
Abstract
Unraveling transcriptional heterogeneity and the labyrinthine nature of neurodevelopment can probe insights into neuropsychiatric disorders. It is noteworthy that adult neurogenesis is restricted to the subventricular and subgranular zones of the brain. Recent studies suggest long non-coding RNAs (lncRNAs) as an avant-garde class of regulators implicated in neurodevelopment. But, paucity exists in the knowledge regarding lncRNAs in neurogenesis and their associations with neurodevelopmental defects. To address this, we extensively reviewed the existing literature databases as well as performed relevant in-silico analysis. We utilized Allen Brain Atlas (ABA) differential search module and generated a catalogue of ∼30,000 transcripts specific to the neurogenic zones, including coding and non-coding transcripts. To explore the existing lncRNAs reported in neurogenesis, we performed extensive literature mining and identified 392 lncRNAs. These degenerate lncRNAs were mapped onto the ABA transcript list leading to detection of 20 lncRNAs specific to neurogenic zones (Dentate gyrus/Lateral ventricle), among which 10 showed associations to several neurodevelopmental disorders following in-silico mapping onto brain disease databases like Simons Foundation Autism Research Initiative, AutDB, and lncRNADisease. Notably, using ABA correlation module, we could establish lncRNA-to-mRNA coexpression networks for the above 10 candidate lncRNAs. Finally, pathway prediction revealed physical, biochemical, or regulatory interactions for nine lncRNAs. In addition, ABA differential search also revealed 54 novel significant lncRNAs from the null set (∼30,000). Conclusively, this review represents an updated catalogue of lncRNAs in neurogenesis and neurological diseases, and overviews the field of OMICs-based data analysis for understanding lncRNome-based regulation in neurodevelopment.
Introduction
N
It has been 55 years since the landmark paper of Jacob and Monod's, titled “Genetic Regulatory Mechanisms in the Synthesis of Proteins” was floated and today, we are aware of regulatory RNAs of all styles and sizes [1]. We now know that the complexity of different species is not only governed by the sheer number of classic protein coding genes and the coding transcriptional diversity, but also by newer types of regulators/players known as non-coding RNAs. Chromatin marks and the birth of high-throughput technologies like tiling microarray and next-generation sequencing further rendered a clearer picture [2 –4].
While the transitional role of RNA in cellular function and biological processes had long been recognized, near recently it has received singular attention from the scientific peers. Several genome-wide analyses have revealed that 66% of the genome is actively transcribed into non-coding RNAs, whereas less than 2% of these sequences encode proteins [5]. Initial studies from the ENCODE (ENCyclopedia Of DNA Elements) project discovered a large group of small non-coding RNA molecules like miRNAs, siRNAs (∼22 nucleotides in length), as well as a few long non-coding RNAs (lncRNAs) those play important roles in gene silencing and posttranscriptional gene regulation [6,7].
Non-coding RNAs have been notably involved in many biological processes. From stem cell proliferation to neuronal differentiation and maturation, ncRNAs like lncRNAs regulate diverse downstream targets, including coding genes, transcriptional factors, and smaller non-coding RNAs [8]. They are also known to be highly tissue-specific and expressed nonuniformly in the mammalian brain [9]. The lower expression of lncRNAs compared with coding transcripts poses a hindrance in their in-vitro and in-silico exploitation and characterization studies. Interestingly, there is a scarcity in the number of lncRNAs annotated in the neurodevelopmental processes. While several reviews have elaborated gene regulatory roles of non-coding RNAs, a more critical analysis on their detection, identification methods, and regulatory mechanisms is lacking across various cell types [10,11].
Similar to inner cell mass of the blastocysts, many stem cells like NSCs also exhibit mosaic aneuploidy, which is evident from processes like lncRNA-mediated transcriptional heterogeneity [12,13]. Insights into the non-coding RNome show several lncRNAs, for example, RMST, which interacts with the transcription factor SOX2, and directly regulates neuronal fate commitment [14]. Additionally, a newly discovered lncRNA Pnky regulates the transition from NSCs to NPCs [15]. In this review, we have chosen a literature mining approach to catalog existing lncRNAs followed by an in-depth analysis of expression profiles to understand their role in neurodevelopment. To further elucidate their association to neuropsychiatric disorders, we have mapped the altered lncRNAs onto web-based brain repositories. Conceivably, this review represents an updated catalogue of lncRNAs, their probable downstream targets, and a novel approach to utilize high-throughput data for understanding lncRNA-based regulation in neural development.
Length (Sequence), Breadth (Function), and Depth (Expression) of Long Non-coding RNAs
LncRNAs are a diverse class of RNA molecules with a length of >200 nucleotides that possess no protein coding potential. These lncRNAs are transcribed, polyadenylated and/or have lesser than 100 amino acid open reading frames. The latest classification of non-coding RNAs based on nucleotide length is grouped into three categories: <50 nucleotides, 50–500 nucleotides, and >500 nucleotides [16]. According to this classification, the specific length classifier for lncRNAs is still obscure.
