
Abstract
Heart development depends on coordinated proliferation and differentiation of cardiac progenitor cells (CPCs), but how the two processes are synchronized is not well understood. Here, we show that the secreted Bone Morphogenetic Protein (BMP) antagonist GREMLIN 2 (GREM2) is induced in CPCs shortly after cardiac mesoderm specification during differentiation of human pluripotent stem cells. GREM2 expression follows cardiac lineage differentiation independently of the differentiation method used, or the origin of the pluripotent stem cells, suggesting that GREM2 is linked to cardiogenesis. Addition of GREM2 protein strongly increases cardiomyocyte output compared to established procardiogenic differentiation methods. Our data show that inhibition of canonical BMP signaling by GREM2 is necessary to promote proliferation of CPCs. However, canonical BMP signaling inhibition alone is not sufficient to induce cardiac differentiation, which depends on subsequent JNK pathway activation specifically by GREM2. These findings may have broader implications in the design of approaches to orchestrate growth and differentiation of pluripotent stem cell-derived lineages that depend on precise regulation of BMP signaling.
Introduction
H
We have previously shown in zebrafish that the secreted Bone Morphogenetic Protein (BMP) antagonist Gremlin 2 (Grem2), also called Protein Related to Dan and Cerberus (PRDC), is expressed in pharyngeal mesoderm during the initial formation of the cardiac tube and is necessary for cardiac laterality and proper differentiation of CMs [14,15]. More recently, we found that Grem2 promotes cardiac lineage expansion during differentiation of mouse ES cells [16,17]. However, whether GREM2 plays a role in the expansion or differentiation of human cardiac progenitor cells (CPCs), or both, is not known.
Using human pluripotent stem cells, we show that GREM2 expression is induced in NKX2.5+ CPCs during the initial steps of their differentiation and promotes the formation of contracting CMs. The GREM2 effect depends on dual positive effects on CPC proliferation and differentiation that depend on sequential inhibition of the canonical, p-SMAD-mediated BMP signaling and activation of the noncanonical JNK pathway, respectively. This novel regulation of BMP signaling provides a unique mechanism to optimize the growth and differentiation of human CMs.
Materials and Methods
Human pluripotent stem cell culture
Human iPS (hiPS) cell lines iMR90 and DF 19-9-11 from WiCell were cultured under feeder-free conditions in mTeSR1 or Essential 8 (E8) media (Stem Cell Technologies) as previously described [18]. Briefly, cells were thawed and seeded onto six-well plates coated with 8.7 μg/cm2 growth factor-reduced Matrigel (Corning) in mTeSR1 or E8 media supplemented with 10 μM Y-27632 dihydrochloride (Tocris Bioscience) to promote cell survival and attachment. Media were exchanged daily until cells reached ∼60%–70% confluence. Cells were passaged using Versene EDTA cell dissociation reagent (Thermo Fisher Scientific/Gibco) and seeded onto Matrigel-coated plates in mTeSR1 or E8 media supplemented with 10 μM Y-27632 dihydrochloride at a split ratio of 1:15–1:20. Cells were kept in a copper-lined humidified incubator (Thermo Fisher Scientific) at 37°C with a 5% CO2 atmosphere.
Human ES (hES) cells WA07 were cultured in conditioned media as previously described [19]. Briefly, cells were cultured in mouse embryonic fibroblast (MEF)-conditioned medium supplemented with basic fibroblast growth factor (bFGF) (8 ng/mL). Cells were cultured for 4–6 days or until colonies occupied 75%–80% of well surface area. Cells were then passaged by rinsing with Dulbecco's phosphate-buffered saline and incubating with collagenase (200 U/mL) for 5–10 min at 37°C to dissociate them into small clumps. Clumps were then plated onto six-well cell culture plates precoated with 1 mL/well of 50 μg/mL Matrigel (Corning). Cells were kept in a copper-lined humidified incubator (Thermo Fisher Scientific) at 37°C with a 5% CO2 atmosphere.
iPS cell differentiation
hiPS cells were differentiated using the “Matrix Sandwich” method, the GiWi method, or the BMP/Activin A method, as already described [11,20,21]. In all differentiation methods, cells were allowed to become 80%–90% confluent and then dissociated by incubating in Versene EDTA cell dissociation reagent (Thermo Fisher Scientific/Gibco) for 10 min at room temperature. After incubation, cells were triturated to dissociate into a single-cell suspension. Single-cell preparations were centrifuged for 5 min at 200 g and cell pellets were resuspended in cell culture media for plating.
For the “Matrix Sandwich” method, cells were resuspended in E8 or mTeSR1 media (Stem Cell Technologies) supplemented with 10 μM ROCKi (Y-27632 dihydrochloride; Tocris) and plated onto Matrigel-coated (8.7 μg/cm2) 12-well culture plates at a density of 500,000 cells per well. Fresh E8 or mTeSR1 media were given daily until cells became 90% confluent. When 90% confluent, cells were overlaid with 8.7 μg/cm2 growth factor-reduced Matrigel (Corning) in E8 or mTeSR1 media. After 24 h, the matrix coating solution was removed and fresh E8 or mTeSR1 media were added until cells were 100% confluent. Cells were then treated with 1 mL/well of day 0 media (RPMI 1640 media supplemented with B27 minus insulin, coated with 8.7 μg/cm2 Matrigel, and 100 ng/mL Activin A; R&D Systems). Exactly 24 h later, day 0 media were aspirated and cells were treated with 1.5 mL/well of day 1 media (RPMI 1640 media supplemented with B27 minus insulin) (Life Technologies), 5 ng/mL of hBMP4 (R&D Systems), and 10 ng/mL human bFGF (Life Technologies).
Four days after addition of day 1 media, cells were treated with 1 mL/well basal differentiation media (RPMI 1640 media supplemented with B27 plus insulin). Cells treated with GREM2 received 1 mL/well of RPMI 1640 media with B27 minus insulin supplemented with 150 ng/mL GREM2 exactly 48 h after adding day 1 media (day 3). At day 5, GREM2-treated wells received basal differentiation medium with 150 ng/mL of GREM2. Media in all wells were replaced daily. Cells were treated in a similar manner with 50 ng/mL NOGGIN or 1.5 μg/mL DAN, based on their specific activities (R&D Systems). GREM2 wild-type protein and mutated versions of GREM2 were synthesized, purified, and measured for activity as previously described [22 –24].
The BMP/Activin A method followed the same protocol as described for the “Matrix Sandwich” method, but without the Matrigel overlay steps.
For the GiWi method, cells were resuspended in E8 media (Stem Cell Technologies) supplemented with 10 μM ROCKi (Y-27632 dihydrochloride; Tocris) and plated onto Matrigel-coated (8.7 μg/cm2) 12-well culture plates at a density of 500,000 cells per well. Once cells were 100% confluent (typically 3–4 days after seeding), differentiation was started by adding 2 mL/well of day 0 media (RPMI 1640 with B27 minus insulin and 12 μM CHIR 99021). Exactly 24 h after adding day 0 media, cells were treated with 2 mL/well early differentiation media (RPMI 1640 with B27 minus insulin). After 48 h, 1 mL/well of conditioned media was removed from differentiating cells and combined with 1 mL early differentiation media and supplemented with 2 μM IWR-1 endo (Tocris). After 48 h, cells were treated with 2 mL/well of late differentiation media (RPMI 1640 with B27 plus insulin). Late differentiation media were then replaced daily.
