
Abstract
P311 is a newly discovered functional gene, and it has been proved to play a key role in blood pressure homeostasis, glioblastoma invasion, renal fibrosis, hypertrophic scar formation, and others. In this study, for the first time, we found that P311 could enhance reepithelialization during wound healing via promoting epidermal stem cell (EpSC) migration through Rho GTPases. P311 expression was highly increased in neo-epidermal cells during human and mouse skin wound healing, and P311was co-localized with 5-bromo-2′-deoxyuridine positive label-retaining cells in a mouse superficial second-degree burn wound model. Furthermore, transfection of human EpSCs with adenovirus encoding P311 significantly accelerated the cell migration in vitro. Moreover, highly expressed P311 could enhance the activities of the Rho GTPases (RhoA, Rac1, and Cdc42) in cultured human EpSCs. P311-knockout mouse EpSCs showed dramatically decreased cell migration and activities of Rho GTPases (RhoA, Rac1, and Cdc42). Besides, both the RhoA-specific inhibitor and the Rac1 inhibitor, not the Cdc42 inhibitor, could significantly suppress P311-induced human EpSC migration. In vivo, the reepithelialization was markedly impaired during wound healing after P311 was knocked out. Together, our results suggested that P311 could accelerate skin wound reepithelialization by promoting the migration of EpSCs through RhoA and Rac1 activation. P311 could serve as a novel target for regulation of EpSC migration during cutaneous wound healing.
Introduction
W
P311, also called PTZ17, was first identified by Studler in 1993. P311 is mainly expressed in the embryonic brain in mice and persisted at a high level in the cerebellum, hippocampus, and olfactory bulb during adulthood [5]. The P311 gene is mapped to the long arm of chromosome 5, which encodes an 8-kDa intracellular protein containing 68 amino acids and does not belong to any known protein family. The N-terminus of P311 protein contains a PEST domain (rich in Pro, Glu, Ser, and Thr) that is highly conserved among humans, mice and chicken. This domain plays a key role in targeted protein degradation by binding to components of the ubiquitin/proteasome pathway via protein–protein interactions and activations. PEST domains were originally detected in short-lived proteins such as transcription factors, cytokines, and signal molecules [6,7]. The half-life of the P311 protein is ∼5 min, with rapid degradation by Met-hepatocyte growth factor/scatter factor (Met-HGF/SF), the lactacystin-sensitive ubiquitin/proteasome system, and an unidentified metalloprotease [5]. P311 plays a role in nerve and lung regeneration [8,9], glioblastoma invasion [10,11], blood pressure homeostasis [12], renal fibrosis [13 –15], myofibroblast differentiation, and amoeboid-like migration [16 –18]. P311-deficient mice display altered behavioral responses in learning and memory [19]. In addition, P311 is involved in the affective, but not the sensory, component of pain [20], which may be consistent with P311 expression in the nervous system.
Our previous studies showed that P311 was highly expressed in the granulation tissue of skin wounds and in the early hypertrophic scars, and P311 was involved in the pathogenesis of hypertrophic scars [16,21]. However, the possible role of P311 in skin wound reepithelialization was still unclear. In this study, we investigated the potential role of P311 in wound reepithelialization by using P311 knockout mice and human skin wound tissue. The underlying molecular mechanism was also studied.
Materials and Methods
Source of specimens and study approval
Foreskin tissue samples were obtained from normal and healthy children during circumcision. Skin wound tissue samples were obtained from the Department of Pathology. Informed consent was obtained from all patients involved in this study. All protocols involving human tissues were approved by the Medical and Ethical Committees of the Southwest Hospital, the Third Military Medical University. All human experiments were performed in accordance with the guidelines of the Third Military Medical University. All the animal experimental protocols were approved by the Animal Experimental Ethics Committees of the Third Military Medical University and were also performed in accordance with the guidelines of the Third Military Medical University.
Animals and the cutaneous excisional wound splinting model
The P311−/− mice were kindly gifted by Professor Gregory A. Taylor [19]. The P311+/+ C57BL/6 mice (Charles River Laboratories) were purchased from the Beijing Vital River Laboratory Animal Company and raised in the Animal Institutes of Daping Hospital, the Third Military Medical University. Sixteen P311−/− mice and 10 age-matched P311+/+ mice (males; 6–8 weeks old; body weight from 18 to 20 g) were used for the wound healing experiments. The mouse cutaneous excisional wound splinting model was generated as previously described [22]. Briefly, the mice were anesthetized and the dorsal hair was shaved. Two symmetrical 3-mm-diameter full-thickness cutaneous biopsy punch wounds were made on the mouse dorsum. A biological membrane (NPWT-1, Negative pressure wound therapy kit) with adhesive dressings was immediately glued on the wound surface to suppress skin contraction. The wound tissue sample was incised and was separated across the center into two equal pieces on day 0, 3, and 5. Then, the skin wound tissue was fixed in 4% formaldehyde overnight, embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin. Sections were photographed under a light microscope (Leica; CTR6000). Wound reepithelialization began at the wound edges and moved toward the center. Low-magnification images of the wound center were acquired, and the distance of reepithelialization was measured by using Image-Pro Plus.
Mouse superficial second-degree burn wound model
Six C57BL/6 mice (males, 6–8 weeks old, body weight from 18 to 20 g) were anesthetized, and the hair on the dorsum was shaved. A temperature-controlled metal plate (YLS-Q5; Shangdong Academy of Medical Science) with a 1.0-cm diameter and 500 g weight was used to produce the burn wound. The metal plate was heated to 65°C and placed perpendicularly on the dorsal skin for 3 s. The superficial second-degree burn wound was confirmed by a histopathological test.
5-bromo-2′-deoxyuridine labeling
The label-retaining cells (LRCs) were detected by using a protocol that was previously described by Bickenbach and Chism and Braun et al., with some modifications [23,24]. Newborn mice were intraperitoneally injected with 5-bromo-2′-deoxyuridine (BrdU) at 50 mg/kg body weight twice daily for 3 days to label skin LRCs. After 7 weeks of injection, the labeled mice were used in the superficial second-degree burn wound model.
Human EpSC isolation and culture
Cells were isolated from the foreskin of children according to the method of Kaur et al. and Liao et al. [25,26]. Briefly, subcutaneous tissue from skin specimens was cut off by using sterile surgical instruments. Epithelial sheets were incubated in 0.5% Dispase II (Roche; 04942078001) at 4°C overnight. The basal keratinocytes were isolated and cut into pastes, which were placed in 0.25% trypsin for 10 min at 37°C. The mixture was stirred and dissociated into individual cells. The trypsin was inactivated in calcium-free Roswell Park Memorial Institute (RPMI) 1640 Medium containing 10% fetal bovine serum (FBS). Cells were filtered through a 70-μm cell strainer and centrifuged at 800 rpm for 10 min. Then, cells were re-suspended in Serum-Free Keratinocyte Medium (K-SFM; Gibco; 17005) with human recombinant epidermal growth factor (0.1–0.2 ng/mL), bovine pituitary extract (20–30 mg/mL), calcium chloride (0.05 mM), and penicillin and streptomycin solution (100 IU/mL; Gibco; 15140122). Cells were seeded in 25-mm dishes that were coated by type IV collagen (Sigma; C5533) and allowed to adhere for 15 min [27]. The rapidly adhering cells were cultured in the K-SFM at 37°C in 5% CO2, non-adhering cells were discarded, and the medium was changed every 2 days.
