
Abstract
Extensive research has been performed to determine the effect of freezing protocol and cryopreservation agents on the viability of adipose tissue-derived stromal/stem cells (ASCs) as well as other cells. Unfortunately, the conclusion one may draw after decades of research utilizing fundamentally similar cryopreservation techniques is that a barrier exists, which precludes full recovery. We hypothesize that agents capable of inducing a subset of heat shock proteins (HSPs) and chaperones will reduce the intrinsic barriers to the post-thaw recovery of ASCs. ASCs were exposed to 43°C for 1 h to upregulate HSPs, and the temporal HSP expression profile postheat shock was determined by performing quantitative polymerase chain reaction (PCR) and western blotting assays. The expression levels of HSP70 and HSP32 were found to be maximum at 3 h after the heat shock, whereas HSP90 and HSP27 remain unchanged. The heat shocked ASCs cryopreserved during maximal HSPs expression exhibited increased post-thaw viability than the nonheat shocked samples. Histochemical staining and quantitative reverse transcription-PCR indicated that the ASC differentiation potential was retained. Thus, suggesting that the upregulation of HSPs before a freezing insult is beneficial to ASCs and a potential alternative to the use of harmful cryoprotective agents.
Introduction
A
Cryopreservation of ASCs is deemed to be successful only if the cells retain their characteristic stem-like properties postfreeze–thaw. Several parameters influence the outcome of a cryopreservation process, including the choice of cryoprotectant (CPA) and the cooling rate [5]. A slow cooling rate of 1°C/min has shown to be optimal for ASCs, and the CPAs such as dimethyl sulfoxide (DMSO), methylcellulose, trehalose, and polyvinlpyrrolidone (PVP) have been reported in the literature to cryopreserve ASCs, bone marrow multipotent stromal cells (BMSCs), and other cells, with varying degrees of success [6 –10]. Currently, 10% DMSO in combination with animal serum is widely used for stem cell cryopreservation [11,12].
In clinical hematology, patients transplanted with hematopoietic progenitor and stem cells cryopreserved in DMSO experience adverse effects, including nausea, chills, hypotension, dyspnea, and cardiac arrhythmia, and in some extreme cases, cardiac arrest, transient heart blockages, neurological toxicity, renal failure, and respiratory arrest [13 –17]. DMSO is a methylating agent that can cause unwanted DNA methylations leading to genotypic changes in cells [18] and affects differentiation potential of stem cells [19]. Furthermore, the presence of animal serum can trigger the production and release of antibodies as the serum-exposed cells are infused into the patient; in addition, the serum can also increase the risk of transmitting microbial infections [20 –22].
Clinical studies involving transfusion of cryopreserved mesenchymal stem cells (MSCs) in human subjects indicate that cryopreservation preserves cell viability but does not guarantee cell functionality, that is, plasticity and their immunomodulatory properties [23 –25]. Specifically, it was found in a recent study that frozen–thawed human MSCs administered in rats displayed altered genomic profile in comparison with freshly isolated cells [26]. Therefore, the development of a cryopreservation protocol free of DMSO and serum that retains ASC post-thaw viability/plasticity/functionality has the potential to reduce patient discomfort and improve the applicability of ASCs in tissue engineering and regenerative medicine applications.
This study postulates that induction of heat shock protein (HSP) expression before cryopreservation will rescue or fortify the ASCs from the freezing damage without impacting their post-thaw functionality. HSPs are often described as the stress proteins that are induced in response to unfavorable conditions encountered by a cell. This phenomenon was first discovered in Drosophila melanogaster and is conserved throughout all living systems [27]. Conditions such as exposure to mildly high temperature (heat shock), heavy elements, toxins, oxidative stress, certain chemicals, and hypoxia trigger the upregulation of HSPs [27 –31]. The elevated HSPs reverse the damage caused by stressful conditions by regulating multiple physiological processes, including the apoptotic pathway, repair of denatured and misfolded proteins, and signal transduction [30,31]. Based on the available literature, it is evident that induction of HSPs, especially HSP90, HSP70, HSP32, and HSP27, offers cytoprotection against various stresses and diseases. Therefore, we were particularly interested in examining the induction of these HSPs with heat shock as a means to enhance the post-thaw cell viability and differentiation potential of cryopreserved ASCs.