The hES cells were differentiated as previously described [25]. Briefly, WA07 hES cells were rinsed with 2 mL DPBS and incubated with 2 mL Versene (EDTA; Life Technologies) for 10 min at 37°C. Versene was aspirated and replaced with 1 mL/well of MEF-conditioned media supplemented with 8 ng/mL of bFGF. hES cells were triturated to produce a single-cell suspension and seeded onto 24-well Matrigel-coated (8.7 μg/cm2) plates at a density of 400,000 cells per well. Cells were given fresh media daily until 100% confluent. Once 100% confluent, cells were given 1 mL/well day 0 medium (RPMI 1640 with 2% B27 minus insulin and 100 ng/mL Activin A). Cells were incubated at 37°C for 24 h and then treated with 1 mL/well of day 1 medium (RPMI 1640 with 2% B27 minus insulin and 10 ng/mL BMP4). After 4 days, the medium was replaced with late differentiation medium (RPMI 1640 with 2% B27). 1 mL/well of fresh late differentiation media was added to each well every other day.
Reverse transcriptase quantitative polymerase chain reaction
Cells were collected from each well using Tryp-LE Select (Thermo Fisher Scientific) and centrifuged at 200 g for 5 min to pellet. Cell pellets were lysed using RLT buffer, and RNA was isolated using the RNeasy Mini Kit following the manufacturer's instructions (Qiagen). cDNA was generated by reverse transcription of 1–3 μg of RNA as we have previously reported [16]. cDNA samples were amplified using GoTaq qPCR Master Mix (Promega) in a Bio-Rad CFX thermocycler. Relative gene expression levels were calculated using the delta-delta Ct method [26,27]. Relative primer efficiencies were determined using the Real-time PCR Miner algorithm and confirmed experimentally using the slope of the standard curve from plotting log(DNA copy number) versus Ct value [28]. Amplification primer sequences are reported in Supplementary Table S1 (Supplementary Data are available online at
Cell quantification
Cells were dissociated into single-cell suspensions using Tryp-LE Express (Life Technologies), stained with trypan blue (diluted 1:2 in PBS or cell culture media) to exclude dead cells and quantified using a Bio-Rad TC-10 automated cell counter. For DAPI-stained cells, cell numbers were quantified by dividing three fields of view from three independent wells per condition into quadrants and manually counting the number of nuclei visible in each quadrant.
Immunofluorescence
Cells were seeded onto Matrigel-coated (8.7 μg/cm2) 12-well plastic culture plates (Thermo Fisher Scientific) at a density of 500,000 cells per well and differentiated as described above for the “Matrix Sandwich” method. Cells were fixed at the desired time points (differentiation days 4, 5, 6, and 10) by rinsing with 500 μL DPBS and incubating in 4% paraformaldehyde in PBS at 4°C for 5 min. The PFA solution was then aspirated and cells were rinsed with 1× PBS five times. Fixed cells were permeabilized by incubating with 400 μL permeabilization buffer (0.2% Triton X-100 in 1× PBS) in each well at room temperature for 1 h. Nonspecific binding was then blocked using 400 μL/well of blocking solution (5% nonfat dry milk in permeabilization buffer) for 2 h at room temperature with gentle rocking. For all subsequent steps, a minimum of 350 μL of solution was added to each well. Cells were then washed 3 × 5 min with 400 μL/well of 1× PBS at room temperature. After washing, cells were incubated with primary antibodies in incubation buffer (0.1% Triton X-100, 1% BSA in 1× PBS) overnight at 4°C with gentle rocking. Next, cells were washed 3 × 3 min each, with PBST (0.2% Tween-20 in 1× PBS) followed by 3 × 3 min each with 1× PBS. Cells were then incubated with secondary antibodies diluted in incubation buffer for 1 h at room temperature in the dark. Finally, cells were then washed 2 times, 3 min each, with 1× PBS and stored in 400 μL 1× PBS for imaging.
Imaging was done using a Leica DM IRB Inverted Microscope or a Zeiss Laser Scanning Microscope (LSM 880). Image analysis and volume rendering were done using NIS Elements, Zen, ImageJ/FIJI, and Imaris software suites. Primary antibodies recognizing phospho-HISTONE H3 (Santa Cruz Biotechnology; Cat. No. sc-8656-R, 1:500), NKX2.5 (Santa Cruz Biotechnology; sc-8697, 1:50), GREM2 (ProteinTech; 13892-1-AP, 1:100), and α-ACTININ (Sigma; A7811, 1:500) were applied. We have also tested anti-GREM2 rabbit polyclonal antibodies from GeneTex (GTX108414), Abcam (ab102563), and R&D Systems (AF2069) at a 1:100 dilution. The fluorescent dye 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) was added during the final 15 min of secondary incubation at a 1:10,000 dilution to stain nuclei.
Flow cytometry
Differentiated hiPS cells were washed once with DPBS minus Mg2+ and Ca2+, incubated with 1 mL/well Accutase at room temperature for 15 min, and triturated to break up into single cells. Accutase was diluted by adding two volumes of late differentiation medium per well and contents were transferred to 15-mL conical tubes through mesh strainer caps to remove large cell clumps. Before centrifugation and after visual confirmation of complete cell detachment and single-cell dissociation with Accutase, the total cell number per well was quantified using the Bio-Rad TC-10 automated cell counter as described above. Cell suspensions were then centrifuged at 200 g for 5 min to pellet cells. Cell pellets were rinsed 1× time with DPBS minus Mg2+ and Ca2+ and repelleted. Washed pellets were resuspended in 500 μL Fix/Perm buffer (fixation/permeabilization solution; BD Biosciences), transferred to 5-mL round-bottomed flow cytometry tubes (BD Biosciences), and incubated for 40 min on ice. Cells were then pelleted at 300 g for 5 min and supernatant was aspirated. Cells were washed 2× times with Perm/Wash buffer (BD Bioscience) and pelleted by centrifugation at 300 g for 5 min. Cells were blocked by adding 100 μL blocking buffer (BD Bioscience) to each tube and incubating on ice with gentle rocking for 30 min.
Primary antibodies recognizing NKX2.5 (Abcam; ab91196) and MYH6 (Abcam; ab50967) at a 1:100 dilution were added directly to the cell suspensions in each tube and incubated for 2 h on ice with gentle rocking. To wash out primary antibodies, 3 mL of Perm/Wash buffer was added to each tube and then centrifuged at 300 g for 5 min. Supernatant was aspirated and cells were washed with 2 mL/tube fresh Perm/Wash solution and centrifuged at 300 g for 5 min. For secondary antibody incubation, cell pellets were suspended in 100 μL blocking buffer with secondary antibodies at 1:400 dilutions. Cells were incubated with rocking for 30 min on ice and then washed twice with Perm/Wash buffer. Cells were then suspended in Flow Assay Buffer (BD Biosciences) and the percentage of NKX2.5+ and MYH6+ cells was determined using FACS Aria flow cytometer and analyzed using FACS Diva (BD Bioscience) or FlowJo (FlowJo, LLC) software in the Vanderbilt University Shared Flow Cytometry Resource Laboratory. The total number of CMs per well was calculated by multiplying the total cell number per well (as described above) by the percentage of NKX2.5+ or MYH6+ cells as determined by flow cytometry.