Mouse EpSC isolation and culture
Cells were isolated from the dorsal skin and the ventral skin of neonatal mice according to the method of Tudor et al. [28]. Briefly, all tissues were immersed in 0.2% chlorhexidine for 10 min and then washed twice in sterile phosphate-buffered saline (PBS). The skin was digested in 0.5% dispase II solution (Roche; 04942078001) at 4°C overnight. The epidermis was removed as intact sheets and dissociated into simple cell suspension by immersion in 0.25% trypsin for 10 min at 37°C. The trypsin was inactivated in calcium-free RPMI 1640 Medium but with 10% FBS, and the tissues were agitated and filtered through a 70-μm cell strainer. Cells were spun down at 800 rpm for 10 min, and the pellets were re-suspended in K-SFM (Gibco; 17005) with human recombinant epidermal growth factor (0.1–0.2 ng/mL), bovine pituitary extract (20–30 mg/mL), 10 ng/mL mouse epidermal growth factor (BD; 354001), 1 × 10−10 M cholera toxin (Sigma; C9903), calcium chloride (0.05 mM), and penicillin and streptomycin solution (100 IU/L; Gibco; 15140122). The cells were counted and cultured in a 5% CO2 and 37°C incubator. Culture medium was changed every 2–3 days.
Immunofluorescence staining
Cells were seeded on 12-well glass slides that had been previously coated with type IV collagen until cells reached ∼50%–60% confluence. Then, cells were washed in PBS and fixed with 4% paraformaldehyde for 10 min. Cells were permeabilized with 0.5% Triton X-100 for 10 min. After being washed, cells were blocked for 30 min at 37°C in goat serum and incubated with either goat anti-mouse Cytokeratin 19 Ab at a 1:100 dilution (Santa Cruz; sc-33119) or rabbit anti-mouse β1 integrin Ab at a 1:100 dilution (Santa Cruz; sc-374429) overnight at 4°C. Cells were then incubated with the secondary antibodies, fluorescein isothiocyanate (FITC)-conjugated immunoglobulin for detecting Cytokeratin 19 and Cy3-conjugated immunoglobulin for detecting β1 integrin for 30 min at 37°C. The samples were mounted in Anti-fade Mounting Medium (Beytime; P0126). Confocal microscopy was performed by using a LSM51 microscope (Zeiss) that was equipped with helium, neon, and argon lasers (Zeiss) and appropriate filters.
Flow cytometry
Cultured EpSCs were used for flow cytometric analysis. Dual staining was performed by using FITC-conjugated CD71 (BD; 561936 and 555536) and phycoerythrin-conjugated α6 integrin (BD; 555736). Flow cytometry data acquisition was performed on an Attune Acoustic Focusing Cytometer (Applied Biosystems, Life Technologies), and data were analyzed by using FlowJo software (Tree Star Incorporation).
Replication-defective adenovirus production and infection
Replication-defective adenovirus was generated as previously described [16]. Briefly, P311 cDNA (pRcCMV, human P311) and GFP cDNA were sub-cloned into an expression plasmid. The pAdTrack-CMV plasmid and the pAdEasy-TMxL Adenoviral Vector System were purchased from Stratagene. The P311 plasmid was digested and ligated with T vector. After the recombinant product was identified by sequencing, pAdTrack-CMV/pGEM T-Easy-P311 shuttle plasmid was constructed and transfected into competent Escherichia coli DH5a (ATCC), and the successful clones were collected. Then, the pAdTrack-CMV/pGEM T-Easy-P311 shuttle plasmid and the adenovirus genome-containing pAdEasy-P311 plasmid were recombined in E. coli BJ5183, and the adenovirus plasmid expressing the target gene was screened. Viruses were packaged and propagated in HEK-293 cells. Then, the viruses were purified with an Adenovirus Purification Miniprep Kit (BIOMIGA; V1160-02), and the titer was measured in plaque-forming units (PFUs) by using a TCID50 kit (AGTC; K-AD0001). Cells were infected at a matched multiplicity of infection. Human EpSCs were seeded at 5 × 104 cells per well and transferred to an incubator at 37°C in 5% CO2. When the cell confluence reached 60%, 2 mL of K-SFM containing 1 × 109 PFU virus was added to each well. Cells were cultured, transfected for 48 h, and examined for the expression of green fluorescent protein by using an inverted microscope (Olympus; IX71).
Wound scratch assay
Cells were seeded in 12-well plates, transfected with replication-defective adenovirus for 48 h, and grown until 90% confluence. Cells were treated with 4 ng/mL mitomycin C (Sigma; M4287) for 2 h to suppress cell proliferation. Then, the cell monolayer was scratched by using a 200 μL disposable plastic pipette tip, and cells were allowed to migrate into the wound surface. Cells were observed 0, 24, and 48 h after the scratching, and cells were photographed by using an inverted microscope (Olympus; IX71). The migration distance and the number of migrating cells were measured in three independent experiments.
RhoA, Rac1, and Cdc42 activity assay
Activity assays for RhoA, Rac1, and Cdc42 were performed by using an RhoA Activation Assay Biochem Kit (Cytoskeleton; BK034), Rac1 Activation Assay Biochem Kit (Cytoskeleton; BK035), and Cdc42 Activation Assay Biochem Kit (Cytoskeleton; BK036), respectively. Briefly, equal amounts of human EpSCs were seeded and infected with replication-defective adenovirus for 48 h. The P311+/+ and P311−/− mouse EpSCs were also seeded in equal numbers. When the cell confluence reached 60%–70%, the cells were dissociated and quantified. Next, 40 μg cell lysis product per group was prepared to detect the total RhoA, Rac1, and Cdc42 by using Western blot. Equivalent amounts of cell lysate (400 μg for each group) were added to a predetermined amount of rhotekin-RBD beads (Rac1 and Cdc42 for PAK-PBD beads) from a bead titration test and were incubated at 4°C on a rotator for 1 h. The beads were pelleted by centrifugation at 5,000 g at 4°C for 5 min. Next, 10 μL of 2× laemmli sample buffer was added to each tube. Then, bead samples were boiled for 5 min and analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Finally, the total and bound proteins were visualized by using a Molecular Imager ChemiDoc™XRS+ Imaging System (BIO-RAD) to detect the chemiluminescence signal.
The specific inhibition of Cdc42, Rac1, and RhoA
Human EpSCs were seeded in 12-well plates, infected with replication-defective adenovirus for 48 h, and grown until 85% confluence. Cells were treated with 4 ng/mL mitomycin C (Sigma; M4287) for 2 h and then scratched with a 200 μL disposable plastic pipette tip. The following inhibitors were added to the cells: the RhoA-specific inhibitor, Rhosin (Calbiochem; 555460) at 30 μM, an IC50 that inhibits the interaction of RhoA with its guanine exchange factors (GEFs); the Cdc42 inhibitor, ZCL278 (Calbiochem; 500503) at 50 μM, an IC50 that selectively suppresses Cdc42 activity; and the Rac1 inhibitor, Z62954982 (Calbiochem; 553512) at 50 μM, an IC50 that selectively inhibits Rac1 activity. Cells were observed 0, 24, and 48 h after scratching. The migration distance was measured to quantify the migration rates.