Materials and Methods
Materials
All materials were obtained from Sigma-Aldrich (
ASC isolation and culture
Protocols were reviewed and approved by the Pennington Biomedical Research Center Institutional Research Board. Lipoaspirate samples were obtained anonymously from deidentified patients undergoing elective liposuction procedures in the clinics of local plastic surgeons with written informed consent. The lipoaspirate samples from subcutaneous adipose tissue sites were processed within 24 h of sample collection. The tissue was washed three to four times with prewarmed phosphate-buffered saline (PBS) to remove erythrocytes and white blood cells. Floating adipose tissue fragments are separated from unwanted erythrocytes by removal of the infranatant solution. The tissue was then suspended in an equivalent volume of PBS containing 1% bovine serum albumin and 0.1% collagenase type I (Worthington Biochemical Corporation,
Heat shock/thermal treatment
Cells at P2 were harvested using 0.25% trypsin–ethylenediaminetetraaceticacid (Invitrogen) digestion, counted using a hemocytometer, and a million cells were plated in T25 cm2 culture flasks. Once the plated cells adhered to the flasks (6–8 h), the flasks were transferred to a cell culture incubator that was set to 43°C with 5% CO2 and incubated for 1 h. After 1 h of heat shock, the flasks were transferred back to 37°C cell culture incubator and allowed to recover for 1, 3, 9, 18, or 48 h periods, respectively, before further experiments.
Freezing
The CPAs, 20% PVP (w/v), 2% DMSO (v/v), and 20% trehalose (w/v), were dissolved in DMEM thoroughly and filtered using 0.2 μm sterile syringe filter (VWR) and stored at 4°C until further use. The heat shocked ASCs after their respective recovery times were suspended in DMEM and diluted by drop wise addition of previously prepared CPA solutions in 1:1 ratio so that the final CPA concentration becomes 10% for PVP and trehalose and 1% for DMSO. The cryovials were then placed at 4°C for 10 min and transferred to control rate freezer (Planner), which was precooled to 4°C. Using a cooling rate of 1°C/min, the cryovials were cooled to −80°C followed by plunging in liquid nitrogen. The samples were stored in liquid nitrogen for 1 day before further processing. The cryovials were thawed at 37°C in a water bath by gently shaking for 1–2 min and adding the stromal medium drop by drop to dilute CPA. The samples were centrifuged at 300 g for 5 min to remove the CPA and replated for 24 h before measuring the cell viability.
Cell viability/apoptosis
The cell viability and apoptosis of post-thawed heat shocked ASCs were determined using annexin V and propidium iodide (PI)-based apoptosis detection kit (BD Biosciences). As a chemically induced apoptotic control, ASCs were incubated in fresh medium enriched with 40 mM etoposide (24 h), as described in earlier publications from our laboratory [6,9,10]. Similarly, for necrotic control, ASCs were incubated for 24 h in fresh medium with 5 mM hydrogen peroxide, as described earlier [6,9,10]. The no-treatment control consisted of ASCs treated in fresh medium, free from inducing agents. The post-thawed cells that were not adhered (dead) to the flasks and the adhered (healthy) cells were pooled together and washed in PBS and suspended in 1× binding solution supplied by the manufacturer. Approximately, 105 cells were stained using 5 μL of each dye, annexin V, and PI, for 15 min in dark and analyzed using a flow cytometer (BD Biosciences) within 1 h. The quadrants were set based on the live and dead control samples as described by Thirumala et al. [6,9,10]. In brief, apoptotic analyses for ASCs were performed on a FACS Calibur flow cytometer (BD Biosciences) utilizing 488-nm laser excitation and fluorescence emission at 530 nm (FL1) and >575 nm (FL3). Forward and side scatter measurements were made using linear amplification, and all fluorescence measurements were made with logarithmic amplification. A total of 20,000 cells per sample were acquired using Cell Quest software (BD Biosciences).