One of the pioneer studies on long ncRNAs was the ENCODE project, which contained minimal information on the lncRNAs when first released in 2003. Following regular updates, it has identified a list of 15,877 lncRNA genes that accounts for 26,414 lncRNA transcripts (GENCODE V7) [17]. Other big data projects like the FANTOM catalogued over 30,000 putative lncRNA transcripts [18]. More recently, Iyer et al. extensively analyzed 7,256 RNA-Seq libraries from various origins (tumor and normal tissue samples, and cell lines) to yield a total of 58,648 expressed lncRNAs and thus, characterized a genome-wide lncRNA landscape. Notably, 79% of these were previously unannotated, thus substantially increasing the total lncRNA population [19]. Currently, the NONCODE database v4.0 stores 167,150 human lncRNA transcripts, of which only 90,062 are annotated [20].
LncRNAs also display less sequence conservation, unlike the highly conserved shorter ncRNAs [21]. However, few exceptions have been identified, including Alu repeats, LINEs, SINEs, which represent the non-coding, ultraconserved lengthy regions of the genome [22]. Another report by Pollard et al. identified human accelerated regions (HAR) having an accelerated rate of nucleotide substitution between human and chimpanzee. Interestingly, almost 96% of HARs were localized to non-coding regions of the genome [23,24].
Unlike the lncRNA transcripts, their promoters are seen to be as conserved as the promoters of many coding RNAs, suggesting a similar selective pressure on them [17,25]. This explains why lncRNAs tend to be functional although having weak coding potentials. However, it is also intriguing that these lncRNA transcripts are rapidly evolving with lesser conservation than protein-coding transcripts. Therefore, understanding the conservation pattern of lncRNAs can be one of the guidance cues to sketch a better lncRNome.
LncRNAs can be found in five genome-specific locations and are named intronic, sense, antisense, bidirectional, and long intergenic. Sense lncRNAs overlap with a transcript's one or more exons on the same strand, whereas antisense lncRNAs overlap with a transcript's one or more exons on the complementary strand. Long intergenic lncRNAs (lincRNAs) are transcribed without any overlapping protein coding genes and are usually located more than 1 kbp away from the coding gene. Intronic lncRNAs originate from an intron and can overlap a messenger RNA in either sense or antisense orientation. Bidirectional lncRNAs are transcribed in an inverted orientation with an mRNA that is less than 1 kbp away and may share its promoter. Currently, there are 593 sense intronic lncRNAs annotated in the human genome, whereas in adult mouse brain 182 are found to be expressed [17,26]. Intronic and intergenic lncRNAs highly outnumber the other types of lncRNAs but, organism-oriented variation exists as in case of mammals and plants. While lincRNAs have shown an abnormal increase in expression in case of both cancerous and normal conditions, a similar skew in expression was seen in case of intronic lncRNAs present in some plant species under abiotic stress [27,28].
Role of lncRNAs in mammalian epigenetics was unraveled during the early 1990s with the discovery of Xist [29,30]. Corroborating studies on lncRNAs have revealed their roles in X-chromosome inactivation and genomic imprinting, including antisense lncRNAs like Tsix, which interacts with chromatin-modifying complexes and affect the neural epigenome [31]. Tsix has been shown to act in combination with Xist to enforce epigenetic modifications through direct association with PRC2 protein complex during embryonic development [32]. Such lncRNAs can act as cellular signals for transient epigenetic states. Currently, it is well accepted that the process of neural development involves extensive cross talks between distinct epigenetic mechanisms like histone modifications and non-coding RNA partners [33].
Although it has been known for a while that lncRNAs regulate genes at the posttranscriptional level, work by Lai et al. identified ncRNA-activating (ncRNA-a) as a new class of lncRNAs that binds to a coactivating complex, Mediator, causing the DNA to coil, thereby modulating gene expression [34]. By definition, ncRNA-a is a type of enhancer lncRNA that activates its neighboring genes using a cis-mediated regulatory mechanism. LncRNAs known to affect genes posttranscriptionally include MALAT1, which is enriched in the neurons and is known to regulate synapse formation by modulating the stage-specific gene expression [35,36]. Recent reports have identified some nuclear lncRNAs acting as “molecular signature codes,” which allow protein complexes and genes to be transported to appropriate locations and subject them to proper activation/deactivation [37]. SINEUPs are another set of modular antisense lncRNAs, which contain an embedded inverted SINEB2 sequence and a second overlapping region for increasing translation in cells and for targeting its sense mRNA, respectively [38]. Although the tertiary structure of lncRNAs allows for recognition that does not involve direct base-pair interactions, their length helps them fold into complex structures with surfaces restricted to protein binding, thus retaining the ability to recognize nucleotide sequences by base-pair interactions.
In this direction, the most recent conceptual framework proposed by Rinn et al. explains how lncRNAs might function analogous to the game “cat's cradle” wherein, the shape of a string is repeatedly changed by opening/closing up new sites for finger placement [39]. In the beginning of the cat's cradle game, the string is folded in a specific structure, which is acted upon by a second force (player) who firmly grips the string at specific locations and pulls the string outward to form a new conformation. In cells, the string is analogous to DNA and the second person implies lncRNAs, which marks spots on the DNA for specific proteins to bind and induce several changes. This analogous model suggests that lncRNA transcription can impose a gripping force on nuclear proteins to generate new 3D epigenomic conformations [39]. Precisely, lncRNAs can determine the switching between active and inactive states of the chromosomes [39]. While several studies have suggested the indispensability of such non-coding RNAs in regulation of overall cell and molecular circuitry, conducting reproducible in-vivo/in-vitro studies on them is utmost essential to elucidate their unexplored mechanisms in neurodevelopment.