Western blotting
Protein samples from differentiated hiPS cells (DF 19-9-11; WiCell) were isolated using the RIPA buffer, including the protease inhibitor cocktail and phosphatase inhibitor cocktail 2 and 3 (Sigma) diluted at 1:100. Cell lysates were shaken at 4°C for 30 min and centrifuged at 12,000 rpm at 4°C for 15 min. Protein concentration in supernatants was measured using the Pierce BCA protein assay (Thermo Fisher Scientific). Fifteen micrograms of protein (adjusted to 7.5 μL with distilled water) was then mixed with 7.5 μL of 2× Laemmli sample buffer (Bio-Rad) containing β-mercaptoethanol (Sigma). Proteins were denatured at 95°C for 5 min and then placed on ice for another 5 min. Electrophoresis was run using sodium dodecyl sulfate polyacrylamide gels with 1× MOPS SDS running buffer (Life Technologies). Size fractioned proteins were transferred to nitrocellulose membranes (Bio-Rad) using the semidry system.
Membranes were blocked with 5% dry milk or 5% BSA (Sigma) in Tris-buffered saline (Corning) containing 1% Tween-20 (Sigma) (1× TBST) for 1 h at room temperature or 4°C, respectively. Next, membranes were incubated overnight with antibodies recognizing phosphorylated SMAD1/5/9 or total SMAD1 and phosphorylated or total JNK 1&2 (Cell Signaling) diluted at 1:1,000, NKX2.5 (Santa Cruz Biotechnology, sc-8697) diluted at 1:500, and β-ACTIN (Sigma Aldrich, A1978) diluted at 1:5,000 in 5% BSA in 1× TBST. Next day, membranes were washed 3 times, 5 min each with TBST and incubated for 2 h at room temperature with peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) diluted 1:5,000 in 5% dry milk-TBST. Three 5-min washes were performed to wash away unbound secondary antibodies. Signals were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and images were taken with ChemiDoc Touch System (Bio-Rad). Protein band intensities were quantified using ImageJ.
Luciferase assays
CGR8 mouse ES cells were transfected with the BRE2-Luc reporter construct with luciferase expression under the control of two canonical BMP signaling responsive elements of the Id2 gene [16,29]. Cells were treated for 8 h with 40 ng/mL BMP4, 50 ng/mL GREM2, and/or 300 ng/mL BMP decoy protein L51P [30]. Firefly luciferase activity was normalized to Renilla luciferase activity to account for transfection efficiency.
Results
GREM2 is expressed in differentiating human pluripotent stem cells during CPC expansion and differentiation
We have previously shown that grem2 expression in zebrafish embryos first appears within the pharyngeal arch mesoderm, adjacent to the cardiac field, during the early stages of heart tube formation [14,15]. In mouse embryos, we found that Grem2 expression appears around embryonic day 8 and overlaps with the nascent cardiac field area [16]. To determine whether GREM2 is associated with early human cardiac development, we cultured and differentiated the hiPS cell line DF 19-9-11, using the “Matrix Sandwich” method that was designed for efficient CM differentiation [20]. We then isolated RNA samples at consecutive days of differentiation and analyzed the temporal program of cardiogenic development by reverse transcriptase quantitative polymerase chain reaction (RT-qPCR). As shown in Fig. 1, expression of the T BRY gene that marks mesoderm formation is transiently induced at days 2–3 after initiation of differentiation, followed by sequential expression of markers specific to early cardiogenic mesoderm (MESP-1), CPCs (NKX2.5, ISL1, and KDR or VEGFR2), and CMs (TNNT2, MYH6). Expression of endothelial and hematopoietic genes (CD31, VE-CADHERIN, HBBY) was at low levels (data not shown), likely because the “Matrix Sandwich” method is optimized specifically for cardiac differentiation.

GREM2 expression during hiPS cell cardiac differentiation follows the expression pattern of cardiac-specific genes. RT-qPCR gene expression analysis in DF 19-9-11 hiPS cells during cardiac differentiation [day 0 (D0) to day 10 (D10)] using the “Matrix Sandwich” method indicates cardiogenic mesoderm specification (MESP-1) and the appearance of cardiovascular progenitor cell gene markers (KDR, NKX2.5, ISL1). GREM2 expression appears concurrently with cardiovascular progenitor cells and increases during differentiation to cardiomyocytes marked by TNNT2 and MYH6 genes. The expression pattern of BMP ligands BMP2 and BMP4, BMP antagonists CERBERUS-LIKE 1 and NOGGIN, as well as BMP receptors type 2 (BMPR2) and type 1 (ALK3 and ALK6) is indicated. n = 3 replicates per condition. ALK3 or 6, activin A receptor type II-like kinase 3 or 6, also BMPR1A or BMPR1B; BMP2 or 4, bone morphogenetic protein 2 or 4; BMPR2, bone morphogenetic protein receptor 2; GREM2, gremlin 2; ISL-1, insulin gene enhancer protein 1; hiPS, human-induced pluripotent stem; KDR, kinase insert domain receptor; MESP-1, mesoderm posterior bHLH transcription factor 1; MYH6, myosin heavy chain 6; NKX2.5, NK2 homeobox 5; T-BRY, T-brachyury; TNNT2, cardiac troponin T 2.
To place GREM2 expression in the context of other BMP signaling components during hiPS cell differentiation, we analyzed expression of select BMP ligands, BMP receptors, and BMP antagonists. Our results show that CERBERUS LIKE 1 and CHORDIN are transiently induced during mesoderm formation, in accordance with their expression patterns in other species [16,31 –33], whereas NOGGIN is expressed at later time points of differentiation. GREM2 expression appears after mesoderm formation, coincidently with early cardiac markers such as NKX2.5, and continues to rise during cardiac differentiation (Fig. 1). The expression of BMP2 and BMP4 is in line with their respective roles in cardiac differentiation in mice, with BMP2 being expressed first during cardiogenic specification and followed by BMP4 [34,35]. In contrast to the dynamic expression of BMP ligands and BMP antagonists during cardiac differentiation, BMP receptors (BMPR2, ALK3, ALK6) appear to be constantly expressed throughout the differentiation process.
A similar expression pattern for GREM2 was also observed in (1) the WA07 hES cells and (2) the independently reprogrammed hiPS cell line iMR90 C4 (Supplementary Fig. S1). Furthermore, because the “Matrix Sandwich” method of cardiac cell differentiation depends on timely activation and withdrawal of BMP signaling, which may influence expression of BMP ligands and antagonists, we used the distinct GiWi differentiation protocol that relies on sequential activation and inhibition of canonical Wnt signaling [11]. Our results show a similar induction profile of GREM2 during cardiac differentiation following the GiWi protocol, indicating that the GREM2 expression pattern is independent of the differentiation method used (Supplementary Fig. S1).