RNA isolation, reverse transcription, and quantitative polymerase chain reaction
RNA was isolated using the purelink RNA kit (Life Technologies) according to manufacturer's instructions. The quality and quantity of isolated RNA were measured using a nanodrop spectrophotometer. The first strand cDNA synthesis was done by using high-capacity cDNA synthesis kit (Applied Biosciences). For real-time quantitative polymerase chain reaction (qPCR), a SYBR green (Applied Biosystems) kit was used as per the manufacturer's instructions for the ABI QPCR machine. The primer sequences for HSPs 90, 70, 32, and 27 were designed using IDT primerquest and are as follows:
HSP90 Forward 5′-CACCACTCTACTCTGTCTCT-3′
Reverse 5′-GTTTCCTCAGGCATCAGTAG-3′
HSP70 Forward 5′-TTGCAGTGTGCCATCTTATC-3′
Reverse 5′-GAGAAAGGAGCAGCATGATT-3′
HSP32 Forward 5′-CCTCCCTGTACCACATCTAT-3′
Reverse 5′-AGCTCTTCTGGGAAGTAGAC-3′
HSP27 Forward 5′-GTCCAACGAGATCACCATCC-3′
Reverse 5′-CGGCAGTCTCATCGGATTT-3′.
The primer sequences for cyclophilin B (internal reference control), osteogenic, and adipogenic genes were described in Thirumala et al. [6]. After the qPCR, the fold change in expression was obtained by calculating 2−(ΔΔCt) [32].
Western blotting
ASCs were washed with PBS twice and lysed using radioimmunoprecipitation assay (Santa Cruz Biotechnology) buffer containing protease inhibitor cocktail (Santa Cruz Biotechnology), phenylmethanesulfonyl fluoride (Santa Cruz Biotechnology), and sodium orthovanadate (Santa Cruz Biotechnology) to extract the proteins. The obtained cell lysate was centrifuged at 8,000 g for 10 min in a refrigerated centrifuge. The supernatant was collected and protein quantitation was performed by BCA assay kit (Thermoscientific). The absorbance of the samples was measured at 535 nm and the amount of protein in the unknown test samples was determined based on a standard curve. The protein samples were then denatured by adding laemmli buffer and boiling at 95°C for 3–5 min. The denatured proteins at concentration of 25 μg per lane were then loaded on to 10% polyacrylamide gels and separated using electrophoresis. The electrotransfer of proteins from polyacrylamide gel to nitrocellulose membrane (Biorad) was carried out at 70 V for 2 h at 4°C. The transfer of proteins on to nitrocellulose membrane was confirmed by Ponceau S staining. The nitrocellulose membrane was washed with Tris buffer saline with tween20 buffer and blocked with 5% nonfat dry milk for 1 h. Next, the nitrocellulose membranes were incubated overnight at 4°C with primary antibodies (Santa Cruz Biotechnology) detecting HSP90, HSP70, HSP32, β-actin in dilutions of 1:200, and for HSP27 in dilution of 1:5,000. After primary antibody incubation, the membranes were washed 4–6 times with TBST buffer and incubated with a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) in dilution of 1:2,000 for 1 h and washed again 4–6 times to remove unbound antibodies. The chemiluminescence substrate (Thermoscientific) was added to membranes and the digital image was captured using a Typhoon digital image scanner (GE Healthcare Life Sciences).
ASC differentiation
Frozen–thawed heat shocked ASCs were plated in 12 well plates at a density of 1 × 104 cells/cm2 and cultured to at least 90% confluence. Adipogenesis was induced by adipogenic induction media consisting of DMEM/F-12 Ham's with 3% FBS, 33 μM biotin, 17 μM pantothenate, 1 μM bovine insulin, 1 μM dexamethasone, 0.5 mM isobutylmethylxanthine (IBMX), 5 μM rosiglitazone, and 100 U penicillin/100 μg streptomycin/0.25 μg fungizone. Three days after induction, media were changed to adipogenesis maintenance media (identical to the induction media, except for the omission of IBMX and rosiglitazone) and cells were fed three times per week [9]. After 14 days, the adipogenic differentiated cultures were fixed in 4% paraformaldehyde and stained using the Oil red O stain. Quantitation of adipogenesis was carried out by eluting the Oil O red stain by 100% isopropanol followed by measuring absorbance of the elution at 510 nm. The amount of Oil O red in the test samples was determined using a standard plot that was generated using known concentrations of Oil O red.
Osteogenic differentiation was carried out using stromal medium supplemented with 10 mM β-glycerophosphate, 50 μg/mL
Statistical analysis
All the values are indicated as mean and standard deviation. Two tailed student's t-test was employed, with P < 0.05 considered significant. The experiments were repeated three times from the ASCs derived from three separate donors (n = 3). For flow cytometry analysis, ∼20,000 cell events were gated and analyzed for each experiment conducted in triplicate.