LncRNA-Based Regulation in Brain Development
Neurogenesis refers to the phrase “origin of nerves.” In humans, two areas of the brain are responsible for neurogenesis, namely the SGZ within the hippocampal DG and the SVZ lining the LV [40]. Neuroblasts are formed in LV region and later migrate through the rostral migration stream to the olfactory bulb gradually decreasing the LV NSC population with age [41,42]. However, the extinction of this neural progenitor population is debatable, as several supportive studies have considered LV as one of the primary residences of neural precursor cells [43,44]. To understand the process of neurogenesis in vitro, human embryonic stem cells are being used as cellular models. Under optimal neurogenic microenvironments, hESCs can generate three major central nervous system lineages: oligodendrocytes, astrocytes, and neuron types [45].
Recent evidences suggest that lncRNAs are a promising class of gene regulators involved in neural cell fate determination, but only few candidates have been characterized so far [46,47]. It has been reported that OTX2 and Nkx2.2-AS play important roles in hESC-derived differentiation of oligodendrocytes and neurons, respectively [48]. In addition to these, Dlx1-AS is selectively required for the SVZ neuronal differentiation, whereas Six3-OS plays a role in both neuronal and oligodendrocyte differentiation [49]. A recently discovered lncRNA linc-ROR has been shown to act as a ceRNA miRNA “sponge” (competing endogenous RNA), regulating all the three essential embryonic stem cell renewal transcription factors Oct4, Nanog, and SOX2 through a feedback loop [50].
In recent past, transcriptomic profiling has revealed interesting lncRNA patterns in embryonic mice brains [51]. This study has concluded that lncRNA-based alternative splicing can act as switch for embryonic stem cell-derived neural cell fate specifications. In the developing neocortex during the gestation period (7–19 weeks) HAR1, which is part of a novel lncRNA gene (HAR1F) is pronouncedly expressed in Cajal–Retzius neurons [24]. Coherent studies on lncRNA-based brain regulatory mechanisms are still in their infancy; one of the reasons being the limited availability of diseased/normal human brain tissue samples. Keeping everything in view, we have tried to understand the scope of all existing lncRNAs in adult neurogenesis and their direct/indirect relevance to neurological impairments.
Mining strategy
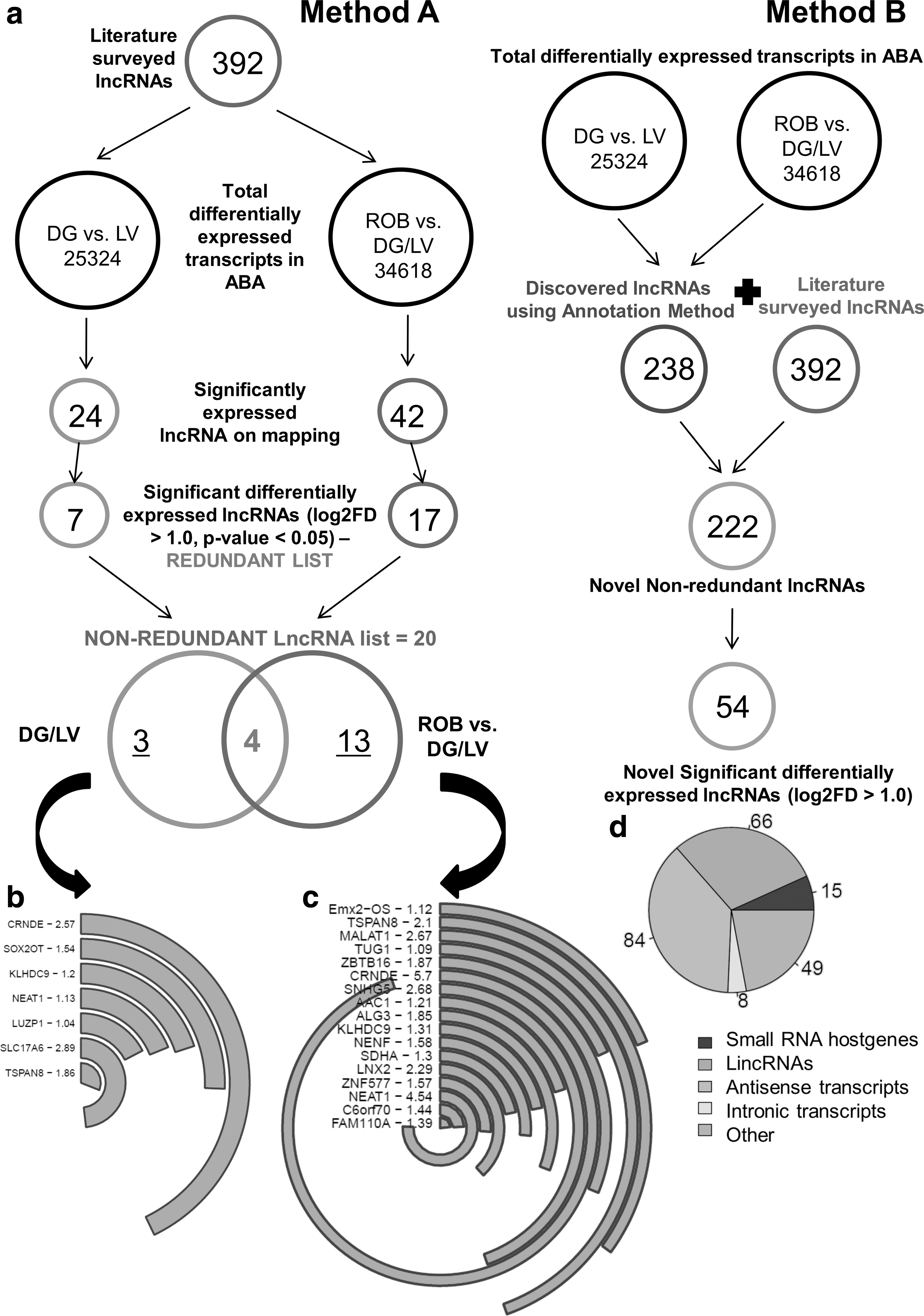
In-depth data mining exercises were conducted using up-to-date e-databases, including PubMed, Google Scholar, Medline, and HighWire Press. The literature survey covered consolidated reviews, original articles of high-throughput nature, and short reports. Publications written in English were solely considered for this search. Specific keywords/phrases used were “lncRNAs OR long non-coding RNAs in neurogenesis,” “long non-coding RNAs OR lncRNAs in neurodevelopment,” “lncRNAs in neurodegeneration,” and “lncRNAs in neurogenesis and disease.” This survey was stopped on 30 May 2016. Finally, we could generate a list containing 392 lncRNAs associated to neurodevelopment and/or neurodegeneration (Fig. 1a, and Supplementary Table S1; Supplementary Data are available online at
Methods employed in the differential expression analysis of neurogenic zones