Immunohistochemistry
Human and mouse skin wound tissues samples were fixed in 4% paraformaldehyde at 4°C for 24 h, embedded in paraffin, and sectioned at 4 μm. Sections were dewaxed in xylene and rehydrated in decreasing ethanol concentrations. Then, sections were incubated with antigen retrieval solution at 37°C for 30 min. After the antigen retrieval, sections were incubated with 3% H2O2 for 20 min at room temperature and then with goat serum at 37°C for 30 min. Then, sections were incubated with rabbit anti-mouse P311 Ab at a 1:400 dilution (Bioss; bs-0427R) overnight at 4°C. Afterward, the sample was washed twice in PBS, and sections were incubated with a biotinylated secondary antibody for 20 min at 37°C. After washing again, sections were incubated with streptavidin–peroxidase complex for 20 min at 37°C. Controlled colorization was performed by using a diaminobenzidine kit (ZSGB-BIO; ZLI-9018) under a microscope (Olympus; CX31). Sections were counterstained with hematoxylin, dehydrated, and mounted with resin.
BrdU were used to label LRCs. The skin wound tissues were harvested from the mouse, which was injected with BrdU after birth, superficial second-degree burn wound model. Paraformaldehyde-fixed sections were dewaxed in xylene and rehydrated in decreasing ethanol concentrations. Tissue sections were microwaved in 10 mM sodium citrate (pH 6.0) for 3 min, incubated for 15 min in a hot solution, and rinsed in PBS. Sections were incubated in 2 M HCl at 37°C, washed in borate buffer, and finally digested in 0.01% trypsin in 0.05 M Tris for 3 min at 37°C. After being blocked in 10% goat serum for 20 min, sections were incubated with rabbit anti-mouse BrdU Ab at a 1:100 dilution (Bois; bs-0489R) overnight at 4°C. Then, sections were washed twice in Tris-HCl buffered saline (TBS), and they were incubated with a biotinylated secondary antibody for 30 min at 37°C. Sections were washed with TBS and incubated with alkaline phosphatase solution for 30 min at 37°C. Colorization by 5-bromo-4-chloro-3-indolyl phosphate/tetranitroblue tetrazolium chloride diluted 1:20 in 0.01 M TBS (pH 9.0–9.5) was examined under a microscope (Olympus; CX31).
Statistical analysis
Statistical comparisons were performed with Student's t-test or one-way analysis of variance. Data are presented as the mean ± standard deviation. In all cases, a P value <0.05 was considered statistically significant.
Results
The expression and distribution of P311 in human and mouse cutaneous wounds
To characterize the P311 expression pattern in clinical samples, we first investigated five samples of human cutaneous wounds from biopsy specimens and normal skin tissue from the same subjects by using immunohistochemical analysis. An abundance of P311 accumulated in the cytoplasm of neo-epidermal cells in skin wound tissues (Fig. 1A). However, P311 was negative in the normal human epidermis (Fig. 1B). To determine whether the increased P311 level in human skin wounds also occurred in mice, we used an excisional wound splinting model and a superficial second-degree burn wound model in mouse skin. The mouse cutaneous superficial second-degree burn wound model was confirmed by pathological methods. The burned skin presented epidermal necrosis, diffuse perivascular infiltration, and collagen degeneration. The damage was limited to the upper third of the dermis (data not shown). Then, we examined the P311 expression in the mouse skin wound tissues by using immunohistochemical staining. Similarly, P311 was also highly expressed in neo-epidermal cells in both the excisional wound splinting model and the superficial second-degree burn wound model (Fig. 1C, D). However, P311 was not detected in the normal mouse skin (Fig. 1E). Moreover, we used BrdU to label the LRCs in the mouse superficial second-degree burn wound model and normal mice [23,24]. It was found that BrdU was accumulated in the nucleus of neo-epidermal cells in burned wounds (Fig. 1F) and epidermal cells in the basal layer of normal skin (Fig. 1G). Meanwhile, the BrdU-labeled LRCs seemed to be co-localized with P311-positive cells in serial sections of burned wounds (Fig. 1D, F). Together, these data indicate that P311 might play a role in human and mouse skin wound reepithelialization.
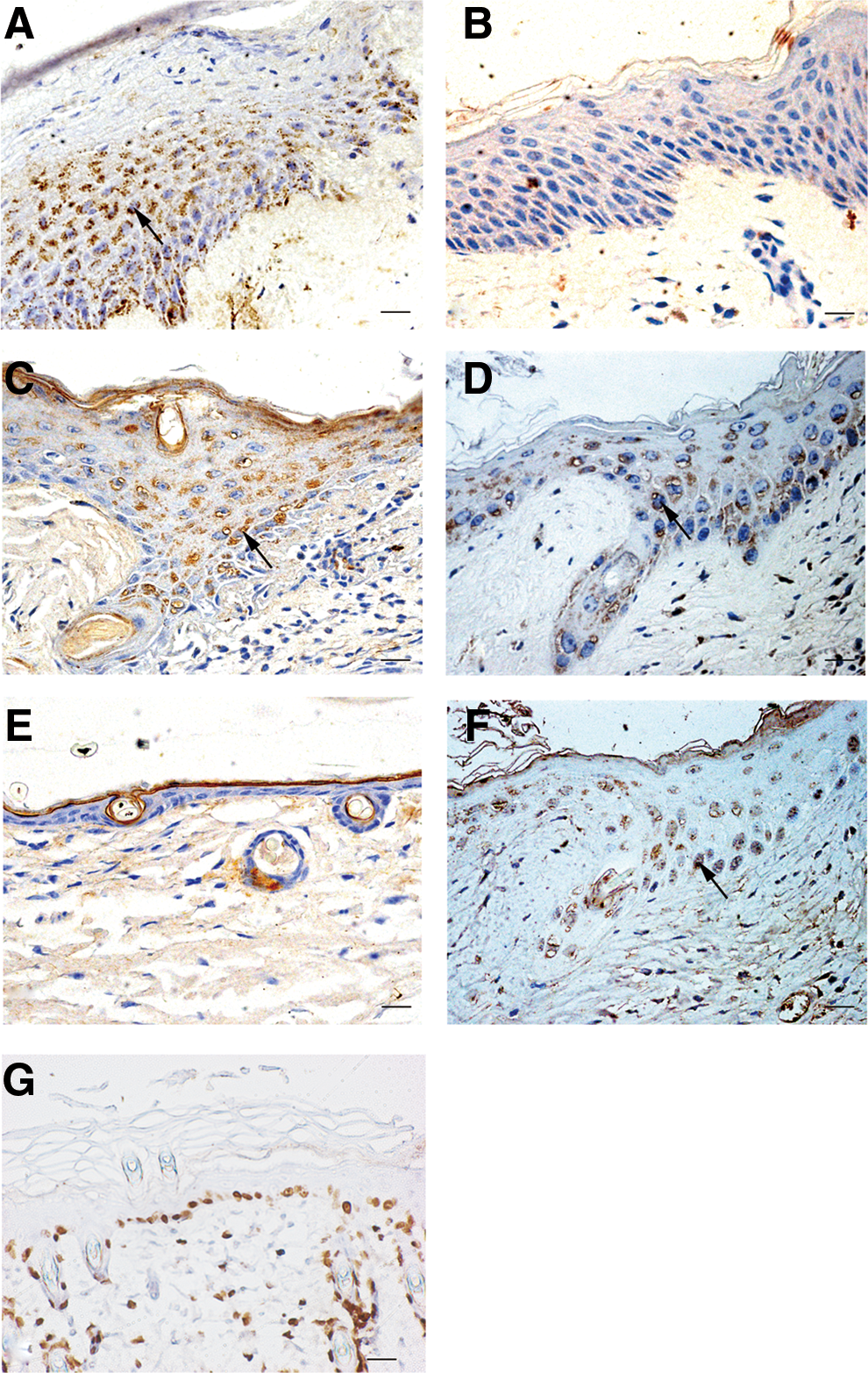
P311 was highly expressed in the neo-epidermal cells of human and mouse skin wounds.