Results
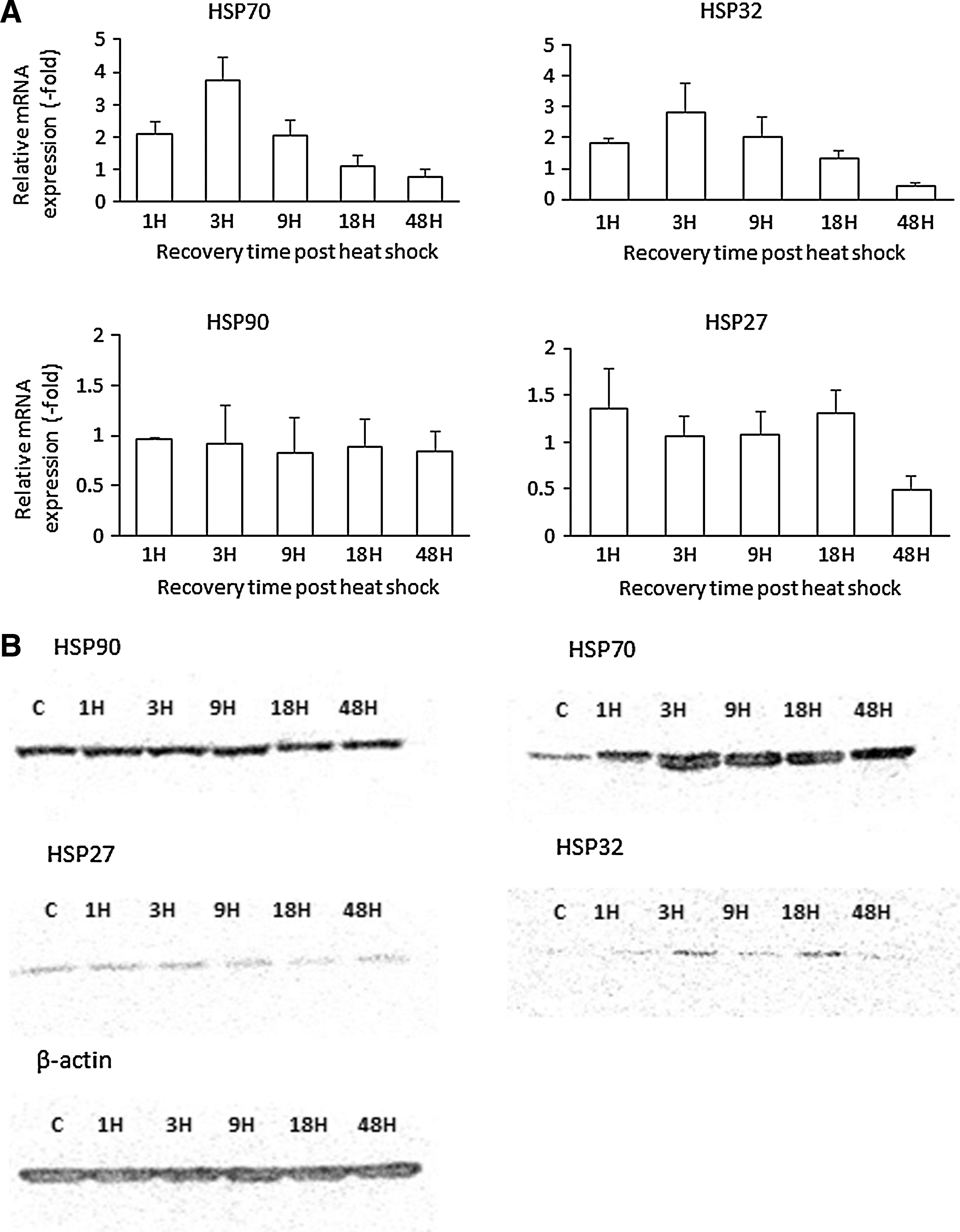
Effect of heat shock on expression of HSPs
To determine the optimal temperature to induce heat shock response, ASCs were exposed to 41°C for 1 h and the HSP70 expression was evaluated by western blot (see Supplementary Fig. S1; Supplementary Data are available online at
Effect of heat shock on cell viability prior cryopreservation
We found that the heat shock treatment at 43°C for 1 h did not hamper cell viability of ASCs. Representative fluorescent dot plots are shown in the Supplementary Data (Supplementary Fig. S3). Heat shocked cells displayed viability equivalent to the nonheat shock controls, indicating that the time and temperature selected had no detrimental effect on ASCs (Fig. 2A).
