMB Core Facility at The University of Texas at Austin (see Supplementary Data for additional details). This allowed us to confirm the presence and type (transition, transversion, small insertion, or small deletion) of the point mutation responsible for the mutant phenotype in each case. A spectrum of point mutations (numbers and percentages of each type of mutation) was determined for each cell type analyzed.
Quantitative assessment of mutation frequencies and statistical significance
The frequency of mutations detected in each cell sample was determined by dividing the number of confirmed, independent (nonclonal) mutant plaques by the total number of plaque-forming units analyzed [18,29,49]. Mutations involving the exact same sequence change at the same position in the lacI gene detected more than once in the same starting cell sample were considered to be clonal and thus representative of a single initial mutation event that was clonally propagated. Thus, regardless of how many times the same mutation was detected in the same starting cell sample, it was counted as a single mutation. The final mutation frequency was calculated by dividing the number of confirmed nonclonal mutations detected from a cell sample by the total number of plaque-forming units (pfu) assayed for that sample (see Supplementary Data for additional details).
Mutation frequencies were analyzed by a Poisson model with parameter estimates obtained by the method of maximum likelihood [48,50]. Because of the low expected frequencies, exact P-values were calculated by the exact conditional test for Poisson variables to compare differences among mutation frequencies, using the Exactci package implemented in R [51,52]. P ≤ 0.05 was considered statistically significant.
We note that whole genome sequencing has the potential to provide a more global assessment of mutation frequencies that would also reveal mutational hotspots within the genome of each cell type. However, genome sequencing is a very costly procedure that must be carefully controlled to distinguish technical sequencing errors from actual biological mutations [53]). The lacI mutation reporter transgene system provides a very convenient and economical system that can be tracked as cells transition from one fate to another and so was ideal for the study reported here.
Results
Mutation frequencies remain low in ESCs maintained in a pluripotent state
We first examined mutation frequencies in two lines of ESCs maintained in a pluripotent state for 35 passages (Line 16A) or 80 passages (Line 26A). In addition, we determined the number of cell doublings that had taken place in each line at the time each sample was recovered. For each cell sample assayed, we examined a minimum of 245,000 pfu, each representing a unique original copy of the mutation reporter transgene recovered from each cell line. This was sufficient to identify a minimum of four nonclonal mutations in each sample. The frequency of confirmed, independent point mutations detected in each sample is shown in Table 1. Mutation frequencies detected in pluripotent ESCs remained consistently low, ranging from 0.60 to 2.22 × 10−5, with values <1 × 10−5 in half of the samples despite being maintained in culture for long periods.
Calculated from time of initial colony formation for ESCs.
Independent, confirmed, nonclonal mutant plaques.
Source of all differentiated cell types.
Derived from 26A/P15 ES cells.
Spontaneous differentiation without addition of RA.
Spontaneous differentiation with addition of RA.
Directed differentiation of macrophages.
ESC, embryonic stem cell.
Mutation frequencies increase in differentiated derivatives of ESCs
We next examined mutation frequencies in three different types of differentiated cells—each derived from the same initial line of pluripotent ESCs (line 26A) at passage 15. These included cells produced by (1) random differentiation in the absence of exogenous retinoic acid (−RA) (Line 26A-RA in Table 1), (2) random differentiation following the addition of RA (+RA) (Line 26A+RA in Table 1), and (3) directed differentiation to specifically derive macrophages (Line 26A-M in Table 1). Once each line became established in culture, mutation frequencies detected in differentiated derivatives of ESCs were significantly higher than those observed in the pluripotent ESCs after a similar number of doublings (eg, P < 0.05 for 26A/P80 vs. 26A−RA/D400, 26A/P80 vs. 26A+RA/D273, and 26A/P80 vs. 26A-M/D400).
Because we maintained an aliquot of ESCs in a pluripotent state at the same time we generated each line of differentiated cells, we were able to conduct a direct comparison of the rate of accumulation of point mutations in otherwise identical cells maintained under pluripotent versus differentiating conditions, respectively (Fig. 1). We actually observed a slight but statistically insignificant decrease in mutation frequency in the pluripotent ESC line 26A as doubling times increased, with frequencies going from 2.03 to 0.76 × 10−5 over a span of 321 cell doublings (P = 0.08442). In contrast, frequencies of mutations increased from 1.04 to 11.47 × 10−5 (11-fold increase) over 253 cell doublings in the presence of exogenous RA (Line 26A+RA in Fig. 1A) (P < 0.05) and from 1.46 to 6.36 × 10−5 (>4-fold increase) in randomly differentiated cells over 362 cell doublings in the absence of exogenous RA (Line 26A-RA in Fig. 1A) (P < 0.05). Similarly, cells that underwent directed differentiation to form macrophages showed an increase in mutation frequency from 1.03 to 4.37 × 10−5 (> 4-fold increase) over 140 cell doublings (Line 26A-M in Fig. 1A) (P < 0.05). Thus, in each case, the frequency of mutations detected in cells subjected to differentiation conditions increased at a significantly greater rate than in cells maintained under pluripotent conditions.