P311 promotes human EpSC migration in vitro
To further elucidate the role of increased P311 expression in neo-epidermal cells, human EpSCs were isolated from the foreskin of children. EpSC-specific markers, such as cytokeratin 19 (K19), β1 integrin, and α6 integrin, were detected. We examined K19 and β1 integrin, and found that they were highly expressed in the cultured cells, as indicated by immunofluorescence staining (Fig. 2A). Flow cytometry showed that the cultured cells had a high level of α6 integrin and a low level of CD71 (Fig. 2B). Then, the P311 replication-defective adenovirus vector (pADEasy-P311) and the control adenovirus vector (pADEasy-Myc) were generated and transfected in human EpSCs for 48 h. The successful transfection of these vectors was confirmed by flow cytometry, real-time polymerase chain reaction, and immunohistochemistry analysis (Supplementary Fig. S1; Supplementary Data are available online at
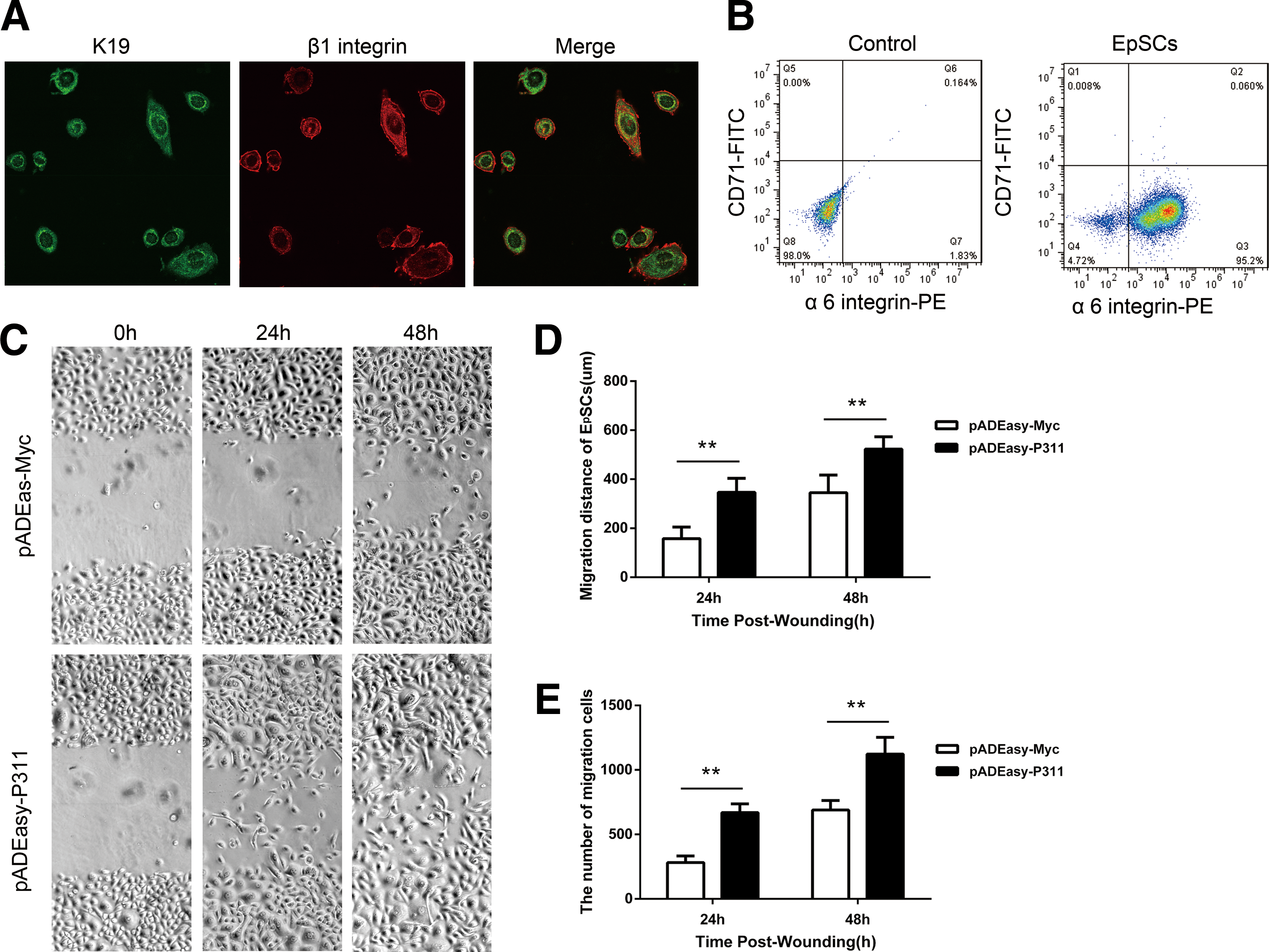
P311 over-expression promoted human EpSC migration in vitro.
P311 deficiency impairs mouse EpSC migration in vitro
The earlier findings suggested a potential functional role for P311 in EpSC migration. To examine this hypothesis, we tested the effects of P311 elimination by using P311−/− mice and P311+/+ mice. We successfully isolated mouse EpSCs from the neonatal mouse skin and identified a high level of α6 integrin expression and a low level of CD71 expression by Flow cytometry (Fig. 3A). A scratch was made in the confluent monolayer of P311−/− and P311+/+ EpSCs, and the cell migration across the scratch was measured after 24 and 48 h. As shown in Fig. 3B and C, the distance of EpSC migration was markedly decreased in the P311−/− group compared with the P311+/+group in in vitro wound models (Fig. 3B, C, 2.22-fold difference, P < 0.01, at 24 h; 1.62-fold difference, P < 0.01, at 48 h). As determined by counting the numbers of migrating cells, the number in the P311−/− group was significantly lower compared with that from the P311+/+ group (Fig. 3D, 208 vs. 608, P < 0.01, at 24 h; 594 vs. 1,038, P < 0.01, at 48 h). Thus, the above data suggest that P311 promotes the migration of EpSCs.