Many study groups have consolidated information on lncRNAs, among which the most prominent databases of relevance are NONCODE, lncRNADb, Allen Brain Atlas, lncRNADisease, LNCipedia, and ncFANs [20,52 –55]. Since we wanted to specifically evaluate the expression of these lncRNAs in two neurogenesis zones of the human brain (DG, LV), we used the extensively curated brain database, Allen Brain Atlas (ABA).
The ABA database contains microarray expression data of all regions and subregions of the human and mouse brains [56,57] and thus, using ABA human brain containing microarray data, we conducted two main “differential” expression searches (Fig. 1a). The first differential search, including rest of the brain (ROB) regions versus exclusive regions, DG and LV, was performed to analyze all transcripts significantly upregulated in the neurogenic regions (Fig. 1b). To solely validate this search and reassure minimal expression of genes in ROB regions, a search involving the exclusive DG and LV versus ROB regions was performed. This search was not considered for further analyses. The next search was conducted between the DG and LV, to look for transcripts that are highly expressed in the hippocampal formation region (HiF), region of SGZ since it is the main hub for newborn neurons and memory formation (Fig. 1c). This kind of search is crucial to understand the significant transcriptional regulatory changes between different regions of the brain.
Thus, we adopted two different methods to ensure maximum data utilization and minimal loss of information. These two Methods A and B are overlapping in a way because even in Method B, we used the literature surveyed list and the ABA generated neurogenic niche expression list of genes, but applied to get novel non-coding genes/lncRNAs, which have not been specifically reported in prior art. In Method A, in-silico mapping of the 392 lncRNAs (degenerate list from data mining) onto above ABA differential expression sets was performed to ascertain their expression and localization specifically in the neurogenesis zones.
All lncRNAs were evaluated for their protein-coding potential based on their PhyloCSF score [58] using LNCipedia database. Stringent PhyloCSF scores lower than 43 were considered to be non-coding. To determine the conservation levels of the final list of lncRNAs, we used the UCSC Genome Browser's Conservation track and extracted their individual PhastCons and PhyloP scores by feeding the genomic coordinates. The PhastCons score is the probability that each nucleotide is conserved with values spanning 0–1, whereas abs (phyloP) is the -log (P-value) under a null hypothesis of neutral evolution, wherein a negative sign indicates faster than expected evolution, and a positive sign implies conservation [59,60]. A high PhastCons score is usually corresponded by a positive PhyloP value, but the correlation is not very strong. In the alternative Method B, we directly filtered the lncRNAs from the ABA transcript null set containing miRNAs, lncRNAs, as well as coding RNAs on the basis of name/annotation of the lncRNAs and their location-specific expression (Fig. 1a). This led to the discovery of a novel lncRNA pool of 222 candidates, including antisense, intergenic, small RNA host genes, and other types of lncRNAs, specific to DG/LV (Fig. 1d). All expression analyses were statistically significant (P < 0.01) and graphs have been made using RStudio (Integrated Development for R).