Dynamic changes in mutation frequencies as a function of changes in cell fate. ), also shown is the frequency of mutations detected in three types of differentiated derivatives of these same ESCs: 26A+RAs ( ), 26A-RAs (
), 26A-RAs ( ), and 26A-Ms (
), and 26A-Ms ( ). All differentiated cell types were derived from aliquots of 26A ESCs at passage 15 ( = 49 cell doublings).
). All differentiated cell types were derived from aliquots of 26A ESCs at passage 15 ( = 49 cell doublings).  ] and 1.5 L2 [
] and 1.5 L2 [ ]) of iPSCs derived from those tail-tip fibroblasts and maintained in a pluripotent state, and one line of fibroblasts derived by directed differentiation from each line of iPSCs (1.2aFs [
]) of iPSCs derived from those tail-tip fibroblasts and maintained in a pluripotent state, and one line of fibroblasts derived by directed differentiation from each line of iPSCs (1.2aFs [ ] and 1.5 L2Fs [
] and 1.5 L2Fs [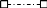 ]) are shown. ESC, embryonic stem cell; iPSC, induced pluripotent stem cell.
]) are shown. ESC, embryonic stem cell; iPSC, induced pluripotent stem cell.
Mutation frequencies decrease during reprogramming of somatic cells to form pluripotent iPSCs
As a complementary approach to examining relative mutation frequencies during the transition of pluripotent ESCs to form differentiated cell types as described above, we also generated iPSCs from somatic cells recovered from double-transgenic mice carrying the 4F2A reprogrammable cassette transgene [46] as well as the lacI mutation reporter transgene [17], which allowed us to examine relative mutation frequencies during the transition from an initial differentiated somatic cell type to a subsequent pluripotent cell type. Thus, in this case, we started with tail-tip fibroblasts from adult mice and detected a relatively high mutation frequency in these cells (8.98 × 10−5), then we induced these cells to undergo reprogramming to form two lines of iPSCs in which we detected initial mutation frequencies at P5 of 0.61 and 1.42 × 10−5 that were each significantly lower than that in the starting tail-tip fibroblast (TTF) line (P < 0.05).
The results of this analysis are shown in Table 2. The significant decrease in mutation frequencies we detected during the transition from differentiated TTFs to pluripotent iPSCs was oriented oppositely from that we observed during the transition from pluripotent ESCs to differentiated cell types as described above. We ascribe this marked drop in mutation frequency following reprogramming of TTFs to the fact that each iPSC line typically emerged from a single reprogrammed cell that did not carry any pre-existing mutation in the lacI mutation reporter transgene. This formed a bottleneck effect similar to the one we previously described in embryos produced by somatic cell nuclear transfer [29] such that each nascent iPSC line began with a mutation frequency of zero in the mutation reporter transgene.
Independent, confirmed, nonclonal mutant plaques.
Tail-tip fibroblasts from adult mice.
iPS cell lines derived by reprogramming TTFs.
Fibroblasts differentiated in vitro from iPS cell lines.
Derived from 1.2a/P15 cells.
Derived from 1.5 L2/P15 cells.
iPSC, induced pluripotent stem cell.
Importantly, the results from our studies of both ESCs and iPSCs are consistent in that cells in the pluripotent state displayed significantly lower mutation frequencies than corresponding differentiated cells that were either derived from the pluripotent cells (as described above for ESCs) or that served as the source of pluripotent cells (in the case of iPSCs) (P < 0.05).
Mutation frequencies remain low in iPSCs maintained in a pluripotent state
We next maintained each of the two iPSC lines (1.2a and 1.5 L2 in Table 2) in a pluripotent state for 25 passages in culture and compared mutation frequencies at early (P5), middle (P15), and late (P25) passages. As was the case for ESCs maintained in a pluripotent state, both iPSC lines preserved relatively low mutation frequencies throughout all 25 passages, ranging from 0.61 to 1.84 × 10−5 for line 1.2a and 1.42 to 3.32 × 10−5 for line 1.5 L2 (Table 2). This is consistent with our general thesis that maintenance of enhanced genetic integrity is a fundamental characteristic of pluripotent cells [5].