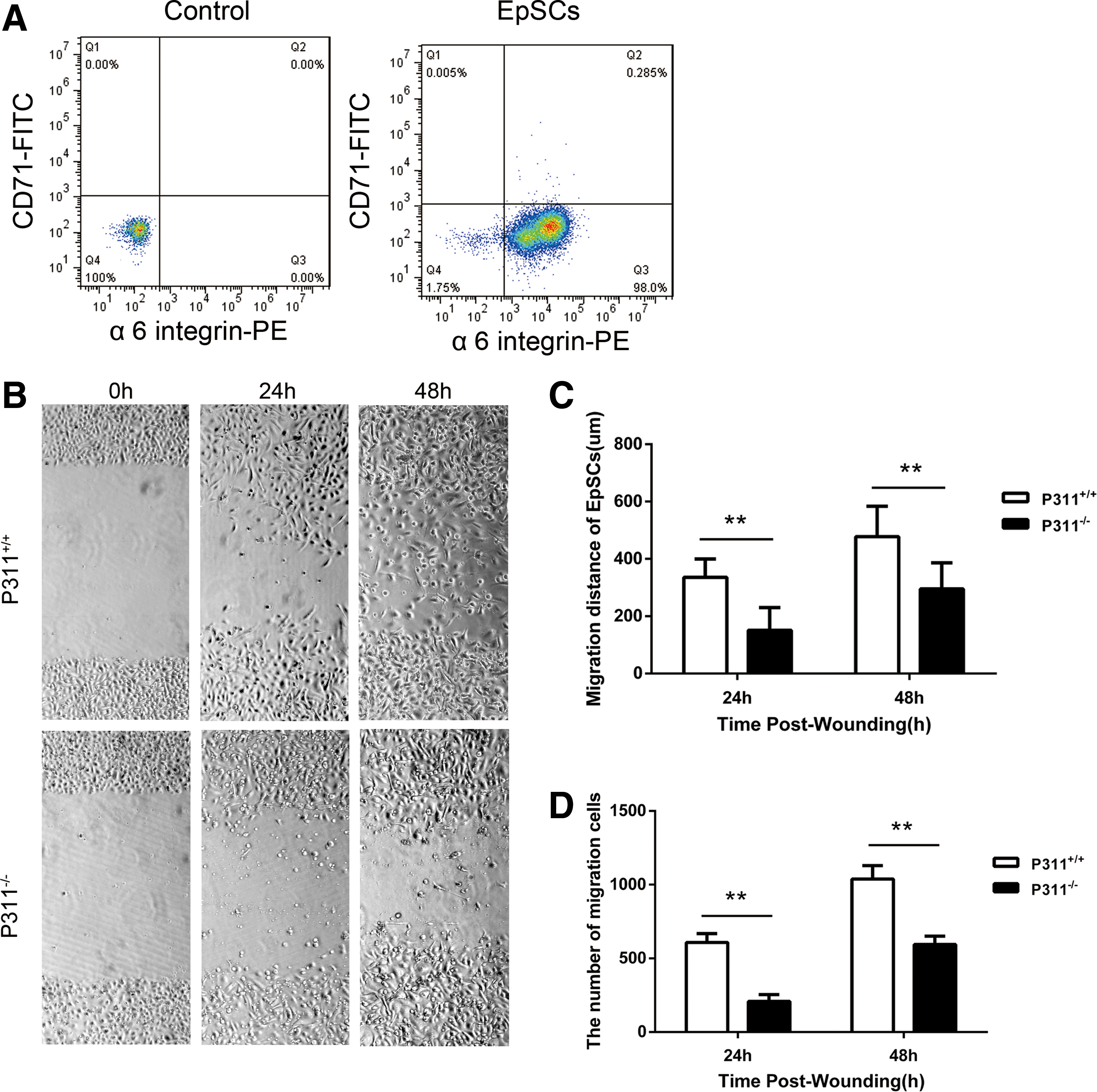
The P311−/− EpSCs showed decreased migration ability.
P311 may promote the EpSC migration through RhoA and Rac1 activation
To evaluate the mechanism responsible for the P311-induced enhancement of EpSC migration, we analyzed the activity of Rho GTPases (RhoA, Rac1, and Cdc42) by using a pull-down assay. As shown in Fig. 4, human EpSCs transfected with pADEasy-P311 showed significantly higher RhoA, Rac1, and Cdc42 activity, compared with that in the pADEasy-Myc control group (Fig. 4A, B, RhoA-GTP in pADEasy-Myc vs. pADEasy-P311, 2.68-fold difference, P < 0.01; Fig 4A, C, Rac1-GTP in pADEasy-Myc vs. pADEasy-P311, 2.61-fold difference, P < 0.01; Fig 4A, D, Cdc42-GTP in pADEasy-Myc vs. pADEasy-P311, 2.59-fold difference, P < 0.01), but the total levels of RhoA, Rac1, and Cdc42 were not significantly different (Fig. 4E–G). We further examined the RhoA, Rac1, and Cdc42 activity in P311−/− EpSCs and P311+/+ EpSCs. Expectedly, P311−/− EpSCs had decreased RhoA, Rac1, and Cdc42 activity (Fig. 5A, B, RhoA-GTP in P311−/− vs. P311+/+, 1.98-fold difference, P < 0.01; Fig. 5A, C, Rac1-GTP in P311−/− vs. P311+/+, 4.68-fold difference, P < 0.01; Fig. 5A, D, Cdc42-GTP in P311−/− vs. P311+/+, 3.60-fold difference, P < 0.01), but the total levels of RhoA, Rac1, and Cdc42 were not significantly different (Fig. 5E–G).