Data extraction and selection
Using Method A, we found 42 lncRNAs upregulated in DG and LV through the first differential search, whereas DG alone showed 24 lncRNAs (second differential search) (Fig. 1a). Finally, we could get a putative, nonredundant list of 20 lncRNAs, which were significantly upregulated (log2FD >1.0) and thus, are specific to neurogenesis (Fig. 1a, and Supplementary Table S1). This list includes lncRNAs and/or non-coding partners of coding genes like CRNDE, SLC17A6, TSPAN8, SNHG5, LNX2, MALAT1, and NEAT1 [35,61 –63] (Fig. 1b). To empathize, CRNDE and SOX2-OT [64] were the lncRNAs with highest expression in both neurogenesis zones whereas, MALAT1, NEAT1, SNHG5, and CRNDE showed higher expression specifically in DG region (Fig. 1b, c). All the noted lncRNAs were found to have protein-coding potential <43 (Supplementary Table S1).
Using Method B, we were able to find 222 novel lncRNAs specific to the neurogenesis zones. Out of these 222, 54 lncRNAs were noted to be significantly upregulated in the neurogenic zones, but further analysis needs to be done for more conclusive data (Fig. 1a, and Supplementary Table S1). These novel lncRNAs can also prove to be potential biomarkers for NSCs and other neurogenic niche cells as these are significantly expressed in the neurogenesis zones. A composition chart suggesting the type of novel lncRNAs discovered showed the majority population to be that of antisense transcripts (Fig. 1d).
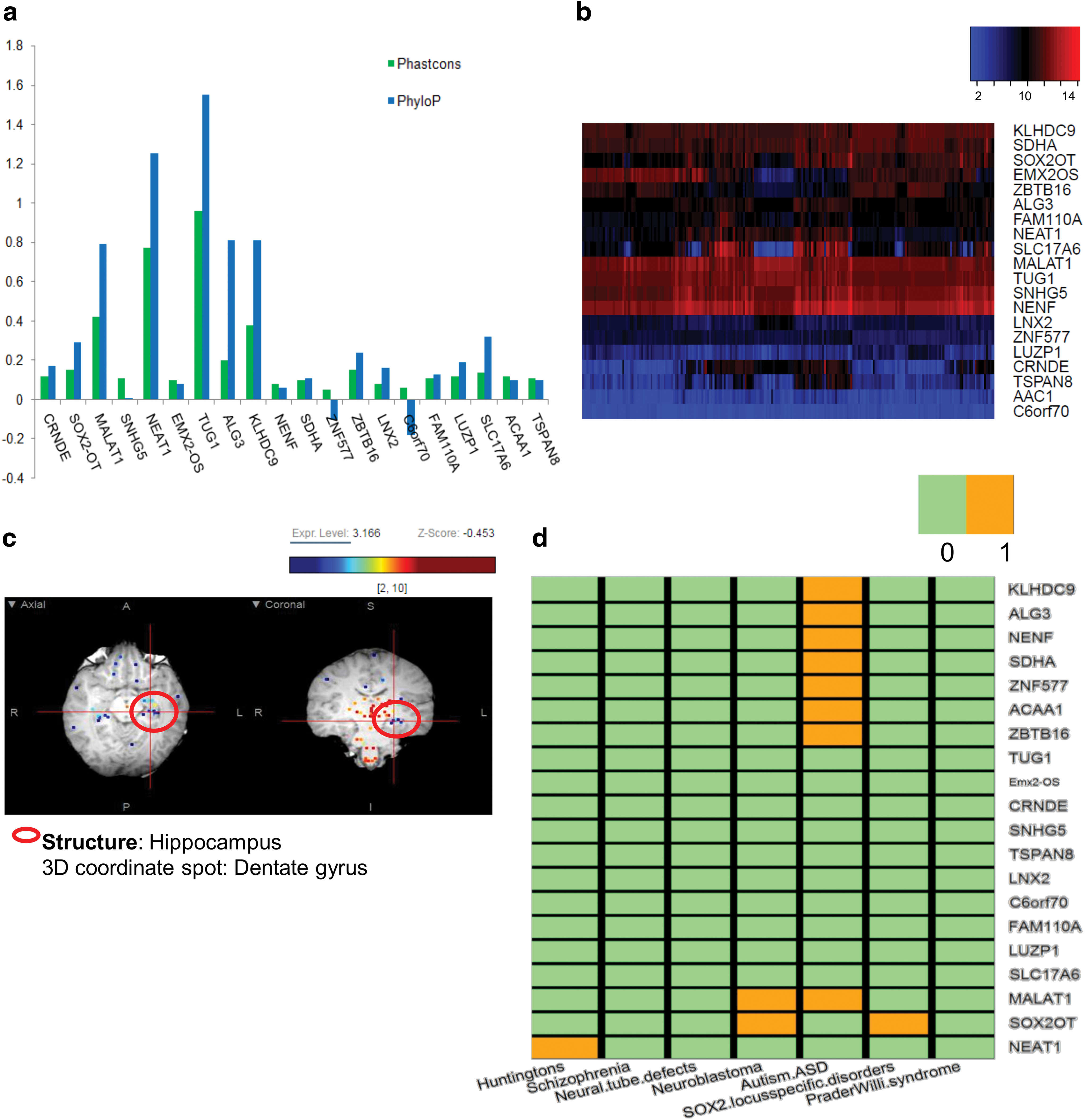
In continuation with the readout from Method A, we performed evolutionary conservation analysis for the set of 20 lncRNAs. Most of the lncRNAs were faintly conserved, except NEAT1 and TUG1 with PhastCons scores of 0.77 and 0.96, respectively (Fig. 2a and Table 1). We also found that the PhyloP scores were mildly positive that is, within the range of 0–0.8, with the exceptions of NEAT1 and TUG1 (Fig. 2a). Conclusively, most of the lncRNAs under strong selective pressure (PhyloP) are assumed to evolve at a faster rate due to their mean low conservation scores (PhastCons). To emphasize, this novel pool of lncRNAs can be further exploited in the future to understand their direct link to onset and progression of neurological diseases. From the data mining readout, we could establish a set of 20 lncRNAs having a potential role in neurogenesis. We also correlated the expression of these 20 important lncRNAs using location-wise expression data from ABA (Fig. 2b). For clear visualization of highly expressed lncRNAs in DG/LV, we used online ABA magnetic resonance imaging viewer tool and generated a visual representation of CRNDE, a highly expressed lncRNA in our study (log2FD = 5.7) (Fig. 2c).