Taken together, our data demonstrate that GREM2 expression starts after mesoderm formation, during the initial phase of CPC specification, and is retained throughout CPC expansion and differentiation to CMs. GREM2 is unique among BMP antagonists to follow the cardiac differentiation pattern, although NOGGIN is also expressed within specific time windows of cardiac differentiation. The GREM2 expression pattern is consistent with our previous studies in zebrafish and mouse development, as well as mouse ES cell differentiation [14 –16], indicating that GREM2 expression and possibly function have been conserved during evolution from lower vertebrates to humans. Furthermore, the GREM2 expression pattern is consistent among different hES and iPS cell lines and independent of the applied differentiation protocol, suggesting that GREM2 is inherently linked to the early stages of the cardiac differentiation process.
GREM2 is expressed in human CPCs and differentiated CMs
To determine the cellular source of GREM2, we performed immunofluorescence staining on fixed cells during early (day 5 and 7) and late (day 10) differentiation stages using primary antibodies against GREM2, and antibodies recognizing typical cardiac-specific proteins, such as the transcription factor NKX2.5 and the sarcomeric component α-ACTININ.
To identify antibodies specific for detection of human GREM2 protein in cultured cells, we screened four distinct, commercially available, anti-GREM2 primary antibodies. To this end, we used HEK293 cells transiently transfected with a plasmid expressing the human GREM2 cDNA under the regulation of the CMV promoter, or empty vector as control. Transfected cells were fixed, stained, and imaged using a fluorescence microscope (Supplementary Fig. S2). Three antibodies gave a specific signal and the one with the best signal to noise ratio was subsequently selected for immunolabeling of differentiating iPS cells.
We found that GREM2 is expressed in NKX2.5+ cells at differentiation day 5 and 7 (Fig. 2A and Supplementary Fig. S3A, B). GREM2 protein expression persists in α-actinin+ CMs at day 10 of differentiation (Fig. 2B and Supplementary Fig. S3C). GREM2 was not detected in NKX2.5− cells, suggesting that GREM2 expression is confined to the cardiac lineage during iPS cell differentiation and acts in an autocrine manner.
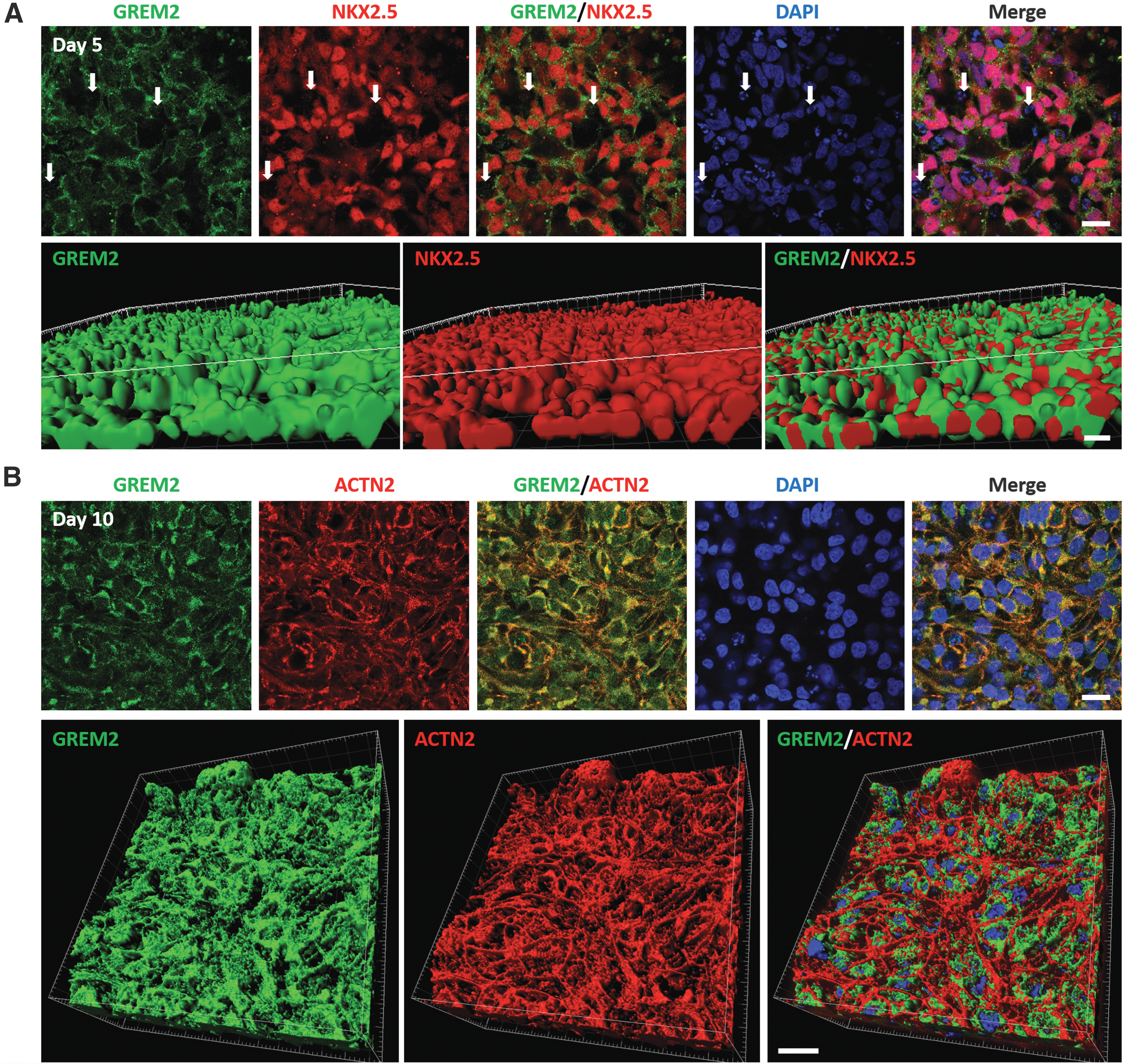
GREM2 is expressed in CPCs and cardiomyocytes. Immunofluorescence analysis of differentiating hiPS cells.