Mutation frequencies increase in differentiated derivatives of iPSCs
Finally, we induced aliquots of each iPSC line recovered at P15 to initiate directed differentiation to revert back to fibroblasts—the same differentiated cell type from which each iPSC line was originally derived. Once again, we observed a significant increase in mutation frequencies in association with cells transitioning from a pluripotent to a differentiated state—similar to the results from our study of ESCs described above. This is evident when mutation frequencies detected in each iPSC-derived fibroblast line are compared with those detected in the corresponding source iPSC line following similar numbers of cell doublings. Specifically, after beginning at a common starting frequency of 1.65 × 10−5 in Line 1.2a iPSCs at P15, the mutation frequency detected in the 1.2aF fibroblast line rose nearly fourfold to 6.43 × 10−5 after another 105 cell doublings (1.2aF/W15 in Table 2) (P < 0.05), whereas that in the 1.2a parent iPSC line that was allowed to remain in a pluripotent state for another 103 cell doublings rose only very minimally (and statistically insignificantly) to 1.84 × 10−5 (1.2a/P25 in Table 2) (P = 1.0).
Similarly, from the 1.5 L2 iPSC line, which showed a mutation frequency of 3.32 × 10−5 at P15 (1.5 L2/P15 in Table 2), the mutation frequency rose 2.5-fold to 8.29 × 10−5 after 93 cell doublings during directed differentiation to generate fibroblasts (1.5 L2F/W8 in Table 2) (P < 0.05), while that in the same iPSC line maintained in a pluripotent state for another 81 cell doublings actually declined slightly (although statistically insignificantly) to 2.48 × 10−5 (1.5 L2/P25 in Table 2) (P = 0.585). This confirms that cells propagated in a pluripotent state maintain enhanced genetic integrity, whereas the same starting cell population maintained under conditions favoring a transition to a differentiated state preserves genetic integrity less stringently, leading to a significant increase in mutation frequencies (P < 0.05).
The rate of accumulation of mutations is lower in pluripotent iPSCs than in differentiated derivatives of iPSCs
The difference in mutation frequencies detected in aliquots of iPSCs that were subsequently maintained in a pluripotent or differentiated state, respectively, reflects a significantly higher rate of accumulation of mutations in the differentiated cells relative to that in the pluripotent cells over a similar number of cell doublings. Thus, just as we did for the ESCs described above, we determined cell doubling times for each line of iPSCs or derived fibroblasts, which allowed us to conduct a parallel comparison of the rate of accumulation of point mutations in each cell type. As shown in Figure 1B, we detected either a very small increase or decrease in the accumulation of mutations in the pluripotent cell lines during this period (1.65 to 1.84 or 3.32 to 2.48 × 10−5), whereas we observed a marked increase in the accumulation of mutations in each line of differentiated cells over a similar period (1.65 to 6.43 or 3.32 to 8.29 × 10−5).
Thus, the rate of accumulation of mutations was significantly higher in the same populations of iPSCs that were subjected to differentiation than those that were maintained in a pluripotent state in each case (P < 0.05 for 1.2a/P25 cells versus 1.2aF/W15 cells and for 1.5 L2/P25 versus 1.5 L2F/W8 cells) (Fig. 1B).
Taken together, our analysis of both ESCs and iPSCs shows a common pattern of enhanced maintenance of genetic integrity in pluripotent cells relative to that in differentiated cells. The extent to which a similar distinction might apply to tissue-specific stem cells relative to the more terminally differentiated cell types derived from those stem cells is yet to be determined.
The spectrum of mutations is similar in pluripotent and differentiated cell types
From each cell sample, each plaque (pfu) displaying a mutant (blue) phenotype was sequenced to confirm the presence of a mutation in the lacI gene, identify and discount any clonal mutations (the same mutation at the same location detected more than once in a single sample), and to determine the type of each mutation. A detailed listing of each specific mutation detected in each cell line at each time point analyzed is shown in Supplementary Table S1 in the Supplementary Data, and a summary of the spectrum of mutations detected in each cell line is shown in Table 3. No consistent difference was detected in the relative occurrence of any type of mutation in pluripotent and differentiated cell types, respectively. Base substitutions (transitions or transversions) were more common than insertions or deletions combined (InDels). The mutation spectra observed among the different cell types investigated support the notion that these reflect random spontaneous mutations rather than an abundance of mutations induced by a particular mutagenic effect.
transitions, btransversions, cinversions, or deletions.
Discussion
We recently showed that enhanced maintenance of genetic integrity in pluripotent cells reflects differential expression of DNA repair and cell death genes, and that pluripotency and genetic integrity gene networks are coregulated at the genomic level [5]. That study revealed elevated levels of multiple DNA repair pathways in pluripotent cells relative to differentiated cell types, including mismatch repair (MMR), double-strand break repair by homologous recombination (DSBR-HR), and double-strand break repair by nonhomologous end joining (DSBR-NHEJ). This predicts that the stringency at which genetic integrity is maintained should change as a function of transitions between pluripotent and differentiated cell states, or vice versa. In this study, we have shown that this is indeed the case. We studied two different types of pluripotent cells (ESCs and iPSCs) and respective differentiated cell-type derivatives from each.