The representative table gives detailed information regarding 20 lncRNAs.
lncRNAs, long non-coding RNAs; HiF, hippocampal formation region.
Do lncRNAs Play a Role in Neurological Disorders?
One of the important inherent features of neurogenesis is neuronal cell migration. Disruption of this process is strongly associated to a heterologous group of neuropathologies, which includes lissencephaly, schizencephaly, and microgyria [65]. In addition, faulty migration of neurons can lead to bilateral periventricular nodular heterotropia, a disease characterized by a specific abnormality around the LV region of the neurogenesis hub. Access to healthy or diseased human neural tissue samples continues to be a daunting task and a barrier to our knowledge regarding the cellular, genetic, and molecular mechanisms in neurogenesis and neurodegeneration.
Till date, there are only few lncRNA biomarkers developed for complicated neurological disorders, although several high-throughput studies have proved the presence of vast proportions of lncRNAs in adult brains. Notably, the regulatory mechanism of lncRNAs and their associated genes is yet to be elaborately studied even in other syndromic and nonsyndromic neurocognitive and structural brain impairments, including autism spectrum disorder (ASD), schizophrenia, bipolar disorder, Parkinson's, Huntington's, Zellweger, and Alzheimer's diseases [66 –70].
One of the few clear examples of lncRNAs altered in neurodegenerative disorders is BACE1-AS that is highly expressed during the Alzheimer's disease progression [71]. This lncRNA is a conserved antisense transcript to beta-secretase (BACE1) gene, an important enzyme involved in the disease pathology [71]. Few other studies describe the role of NEAT1 and Gomafu/MIAT in Huntington's disease and Schizophrenia, respectively [72 –74]. Recently, another interesting lncRNA Uchl1-AS was discovered, which contains an invSINEB2 element to upregulate translation of UchL1 protein, thus, leading to the discovery of a novel class of gene-specific inducers of protein synthesis [38,75]. Based on this information, we decided to look into disease associations of the total 20 significantly expressed lncRNA candidates from Method A by screening online repositories like Simons Foundation Autism Research Initiative and lncRNADisease (Fig. 2d) [52,76].
Disease mapping
Using databases containing brain disease information we constructed a Boolean matrix, wherein the absence and presence of lncRNA association to a disease was recorded by numbers 0 and 1, respectively. We found almost a dozen lncRNAs, which were not associated with any disorders. Precisely, we could detect involvement of lncRNAs in few neurodegenerative disorders, namely, Huntington's disease, neural tube defects, neuroblastoma, schizophrenia, ASD, SOX2 locus-specific disorders, and Prader–Willi syndrome. The Boolean matrix showed 8 out of 20 lncRNAs to be specifically associated with ASD when mapped onto the disease databases (Fig. 2d). Sox2-OT and MALAT1 showed distinct expression in neuroblastoma, which were also found to be involved in neurogenesis (Fig. 2d). Similarly, in case of Huntington's disease, only NEAT1 showed association, which has earlier been linked to neurogenesis (Fig. 2d). We conclude that out of 20 putative lncRNAs showing high expression in neurogenesis, 10 lncRNAs displayed putative roles in neurological disorders.