BMP signaling antagonism is required for CM differentiation
The GREM2 expression data described above suggest that BMP signaling antagonism is necessary for CM differentiation. To test this possibility, we treated differentiating iPS cells with an L51P mutant of BMP2 that was shown to bind the BMP antagonist NOGGIN, but unable to bind BMPRI and activate BMP signaling, thus acting as a BMP ligand decoy [30]. To test the ability of the BMP decoy to block GREM2, we first transfected CGR8 mouse ES cells with a plasmid containing the firefly luciferase gene under the control of the BMP signaling response elements from the Id2 gene promoter (BRE2-Luc) [16,29]. Optimal concentrations for effective BMP signaling inhibition by GREM2 were first determined by titrating increasing concentrations of GREM2 in cells exposed to a constant BMP4 amount (Supplementary Fig. S4). The cells were then treated with BMP4, GREM2, and/or the BMP decoy. The results show that BMP4 induces canonical BMP (BRE2-Luc) signaling, whereas the BMP decoy alone does not signal nor does interfere with BMP4 signaling activation (Fig. 3A). GREM2 inhibits BMP4 activity, where GREM2 alone does not induce BRE2-Luc activity, as expected. Moreover, coincubation of BMP4, GREM2, and the BMP decoy restored canonical BMP signaling, indicating that the BMP decoy effectively blocks the inhibitory effect of GREM2 (Fig. 3A).
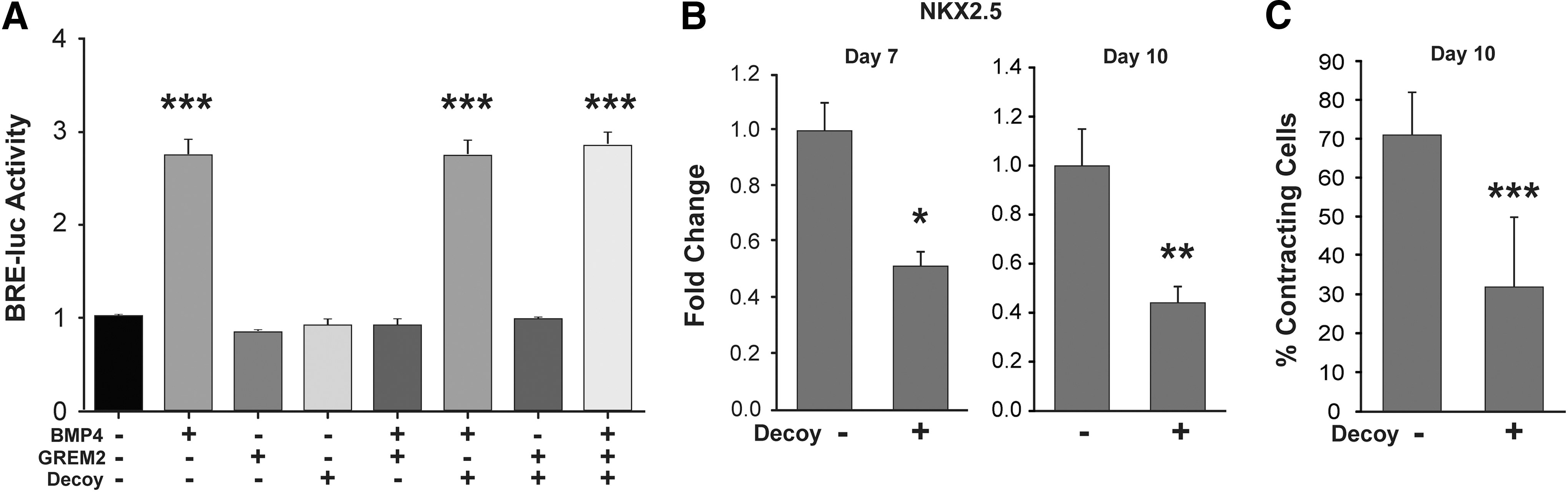
BMP ligand antagonism is required for cardiac differentiation of hiPS cells.
Treatment of differentiating human pluripotent stem cells with the BMP decoy protein starting at day 4 of differentiation, when CPCs first appear and GREM2 expression is induced, caused a significant reduction in cardiac gene expression levels and reduced the percentage of contracting cells in culture (Fig. 3B). These data suggest that BMP signaling inhibition is required for cardiogenic differentiation of human pluripotent stem cells.
GREM2 enhances the cardiogenic potential of differentiating iPS cells
The results described above suggest inhibition of BMP signaling is necessary for CM differentiation of hiPS cells. To test whether GREM2 can further enhance human CM differentiation, we added GREM2 protein to differentiating iPS cells from day 3 onward, when cardiogenic mesoderm appears and endogenous GREM2 expression begins to rise. The “Matrix Sandwich” protocol (SP) served as a positive control, whereas a modified, truncated “Matrix Sandwich” differentiation protocol (MP) without any factor addition after mesoderm induction at day 3 served as the baseline or negative control (Fig. 4A).

GREM2 enhances the cardiogenic potential of differentiating iPS cells.
Visual comparison of cultures revealed that GREM2 protein addition accelerated the appearance of contracting cells, indicating the presence of CMs at day 7, which is 1 day earlier than the “Matrix Sandwich” method and 3 days earlier than the negative control. In addition, GREM2 also increased the overall areas of contracting cells, suggesting further expansion of the cardiac lineage (Fig. 4B, C).
In agreement with the visual observations, analysis of RNA samples prepared at days 10 and 14 of differentiation showed that addition of GREM2 leads to a further increase in cardiac-specific gene expression levels compared to the “Matrix Sandwich” method (Fig. 4D). To quantify the long-term effects of GREM2 on CM yields, we performed flow cytometry analysis at differentiation day 30 using antibodies recognizing the cardiac-specific myosin heavy chain MYH6. We also used antibodies recognizing NKX2.5, the expression of which is maintained in mature CMs, thus providing an independent marker to quantify cardiac cells. The data showed that GREM2 treatment gives rise to increased numbers of CMs compared to the “Matrix Sandwich” method (Fig. 4E). Specifically, GREM2 treatment increased the percentage of MYH6+ cells from ∼83% in the optimal Sandwich protocol (SP) to ∼93% and approximately doubled the total number of cells, thus increasing the total number of CMs/well, as shown in Fig. 4E. Similarly, the percentage of NKX2.5+ cells increased from ∼70% to ∼88%.
Using engineered GREM2 mutants that retain or lose BMP binding activity showed that BMP binding is required for cardiogenic differentiation (Fig. 4F). The positive effect of GREM2 was also evident using an independent hiPS cell line, iMR90 C-4, indicating that the procardiogenic properties of GREM2 are not limited to a single pluripotent stem cell line (Supplementary Fig. S5). Taken together, these data demonstrate that GREM2 protein addition during cardiac differentiation accelerates and enhances the cardiogenic potential of differentiating hiPS cells.
GREM2 promotes proliferation of CPCs
Using western blotting analysis of NKX2.5 protein, we found that GREM2 does not affect the initial induction of early cardiac genes, but leads to expansion of cardiac lineage markers at subsequent differentiation stages (Supplementary Fig. S6). Instead, the GREM2 expression during the initial appearance of CPCs and its stimulatory effect on cardiac differentiation raised the possibility that GREM2 promotes proliferation of CPCs. To directly test this possibility, we stained GREM2-treated differentiating iPS cells with antibodies recognizing the phosphorylated form of HISTONE H3 (pHH3) that is specifically phosphorylated during the mitosis and meiosis phases of cell division, thereby marking proliferating cells. The “Matrix Sandwich” (SP) and baseline (MP) protocols as outlined above in Fig. 4A served as positive and negative controls, respectively.