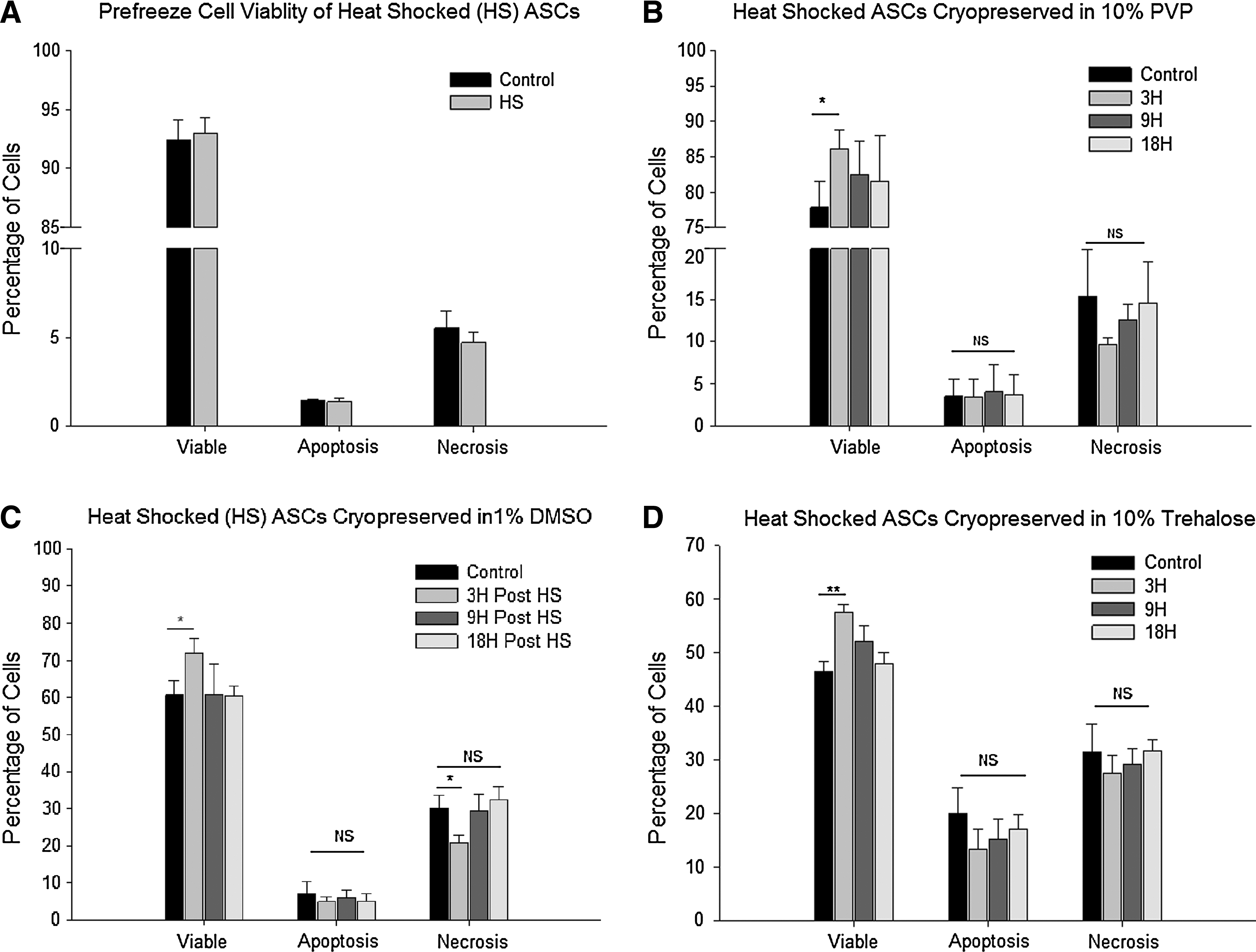
Effect of heat shock on cryopreservation
Heat shocked ASCs that were cryopreserved in the absence of CPAs displayed extremely poor post-thaw viability. Preliminary experiments conducted with heat shock and without CPAs resulted in a viability that was in the range of 32.2% ± 5.3%, whereas the nonheat shocked samples showed 34.5% ± 7.3%. This value was deemed to be unacceptably low and further experiments were carried out exclusively in the presence of CPAs to determine whether the heat shock offers any beneficial effect on cryopreservation outcome. Three different CPAs (10% PVP, 1% DMSO, and 10% trehalose) were used. Heat shocked ASCs cryopreserved at 3 h postheat shock (maximal HSP expression) showed higher viabilities (P < 0.05) than controls (no heat shock) or ASCs at 9 h or 18 h postheat shock (Fig. 2B–D). The increase in cell viabilities of heat shocked ASCs cryopreserved in 10% PVP, 1% DMSO, or 10% trehalose was 86.1% ± 2.7%, 72.0% ± 3.9%, and 57.4% ± 1.6%, respectively, as compared with those of nonheat shocked ASCs of 77.8% ± 3.8%, 60.6% ± 3.9%, or 46.5% ± 1.9% with the same cyroprotective agents (see Supplementary Table S1). Interestingly, no significant changes in the number of apoptotic cells between the samples were observed (Fig. 2B–D).