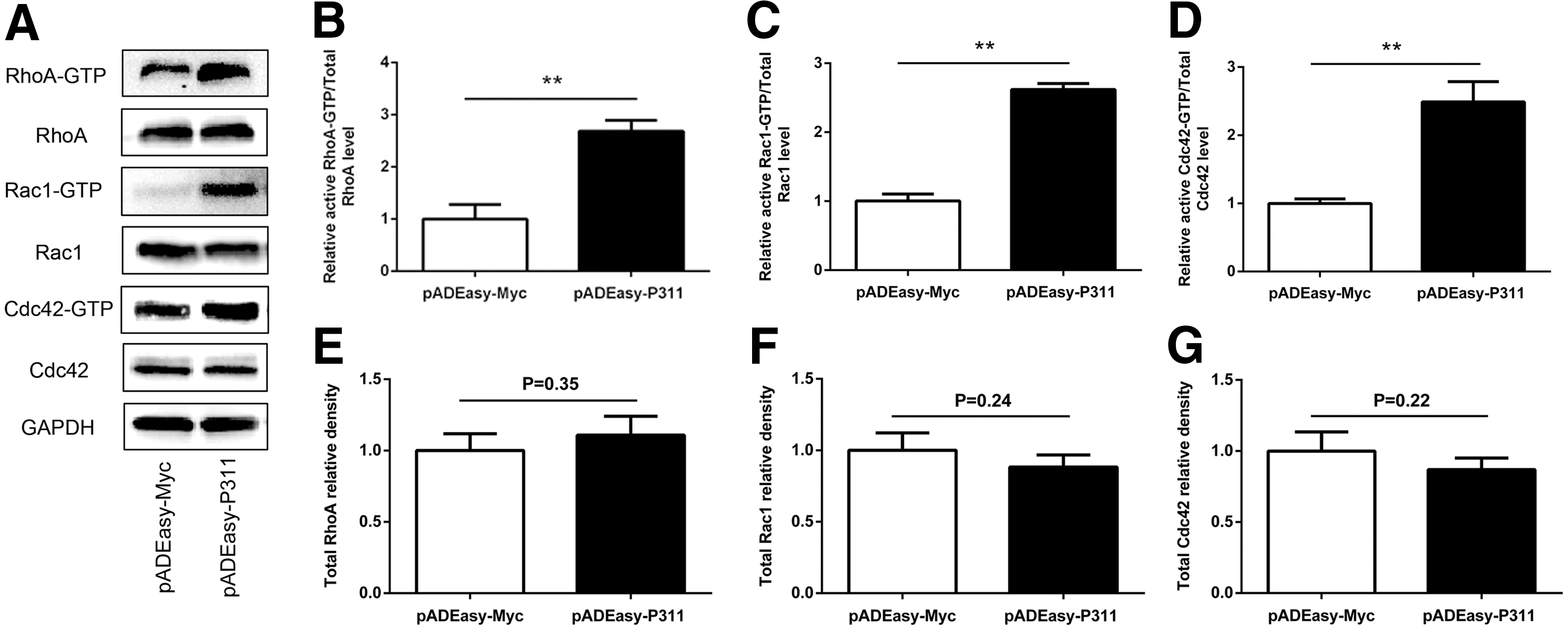
P311 over-expression increased the activities of Rho GTPases in human EpSCs.
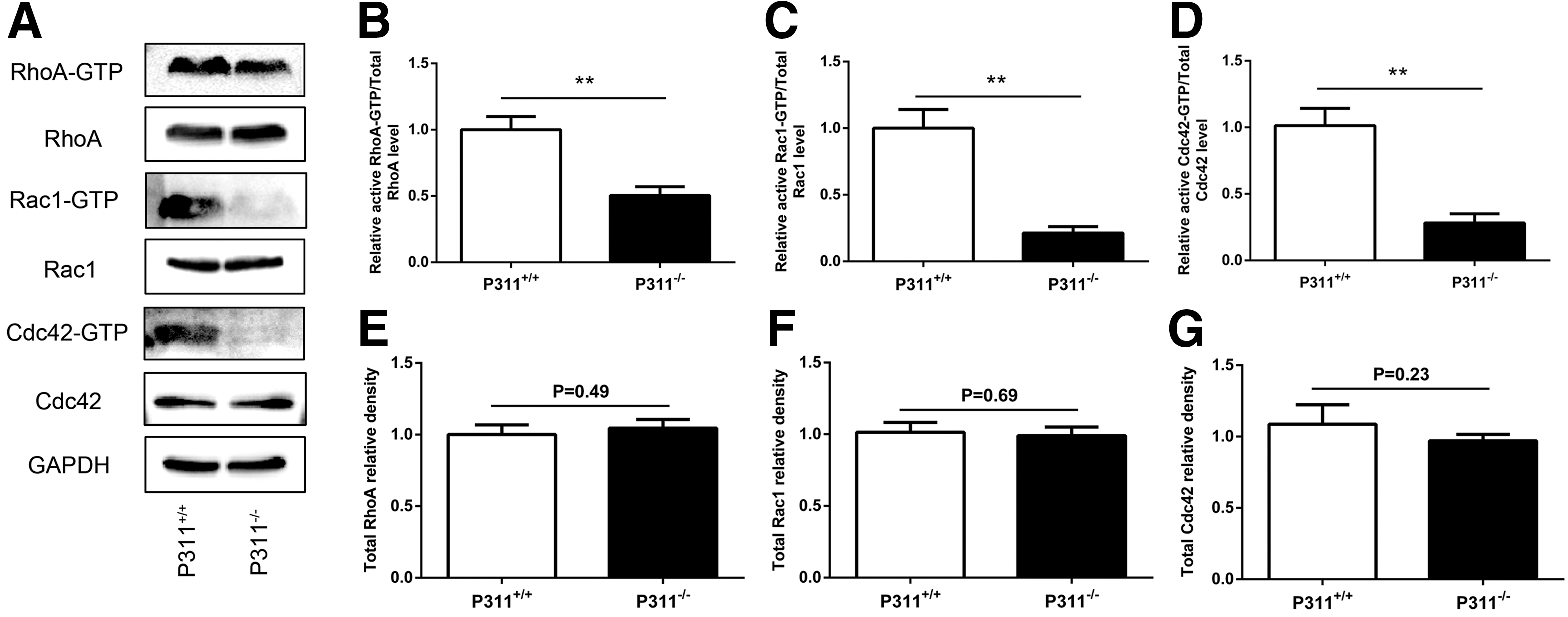
The P311−/− EpSCs exhibited decreased activities of Rho GTPases.
To determine whether the specific migratory responses induced by P311 were affected by the activity of Rho GTPases (RhoA, Rac1, and Cdc42), we further evaluated the effect of the Rho GTPase inhibitors on cell migration. Human EpSCs were transfected with pADEasy-P311 for 48 h, then scratched with a sterile pipette tip, and finally with or without the addition of the RhoA-specific inhibitor Rhosin or the Rac1 inhibitor Z62954982 or the Cdc42 inhibitor ZCL278 for 48 h. As shown in Fig. 6 and Supplementary Table S1, the P311-transfected group showed an increase in migration distance at 24 and 48 h, and Rhosin or Z62954982 significantly inhibited this effect (Fig. 6, pADEasy-P311 vs. pADEasy-P311 + Rhosin, 1.58-fold difference, P < 0.01, at 24 h; 1.46-fold difference, P < 0.01, at 48 h; pADEasy-P311 vs. pADEasy-P311 + Z62954982, 1.26-fold difference, P < 0.05, at 24 h; 1.28-fold difference, P < 0.01, at 48 h). However, no significant difference in migration was observed between the P311-transfected group and the ZCL27-treated group (Fig. 6). Altogether, these results suggest that P311 may promote the EpSC migration through RhoA and Rac1 activation, not Cdc42 activation.