Our results demonstrate that addition of GREM2 protein at day 3 leads to a five- to sixfold increase in pHH3+ cells at day 4 (Fig. 5A and Supplementary Fig. S7). At this stage, pHH3 colocalizes with cell colonies expressing the early cardiovascular progenitor-specific membrane receptor KDR (or VEGFR2) [10]. Notably, KDR expression diminishes in SP cultures compared to MP and even more in GREM2-treated cells, likely due to acceleration of cardiac differentiation and loss of vascular markers, which is consistent with the timing of appearance of contracting cells under the different protocols at later stages (Fig. 4B). At day 5, pHH3 is specifically found in the nuclei of NKX2.5+ CPCs (Fig. 5B). The immunofluorescence analysis results show that GREM2 promotes proliferation of CPCs. Interestingly, the “Matrix Sandwich” protocol, which requires BMP4 addition at differentiation days 3–5, initially generates less proliferating CPCs than the other conditions (Fig. 5A, B, and Supplementary Fig. S7). However, the “Matrix Sandwich” protocol eventually promotes CM differentiation after BMP4 removal at day 5, but not to the same extent as addition of GREM2.

GREM2 promotes proliferation of CPCs. Immunofluorescence staining of CPCs during iPS cell differentiation. Cells were treated with GREM2 and compared to “Matrix Sandwich” (SP) and baseline negative controls (MP) as described in Fig. 4A.
BMP inhibition promotes proliferation of CPCs
The results described in the previous sections show that GREM2 promotes proliferation of CPCs. To test whether GREM2-mediated inhibition of BMP is sufficient to also stimulate cardiac differentiation, we compared the effects of GREM2 to two additional, distinct BMP antagonists, namely NOGGIN and DAN (Fig. 6A). All three BMP antagonists led to suppression of the canonical BMP signaling target gene ID2, respectively, showing they function as expected (Fig. 6B). Treatment with any of the three BMP antagonists led to a significant increase in overall cell numbers (Fig. 6C). Costaining of differentiating iPS cells at day 6 with antibodies recognizing NKX2.5 and pHH3 showed that GREM2-treated cells had higher proliferation rates than baseline. NOGGIN and DAN also increased CPC proliferation, compared to baseline (Fig. 6D). Of note, there were comparable numbers of NKX2.5+ cells at day 6 in control and BMP antagonist-treated differentiating iPS cells, further suggesting that GREM2 and BMP signaling antagonists do not affect the specification of CPCs.
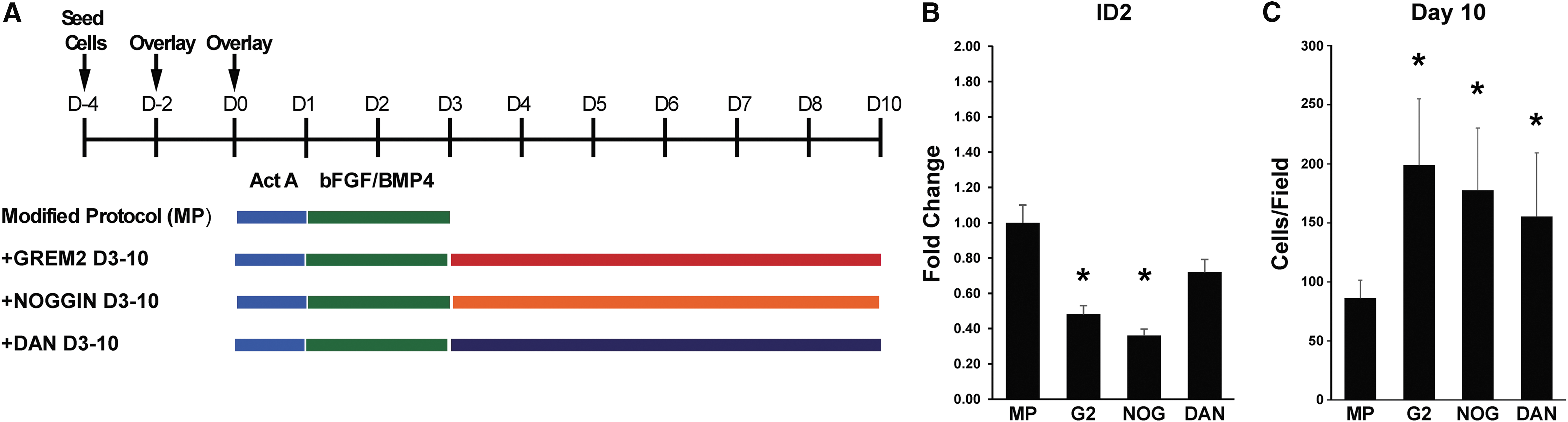
BMP inhibition promotes proliferation of CPCs but is not sufficient to induce cardiac-specific genes.
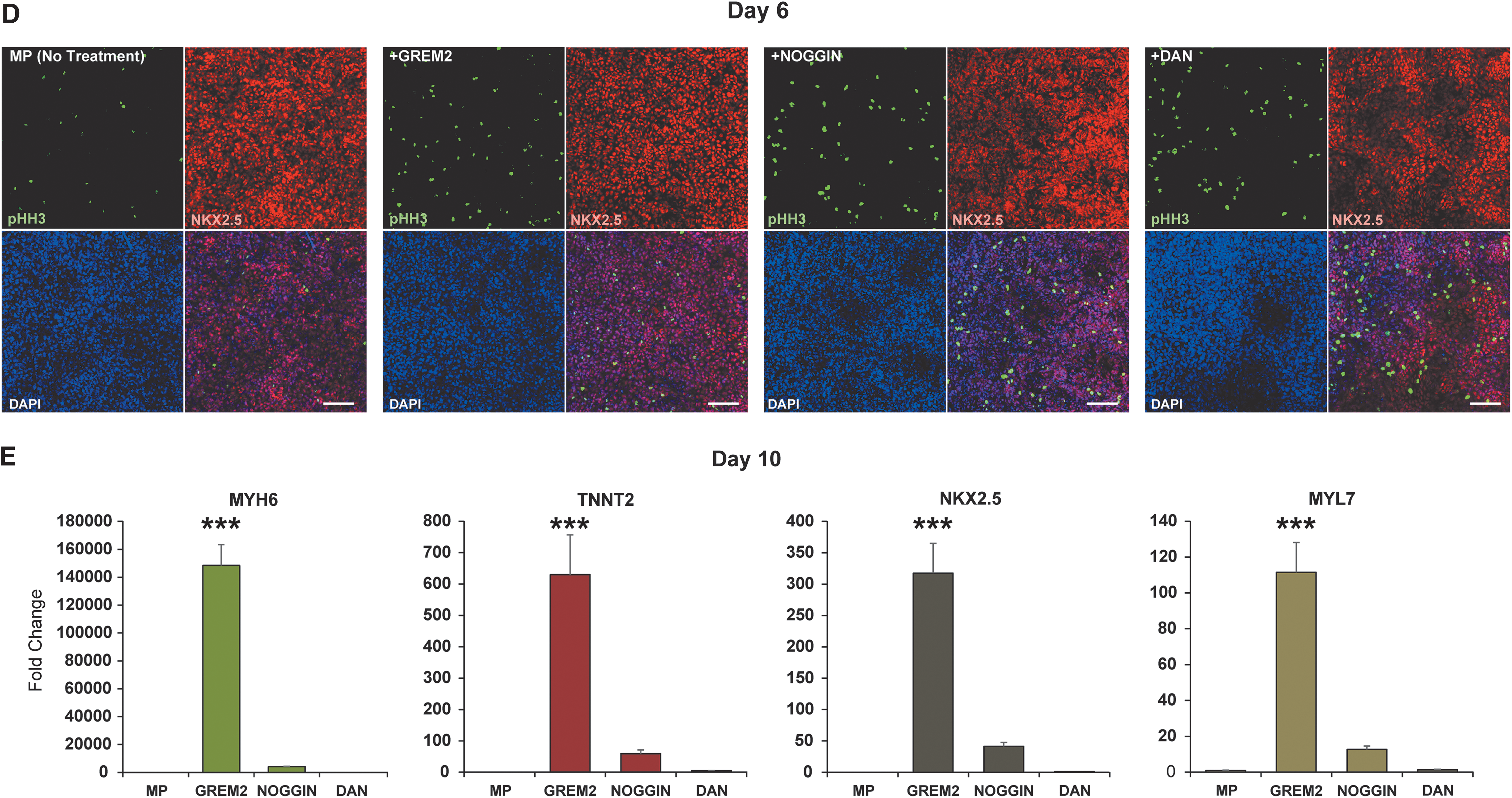
To test whether BMP signaling inhibition is sufficient to promote CM differentiation, we isolated RNA samples at day 10 of differentiation from cells treated as shown in Fig. 6A. qPCR analysis showed that GREM2 led to induction of cardiac-specific gene markers characteristic of contracting CMs such as MYH6 and TNNT2, but NOGGIN and DAN had minimal or no effect (Fig. 6E). Therefore, although all three BMP antagonists enhanced proliferation of NKX2.5+ cells, induction of CM differentiation was a characteristic specific to GREM2.