Correlation analysis
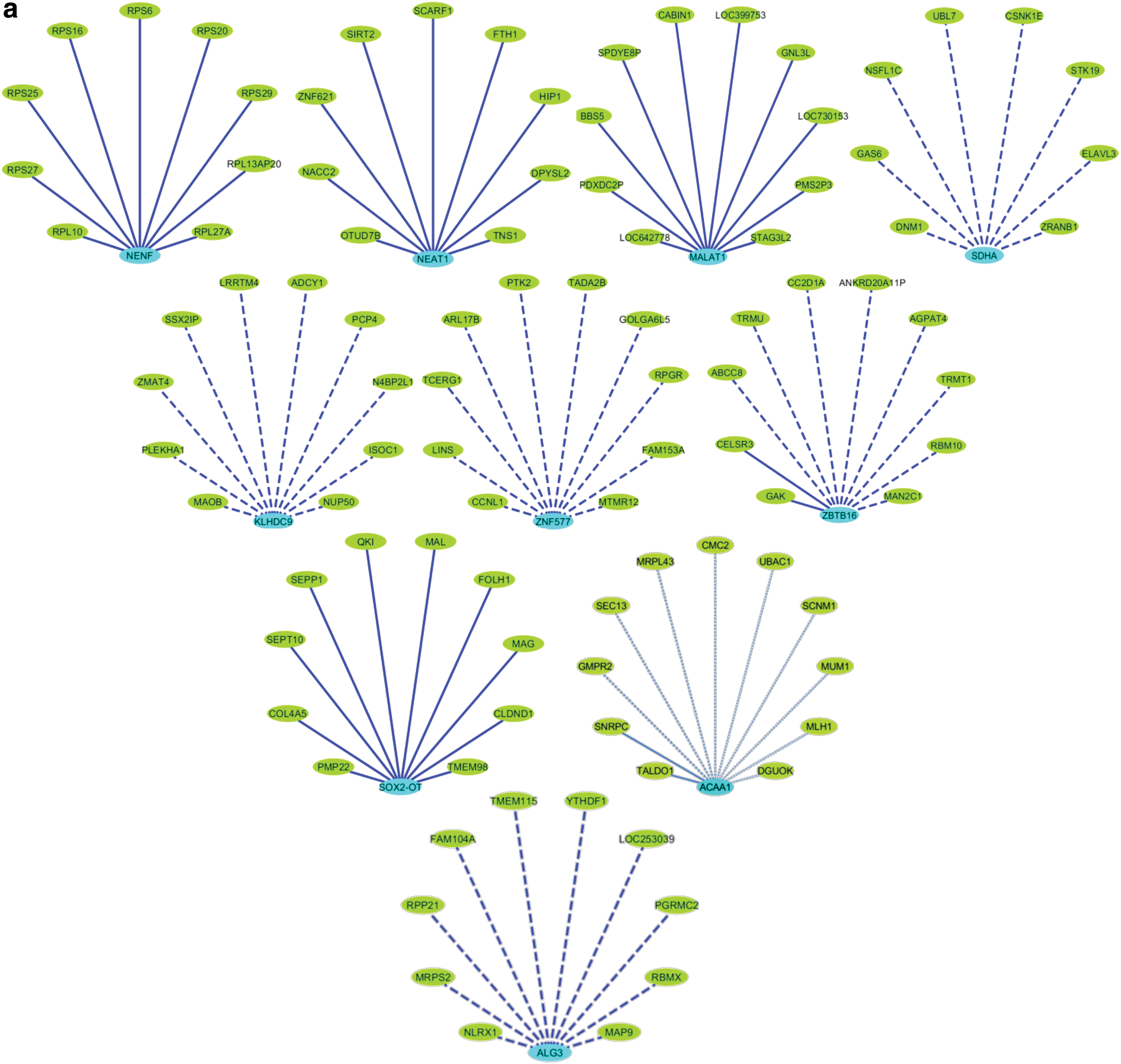
This pool of shortlisted 10 lncRNAs was further evaluated for coexpressed genes. For finding closest correlating gene counterparts, we used the ABA-based expression gene correlation maps. This correlative search of ABA returns probes with a similar expression profile to brain regions of interest. Therefore, we collected the top 10 gene correlates for each lncRNA in the HiF and constructed individual networks using Cytoscape [77]. All the networks were built in a Degree Sorted Layout. The dashed lines represent relatively weaker correlates, whereas the dark solid lines indicate stronger correlates. The edges of the networks grow from dashed to solid lines with a concomitant increase in the Pearson's correlation value (R value) (Fig. 3a). We could infer that three lncRNAs, namely MALAT1, SOX2-OT, and NENF possess relatively stronger correlates compared with ACAA1, ZNF577, and SDHA.
Functional significance of candidate lncRNAs
The timeline of discovery of RNAs (coding and non-coding) are strongly supportive of the diversity of RNAs working in biological regulation [1]. Following in-depth analysis of localization and expression of lncRNAs in brain, we resolved to shed light on the functions of resultant 10 candidate lncRNAs. Some of these lncRNAs have already been annotated and have known functions in neurodevelopment, like NEAT1, MALAT1, and SOX2-OT. Earlier reports suggest how MALAT1 regulates the process of alternative splicing by modulating SR splicing factor phosphorylation resulting in a variety of proteins [78,79]. Notably, MALAT1 plays an important role specifically in neurogenesis by regulating synapse formation and modulating the expression of genes involved particularly in synapse formation and maintenance [35]. NEAT1 is constitutively dysregulated throughout the process of neurogenesis and forms a crucial signal for neurodifferentiation [63,80]. SOX2-OT does its namesake-specific role of regulating the set of SOX2 and other SOX gene family cluster along with other miRNAs at a posttranscriptional level [64]. Other lncRNAs like EMX2-OS, NENF, and LUZP1 only displayed probable roles in neurogenesis according to prior literature (Table 1) [81 –83].