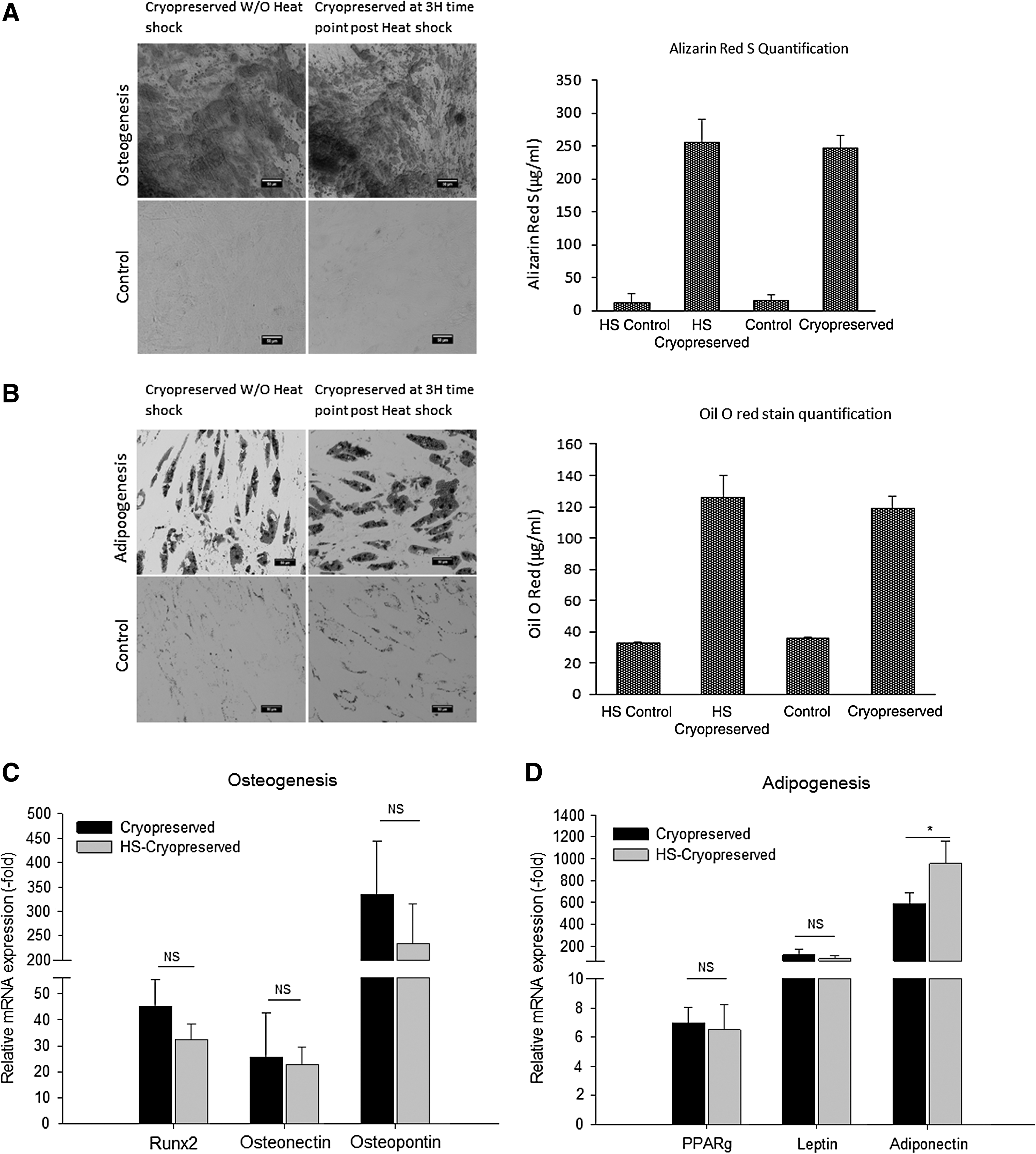
Differentiation potential of heat shocked cryopreserved ASCs
To determine the differential potential, heat shocked ASCs were cryopreserved in 10% PVP and induced to undergo osteogeneic or adipogenic differentiation. Histochemcial staining with Alizarin red S for osteogenesis (Fig. 3A) or Oil O red for adipogenesis (Fig. 3B) and qPCR analyses for the expression of osteogenic and adipogenic genes indicated that the heat shocked cryopreserved ASCs retained both osteogenic and adipogenic differentiation potential (Fig. 3C, D) relative to nonheat shocked cryopreserved controls (P > 0.05).
Discussion
In the pursuit of developing DMSO and serum-free cryopreservation protocol, our group has previously demonstrated PVP as an alternative CPA to DMSO for ASCs [6]. In this study, we explored the beneficial effect of inducing the cell's heat shock defense mechanisms on cryopreservation in the absence of serum and reduced DMSO concentrations. We found that heat shocked ASCs cryopreserved during the higher expression period of HSP70 and HSP32 (3 h after heat treatment) increased post-thaw cell viability by 10.6%, 18.8%, and 23.4% in the presence of CPAs 10% PVP, 1% DMSO, and 10% trehalose. Previously, Thirumala et al. cryopreserved ASCs with 10% PVP in 90% DMEM using an ethanol jacketed container in a −80°C freezer and obtained 69.7% viability [6]. Using the programmable control rate freezer and heat shocking ASCs before cryopreservation with 10% PVP in 90% DMEM, we were able to achieve maximum cell viability of up to 86.08%. In addition, we have reduced DMSO to 1/10th of the concentration used in traditional cryopreservation protocols and obtained up to 72.02% viability with the heat shock induction approach. Irrespective of the nature of CPA, whether it is intracellular (DMSO) or extracellular (PVP and trehalose), heat shocking ASCs resulted in improvement of post-thaw viability.