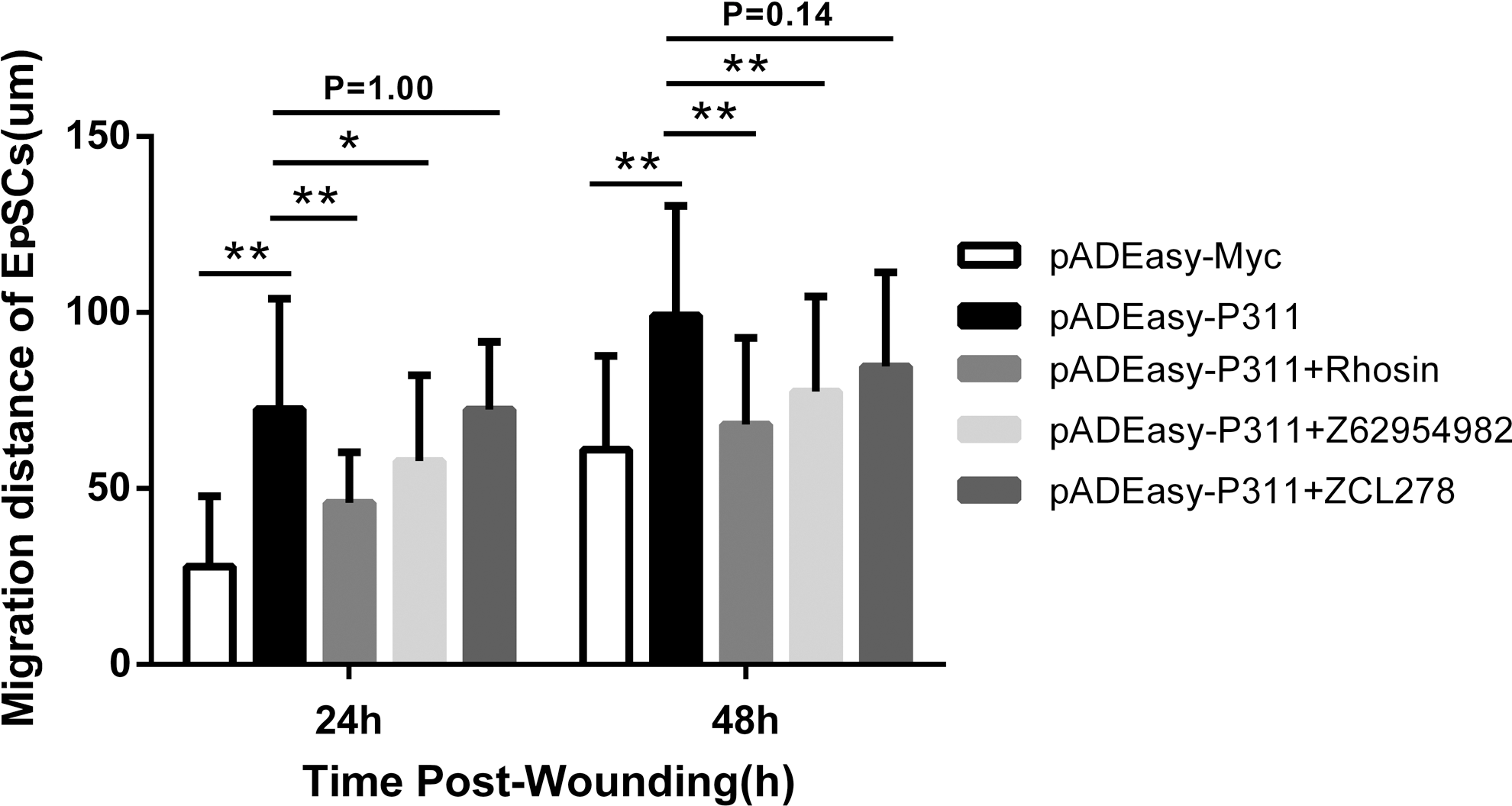
The Rho-specific inhibitor Rhosin and the Rac1 inhibitor Z62954982 impeded the cell migration induced by P311. Columns represent the distance of cell migration. Results are averaged from three independent experiments. Data are presented as the mean ± SD. It is representative of at least three similar experiments.
P311 deficiency impairs skin wound reepithelialization in vivo
Considering the high expression of P311 in the neo-epidermal cells of skin wound tissue samples from human and mice, we attempted to investigate the effect of P311 deficiency on wound repair. Full-thickness 3-mm-diameter cutaneous biopsy punch wounds were made on the mice dorsum, and then a biological membrane with adhesive dressings was used to efficiently prevent skin contraction. Because reepithelialization is a key step in wound healing, we tested the possible role of P311 deficiency in reepithelialization. The distance of reepithelialization (measured as the length of neo-epidermis) was histologically assessed under a microscope 0, 3, and 5 days after injury. The migration distance of neo-epidermis in P311+/+ mice was significantly greater than that in P311−/− mice on day 3 (Fig. 7A, B, 1.92-fold difference, P < 0.01) and on day 5 (Fig. 7A, B, 1.27-fold difference, P < 0.01, on day 5). Five days after injury, as shown in Fig. 7A, the wounds in the P311+/+ mice appeared to be completely epithelialized. In contrast, the wounds showed only partial epithelialization in P311−/− mice. These results indicate that P311 deficiency impairs skin reepithelialization.
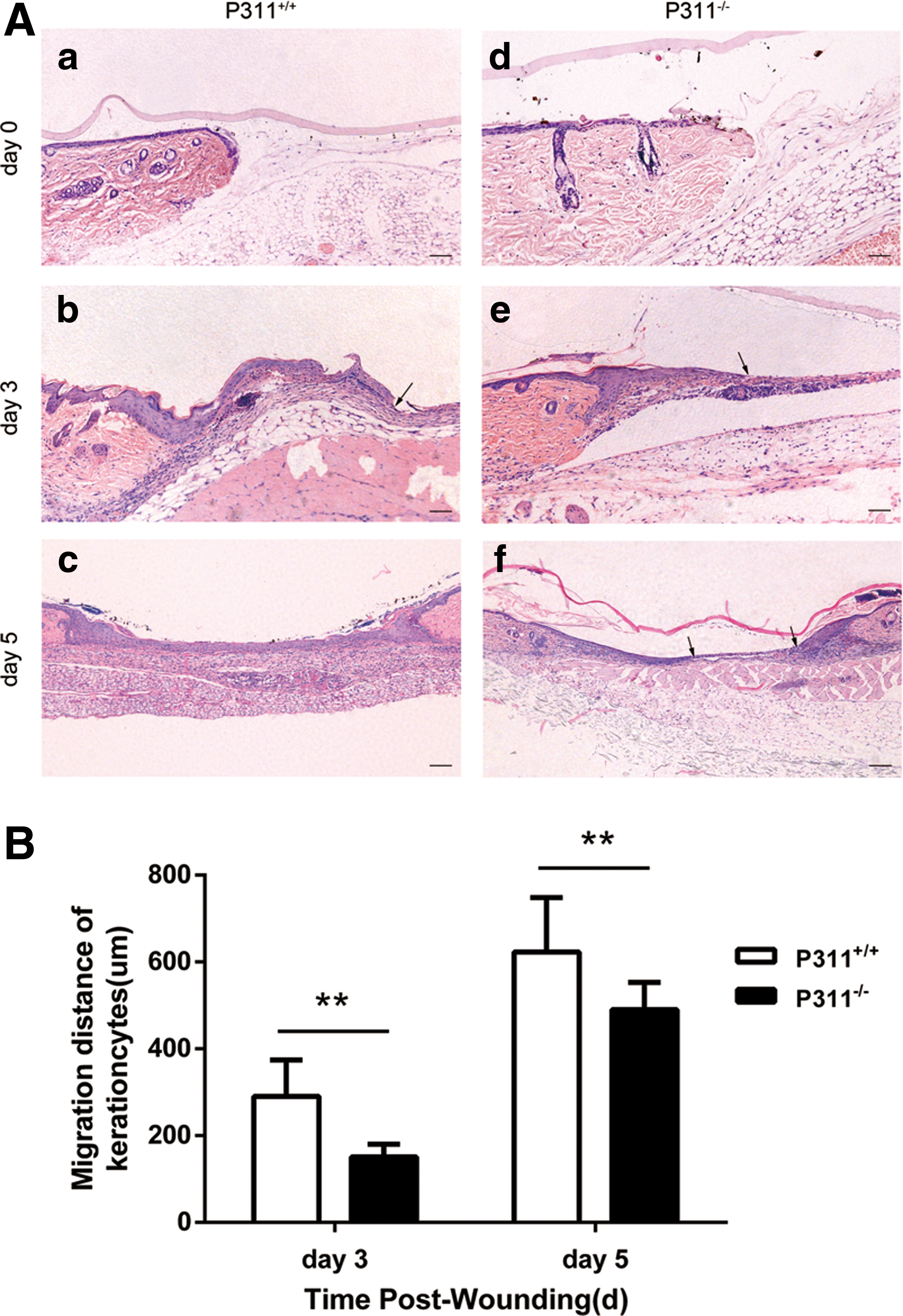
P311 deficiency impaired mouse skin reepithelialization.