In addition, we studied a differentiated cell population and pluripotent iPSC lines derived from those cells. This approach facilitated direct comparisons of mutation frequencies in matched pluripotent/differentiated cell populations that shared a common origin and genetic identity and therefore differed only with respect to having attained a pluripotent or differentiated cellular state, respectively. This provided a tightly controlled assessment of the correlation between pluripotency and enhanced maintenance of genetic integrity. In addition to showing that pluripotency correlates with maintenance of enhanced genetic integrity, whereas differentiated states are associated with less stringent maintenance of genetic integrity, our data confirm that transitions between pluripotent and differentiated cellular states, or vice versa, are accompanied by dynamic statistically significant transformations in the relative stringency at which genetic integrity is maintained.
We examined two types of pluripotent cells, ESCs and iPSCs, as well as multiple types of differentiated cells—randomly differentiated cells, fibroblasts, and macrophages— and observed maintenance of genetic integrity at consistently enhanced levels in the pluripotent cell types relative to the differentiated cell types as reflected in mutation frequencies that were significantly lower in the pluripotent cells than in each matched cell population that had been maintained in a differentiated state. However, beyond a simple static comparison of mutation frequencies in populations of pluripotent and differentiated cells, our study also facilitated a dynamic assessment of mutation rates as a function of either passage number or number of cell doublings in either pluripotent or differentiated cell types. This allowed us to observe the rate of accumulation of mutations during prolonged maintenance of cells in the pluripotent or differentiated state or during transitions from the pluripotent state to a differentiated state, or vice versa.
Our previous study of mutation frequencies in donor somatic cell populations and embryos produced from those cells by somatic cell nuclear transfer (cloning) revealed that mechanisms regulating the relative maintenance of genetic integrity in pluripotent versus differentiated cell types are subject to epigenetic reprogramming [29]. In this study, we show that a similar reprogramming of mechanisms regulating genetic integrity occurs during derivation of iPSCs from differentiated adult fibroblasts. Enhanced maintenance of genetic integrity in both germ cells and pluripotent cells has been attributed to upregulation of genetic integrity genes encoding proteins involved in DNA repair and/or cell death activities [4,5,27,54 –58]. This suggests that reprogramming of mechanisms regulating expression of genetic integrity genes occurs in conjunction with transitions between pluripotent and differentiated states.
In our recent study of interrelationships between the pluripotency and genetic integrity gene networks at the genomic level, we found that master regulators of pluripotency (eg, OCT4, SOX2, NANOG)—all of which are transcription factors—directly interact with multiple genetic integrity genes that show differential expression in pluripotent cells versus differentiated cells [5]. These regulators were also found to interact with additional genes that encode other transcription factors that are also differentially expressed in pluripotent and differentiated cell types and interact with additional downstream genetic integrity genes.
These types of interactions between the pluripotency and genetic integrity gene networks suggest a direct mechanism by which the gain or loss of pluripotency can influence the relative stringency of maintenance of genetic integrity in stem cells and/or their differentiated derivatives. These results support our contention that pluripotent cells likely eliminate cells carrying large genetic aberrations by elevated cell death activities while simultaneously limiting the accumulation of point mutations by elevated DNA repair activities.
Conclusion
The maintenance and manipulation of cells in culture will be central to the proposed use of stem cells for either cell-based therapeutic approaches or patient-specific modeling of disease states in vitro (eg, disease in a dish)[59 –64]. Our results provide insight into basic biological distinctions between pluripotent and differentiated cell types that impact genetic integrity in a manner that is directly relevant to the potential clinical use of these cell types. In particular, these results demonstrate that transitions in cell states commonly induced when stem cells are manipulated for therapeutic purposes will be accompanied by corresponding changes in the stringency at which genetic integrity is maintained.
During the preparation of this article, a related study by Rouhani et al. [65] was published that reached similar conclusions regarding the enhanced maintenance of genetic integrity in pluripotent cells.
Footnotes
Acknowledgments
The authors thank Ms. Jacey Hornecker for assistance with derivation of the ES cells, Dr. Rogelio Zamilpa for assistance with teratoma procedures used with the iPS cells, and Dr. Christopher Navara for assistance with immunocytochemistry. This work was supported, in part, by a grant from the Kleberg Foundation to J.R.M.
Author Disclosure Statement
The authors declare no competing or conflicting interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.