GREM2 enhancement of cardiac differentiation requires JNK activation
We have previously shown that GREM2 inhibited canonical BMP, that is, SMAD1/5/8-mediated signaling shortly after treatment of differentiating mouse ES cells, followed by JNK signaling activation at later stages. Moreover, we found that this dual effect on canonical BMP signaling inhibition and the subsequent noncanonical JNK signaling activation is a unique property of GREM2 among BMP antagonists [16]. To test whether JNK activation drives cardiogenic differentiation of hiPS cells by GREM2, we prepared protein samples at differentiation days 4, 5, 6, 7, and 8 of GREM2- and NOGGIN-treated cells. The “Matrix Sandwich” and baseline protocols served as positive and negative controls, respectively (as described in Fig. 4A). Western blotting analysis using antibodies recognizing activated phospho-SMAD1/5/8 showed that both GREM2 and NOGGIN effectively blocked SMAD phosphorylation 24 h after treatment (Fig. 7A). Phospho-SMAD1/5/8 levels became undetectable in all samples at subsequent time points (not shown). We did not detect JNK phosphorylation at days 4–7 (not shown). In contrast, western blotting showed that phospho-JNK1/2 protein was specifically activated by GREM2 at day 8, but not NOGGIN (Fig. 7A). Of note, we did not detect JNK activation by the Matrix Sandwich method at this stage, suggesting that either alternative procardiogenic mechanisms are induced by this method or that GREM2 accelerates JNK signaling activation.
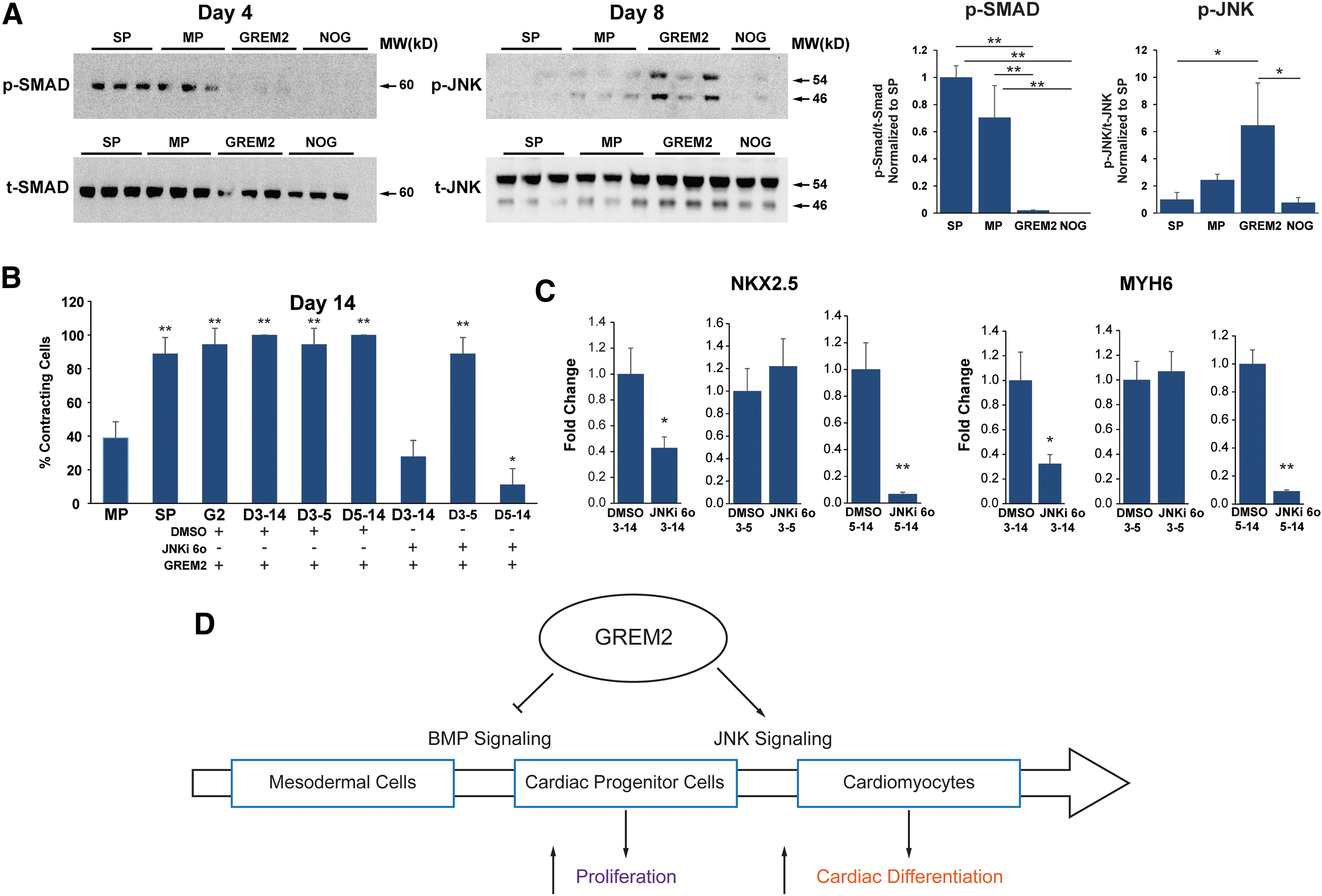
JNK signaling is required for GREM2-induced cardiac differentiation.