Discussion
In this study, for the first time, we found that P311 could promote EpSC migration through Rho GTPases to enhance skin wound reepithelialization, which was supported by the following facts. (i) P311 was significantly increased in the cytoplasm of neo-epidermal cells in skin wound tissues from both humans and mice, and P311 was co-localized with BrdU-labeled LRCs in mouse burned wounds. (ii) P311 overexpression was able to promote EpSC migration and to activate Rho GTPases (RhoA, Rac1, and Cdc42). P311-deficient EpSCs had decreased cell migration and reduced activation of Rho GTPases (RhoA, Rac1, and Cdc42). (iii) Both the RhoA inhibitor and the Rac1 inhibitor, not the Cdc42 inhibitor, could suppress the P311-induced human EpSC migration. (iv) P311−/− mice showed impaired reepithelialization.
The skin epidermis provides the first line of defense against harmful elements and prevents the loss of essential body fluids. EpSCs reside either in the basal epidermis or in the bulge region of HFs, which is a niche of multipotent stem cells [29]. We successfully isolated human and mouse EpSCs and examined how forcing or knocking out the expression of P311 affected EpSC migration in vitro. Forcing expression of P311 in human EpSCs promoted EpSC migration, whereas knocking out P311 in mouse EpSCs decreased EpSC migration. Our previous study showed that HaCaT cells transfected with P311 also had increased cell motility. P311 overexpression enhanced glioma cell migration and induced ameboid-like migration in NIH3T3 cells [10,17]. P311 was central to the reactive oxygen species-mediated hepatic stellate cell migration induced by different chemokines [30]. These findings were consistent with our study, which indicated that P311 could enhance EpSC migration.
Rho GTPases are critical signal transducers for inducing the formation of lamellipodia, filopodia, invadopodia, and blebs during cell migration. Our study showed that P311 might increase RhoA and Rac1 activation to accelerate EpSC migration. This was supported by the following findings. Overexpression of P311 could induce RhoA, Rac1, and Cdc42 activation in human EpSCs and P311−/− mouse EpSCs decreased RhoA, Rac1, and Cdc42 activation. Furthermore, the Rho-specific inhibitor Rhosin and the Rac1 inhibitor Z62954982 could significantly inhibit the EpSC migration induced by P311, whereas the Cdc42 inhibitor ZCL278 exerted no obvious effects. However, different members of Rho GTPases might mediate the P311-induced migration of different cells. In the migration of glima cells, overexpression of P311 activated Rac1, but had no effect on Cdc42 activation [10]. In the ameboid-like migration of myofibroblasts, P311 could activate RalA, not the RhoA, Rac1, and Cdc42 [17]. Most Rho GTPases are activated by GEFs, which could induce exchange of GDP for GTP, and are inactivated by GTPase-activating proteins (GAPs), which could change the GTP to GDP. The common GEFs of Rac1 and RhoA include β-PIX, DOCK180, Trio, and Tiam1. The common GAPs of Rac1 and RhoA include srGAP1 and p190RhoGAP. Moreover, Mst3 could directly phosphorylate RhoA [31]. To our knowledge, there was no evidence of a direct interaction between P311 and Rho GTPases [10,17]. Therefore, it is likely that P311 might regulate the GEFs, GAPs, or Mst3 to activate RhoA and Rac1, and then promote the migration of EpSCs. Further study is still required to clarify this issue.
To explore the potential role of P311 in wound healing, we then introduced P311−/− mice that had normal skin architecture. We established a mouse excisional wound splinting model to exclude the influence of mouse skin wound contraction on wound closure. We found that P311−/− mice had impaired reepithelialization. In the epidermis, EpSCs generate daughter cells by asymmetric division mitosis and maintain homeostasis [32]. In wounds, EpSCs proliferate, migrate to the wound site, and repair the damaged epithelium. We used BrdU-labeled LRCs to trace EpSC migration in a mouse superficial second-degree burn model; these LRCs co-localized with the P311-positive region in neo-epidermal cells. These observations suggest that P311 might play a role in accelerating skin wound reepithelialization.
In our study, we confirmed that P311 was abundantly expressed in the cytoplasm of neo-epidermal cells in both humans and mouse skin wound tissue, whereas the normal human and mouse epidermis was negative for P311. P311 was detectable in the activated fibroblasts and myofibroblasts in human wounds during healing, but it disappeared after the wound healed [18]. P311 was upregulated in axotomized facial motor neurons, but few signals could be detected in the control side [8]. We previously observed that P311 was also highly expressed in a subset of tubular epithelial cells in both human and mouse renal fibrosis, whereas it was negative in normal human and mouse tubular epithelial cells [13]. The above studies together showed that P311 expression was little in cells from normal tissue and was obviously increased in cells from injured tissue, implying that P311 might be the microenvironment-dependent protein. Our unpublished data also showed that the P311 level in mouse EpSCs could be increased by the injury microenvironment, such as hypoxia, interleukine-1β, interleukine-6, and tumor necrosis factor α. More studies are needed to elucidate the underlying mechanism.
In summary, our work is the first to find that P311 could accelerate skin wound reepithelialization by promoting the migration of EpSCs through RhoA and Rac1 activation. P311 could serve as a novel target for promoting cutaneous wound healing.
Footnotes
Acknowledgments
The authors thank Gregory A. Taylor for providing P311−/− mice. This work was supported by grants from China's NSFC grants program (81471870, 30957768, 30973116, and 81171809) and the Key Project of Military Medical Plan (AWS11J012-05, BWS11J039).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.