A study conducted on cryopreserved MSCs found that the frozen–thawed cells displayed increased susceptibility to complement mediated cell lysis and reduced therapeutic potential than the fresh cells [24]. Our results suggest that heat shock enhances post-thaw viability and recovery of cryopreserved ASCs and, therefore, it could be possible that heat shocked cryopreserved ASCs may be less susceptible to complement mediated lysis with an improved therapeutic potential than the nonheat shocked cryopreserved ASCs; however, further research is required to address whether heat shock enhances the immunomodulatory properties of ASCs. Nonetheless, a heat shock induction protocol may provide an alternative mechanical approach to optimizing ASC cryopreservation.
The most extensively studied HSPs are HSP90, HSP70, HSP32, and HSP27. These HSPs are reported to inhibit the apoptosis pathway, reduce oxidative stress, and repair of misfolded proteins. HSP90 negatively regulates the apoptosis pathway by interfering with the interaction between cytochrome c and the apaf-1-capase-9 apoptosome complex, thus preventing the activation of caspases [33]. HSP90 also enhances cell survival by binding to inositol hexakisphosphate kinase-2, an enzyme that mediates apoptosis, and inhibits its enzymatic activity [34]. Furthermore, overexpression of HSP90 in pigs' hearts reduces the ischemic reperfusion injury by enhancing nitric oxide through endothelial nitric oxide synthase [35]. HSP70 plays an instrumental role in the folding and assembly of newly synthesized non-native polypeptides, repair/renaturation/degradation of misfolded and aggregated proteins [36]. More importantly, upregulation of HSP70 reduces the apoptotic cell death by inhibiting the activation of cytosolic phospholipase A2 and other downstream caspase-dependent events, whereas downregulating HSP70 makes the cells more susceptible to apoptosis when exposed to apoptotic-inducing chemicals [37].