To test whether JNK activation by GREM2 drives human CM differentiation, we exposed GREM2-treated differentiating iPS cells to the JNK signaling-specific chemical inhibitor TCS JNK 6o (JNKi 6o) [36,37] or vehicle control. Visual observations and RNA analysis by qPCR showed that JNK signaling inhibition abolished the stimulatory effect of GREM2 on the cardiac lineage expansion (Fig. 7B, C). Of note, JNK inhibition was effective when applied during differentiation of CPCs after day 5. In summary, our data show that GREM2 coordinates cardiogenic output in a 2-step mechanism, initially inhibiting canonical BMP signaling to promote CPC proliferation followed by induction of differentiation through JNK signaling activation (Fig. 7D).
Discussion
Our data demonstrate that expression of the secreted BMP antagonist GREM2 is induced in pluripotent stem cell-derived human CPCs shortly after the specification of cardiac mesoderm. GREM2 expression is maintained in CPCs throughout their differentiation and persists in differentiated CMs. BMP signaling antagonism is required for cardiac lineage development, since BMP ligand decoys that bind antagonists, but do not activate BMP receptors, abort CM differentiation. Our results further show that GREM2 increases cardiac gene expression and CM differentiation, a process that depends on JNK signaling activation. Interestingly, although BMP antagonists such as NOGGIN and DAN equally stimulate CPC proliferation, only GREM2 efficiently promotes differentiation to CMs.
BMP ligands play crucial roles at various stages of cardiac development [38], starting from the initial cardiogenic specification of mesoderm [39], to subsequent cardiac tube assembly, asymmetric looping and jogging [40], ventricular identity [41], outflow track and cushion formation [35], and CM differentiation [42]. Although canonical BMP signaling induces NKX2.5 gene in the cardiac crescent to generate CPCs, BMP signaling needs to be suppressed to allow proliferation and expansion of CPCs. Interestingly, during mouse secondary heart field development, Nkx2.5 suppresses BMP signaling by blocking pSmad1, forming a negative regulatory loop that promotes proliferation of CPCs [43]. Our results show that this mechanism likely applies to human CPCs as BMP signaling inhibition by NOGGIN, GREM2, or DAN proteins promotes proliferation of CPCs.
On the contrary, elegant gain- and loss-of-function studies in mouse embryos have shown that persistent BMP signaling inhibition prevents differentiation of CPCs [43]. Our data confirm that NOGGIN and DAN prevent expression of genes specific to differentiated CMs, including sarcomere proteins such as MYH6 and TROPONIN T2. In contrast, GREM2, after the initial stimulation of CPC proliferation, leads to superinduction of cardiac differentiation. We have previously shown that GREM2 has the unique property among BMP antagonists, after its prescribed canonical BMP signaling inhibition, to subsequently induce JNK signaling activation that is required for cardiac differentiation [16]. Our results indicate that this GREM2 property is responsible for promotion of human cardiac cell differentiation as well, since JNK signaling inhibition aborts the procardiogenic effect of GREM2. These findings are consistent with previous reports that have implicated JNK signaling in cardiogenic differentiation of pluripotent stem cells [44,45].
Recent crystallographic evidence revealed that GREM2 folds into a unique tertiary shape that has not been described before. Specifically, GREM2 dimerizes in a head-to-tail manner, unlike the head-to-head pairing of NOGGIN [24,46,47]. This head-to-tail arrangement gives rise to large, constrained, and arching hydrophobic surfaces on the three-dimensional structure, which precludes Grem2 from wrapping around BMP dimers as NOGGIN does [24,46,47]. We currently test whether this unique structural arrangement is also critical for the function of GREM2 in regulating both proliferation and differentiation of CPCs. Future biochemical analyses may further identify critical structural motifs that could be exploited to design molecules that mimic the biological effects of GREM2, such as the ability to selectively alter its BMP inhibitory and JNK activating properties. Recognizing these mechanisms may offer additional novel insights in the cardiac differentiation process. Moreover, due to the wide interest in regulating BMP signaling in a number of human diseases, BMP signaling inhibitors are being developed for clinical use [48,49]. Thus, our findings may facilitate future repurposing of these new pharmacological resources for coordinated growth and differentiation of stem cell populations.
We recently established that during embryonic development in zebrafish, grem2 first appears in the pharyngeal mesoderm next to the forming heart tube [14,15]. Loss- and gain-of-function approaches demonstrated that Grem2 is necessary for proper cardiac tube jogging and looping, cardiac laterality, and CM differentiation by suppression of Smad1/5/8 phosphorylation [15]. Although left/right asymmetry and complex morphogenetic processes cannot be replicated in cell culture, our data show that the critical role of GREM2 in CM differentiation has been conserved across species and applies to human pluripotent stem cells. Moreover, we found that Grem2 is critical for atrium formation in zebrafish and promotes differentiation of pluripotent mouse ES cells to atrial-like CMs [16]. We did not detect preferential induction of atrial- versus ventricular-specific genes in the examined early stages of GREM2-treated hiPS cells (data not shown). Because human CMs take longer to mature in culture than mouse cells [50], it is likely that longer treatment periods might be required to test whether GREM2 can regulate chamber identity in iPS cell-derived CMs.
In the adult mouse heart, we recently discovered that Grem2 is highly induced in peri-infarct CMs at the end of the inflammatory phase after myocardial infarction. Using genetic gain- and loss-of-Grem2-function mouse models and chemical compounds that inhibit BMPs, we found that Grem2 modulates the magnitude of the inflammatory response and keeps inflammation in check through suppression of canonical BMP signaling [51]. Grem2 levels after myocardial infarction correlate with functional recovery, suggesting a new strategy to control inflammation of cardiac tissue after acute ischemic injury and improve cardiac function. It is intriguing that the BMP developmental pathway that initiates cardiac specification is also among the earliest induced after ischemic injury in the adult heart, but assumes a very different role. It is also intriguing that Grem2 induction takes place in peri-infarct CMs indicating that reactivation of CM-specific expression of Grem2 takes place under pathological conditions. However, unlike in developing CMs, Grem2 does not induce cell proliferation in the adult heart. Understanding how this property is lost in adult CMs might open the way to unlock cardiac regenerative mechanisms.
Footnotes
Acknowledgments
We thank the Vanderbilt FACS Core and the Cell Imaging Shared Resource for expert assistance. We are grateful to Gokhan Unlu and Dr. Ela Knapik for their assistance and advice with immunofluorescence imaging. This work was supported by NIH grants HL100398 to A.K.H and GM114640 to T.B.T. and A.K.H.; and T32 Program in Cardiovascular Mechanisms: Training in Investigation (HL007411) fellowships to J.B.B. and D.T.P.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.