HSP32, also known as hemeoxygenase-1, is an early stress response gene, whose upregulation enhances cell survival against oxidative stress [38]. HSP32 acts as an antioxidant enzyme by degrading heme from hemeproteins to synthesize antioxidants [38]. Furthermore, HSP32 has been distinguished from other HSPs based on its cytoprotective effects against ischemic reperfusion injuries, acute renal failures, and gastrointestinal diseases [39 –42]. HSP27 is a small HSP that promotes the recovery process in cells subjected to stressful conditions by regulating several crucial cellular processes, which include proteasomal degradation of unwanted proteins and minimizing the aggregation of misfolded proteins [43 –45]. HSP70 and HSP32 are also noteworthy for their antiapoptotic functions and cytoprotective effects [30,31,36 –42]. Therefore, it could be possible that the mechanism by which these two HSPs improve post-thaw viability is by reducing the damage caused by oxidative stress and apoptosis; however, the number of apoptotic cells did not reach statistical significance between the experimental groups.
There was a significant difference in the number of necrotic cells (Fig. 2C), suggesting a possible role for these HSPs in the inhibition of cell necrotic pathways. It is feasible that other HSP family member proteins, outside the scope of this study, contribute to this mechanism. Nevertheless, the current data support the conclusion that HSP70 and HSP32 are responsible for the enhancement of cryopreservation. Further studies will be required to investigate the specific mechanism(s) by which HSP70 and HSP32 enhance cryopreservation.
The post-thaw functionality of heat shocked ASCs to differentiate along the osteogenic and adipogenic lineages was retained and no statistical difference was found relative to the nonheat shocked controls. This result is also in agreement with several findings that cryopreserved MSCs retain differentiation potential as reported in review [23]. Independent published studies of human bone marrow derived stem cells have reported enhancement of osteogenesis with a periodic heat shock treatment during differentiation [46]. The supplementation of extracellular HSP70 in the osteogenic medium promoted osteogenic potential of human MSCs [47]. Throughout the length of the differentiation process, these studies either supplemented HSP70 in the medium or performed periodic heat shock to maintain high HSP70 expression levels. In contrast, in our study, we heat shocked the ASCs only once before cryopreservation and the derived HSP temporal profile indicates that by 48 h, HSP mRNA expression had returned to basal level. Thus suggesting that the initial expression of HSP was sufficient to confer cryoprotection to ASCs.
Overall, our results indicate that heat shocking ASCs and cryopreserving at the point of maximal HSP expression using 10% PVP in a control rate freezer at a cooling rate of −1°C/min will enhance the post-thaw viability without impacting their differential potential even in the absence of serum and DMSO. Therefore, this protocol can be used to replace the conventional cryopreservation protocol that routinely employs 10% DMSO and high concentration of animal serum to cryopreserve ASCs for in vitro and in vivo tissue engineering applications.
Conclusion
Exposing ASCs to 43°C for 1 h is innocuous and upregulates the expression of HSP70 and HSP32; however, the HSP90 and HSP27 expression levels are unchanged. The peak expression levels of HSP70 and HSP32 are found at 3 h post the heat shock, and cryopreservation of ASCs during this time point improves the post-thaw viability and retains stemness regardless of nature of CPA. Further studies need to be done to conclusively determine whether HSP70 and HSP32 are responsible for the observed cytoprotective effect. Nevertheless, this protocol could replace traditional cryopreservation protocols that require the presence of DMSO and animal serum to maintain ASCs for in vitro and in vivo clinical applications.
Footnotes
Acknowledgments
Research reported in this publication was supported by the National Institute Diabetes, Digestive and Kidney Diseases of the National Institutes of Health under award R21 DK 91852. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank Drs. Clubb and Wade at the Pennington Biomedical Research Center (PBRC) for supplying the liposuction aspirates, their many patients for consenting to participate in this protocol, and M. Dietrich, of the LSU School of Veterinary Medicine Flow Cytometry Core Facility, for her technical assistance.
Author Disclosure Statement
During the study period, Dr. J.G. was employed at LaCell LLC, a for-profit biotech company cofounded and coowned by Dr. J.G. and Dr. Wu. The remaining coauthors have no conflicts to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.