Detection of putative downstream targets
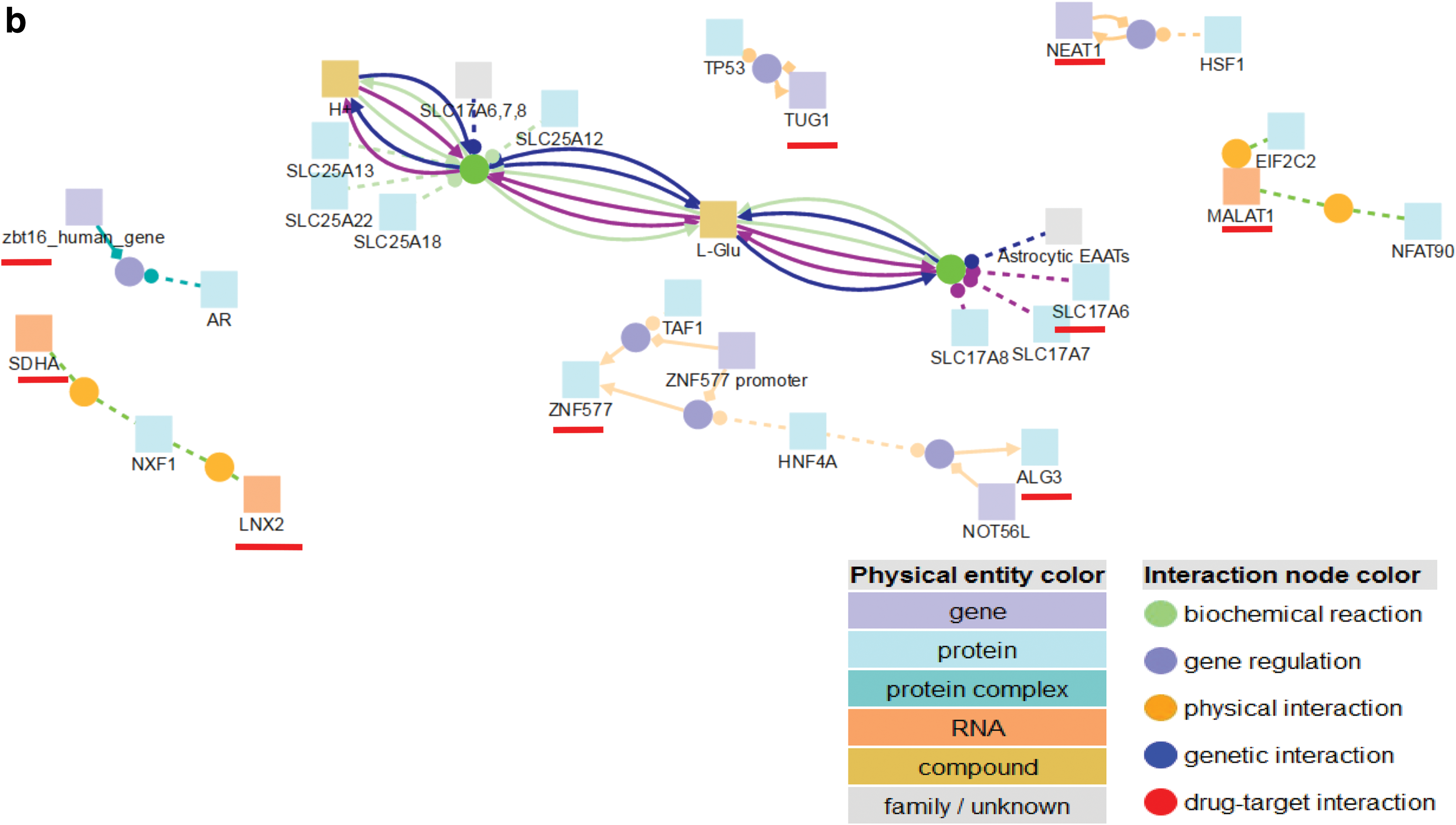
We further looked for pathway associations of these 10 lncRNAs using CpDB database, a database containing integrative information regarding human interaction networks including binary and complex protein–protein, genetic, metabolic, signaling, gene regulatory, and drug–target interactions, as well as biochemical pathways from 32 public resources [84]. We utilized the Interactions of Molecules/Pathways module of this database and inputted the 10 lncRNAs.
The results showed cellular pathway associations for 9 out of 10 lncRNAs (Fig. 3b). Physical interactions were noticed for three lncRNAs namely, LNX2, SDHA, and MALAT1 (Fig. 3b). While MALAT1 was interacting with two genes, NFAT90 and EIF2C2, and LNX2 and SDHA were seen to interact with a common gene, NXF1. Prior literature had indicated that LNX2 and SDHA have probable functions in neuronal differentiation and pituitary adenoma, respectively [85,86]. Gene regulatory interactions were exhibited by five lncRNAs namely, ZNF577, ZBT16, ALG3, TUG1, as well as NEAT1 (Fig. 3b). Interestingly, TUG1 showed association with TP53, a crucial gene for brain glioma and other cancers, and NEAT1 was linked to another essential brain transcription factor, heat shock protein transcription factor-1. An interesting biochemical interaction was noticed for SLC17A group of genes, of which one of them has no coding potential that is, SLC17A6 lncRNA (Fig. 3b). Such interactions are suggestive of the direct/indirect roles of lncRNAs in neurodevelopment. The overall workflow has been compiled (Fig. 4). Thus, the above lncRNA signatures can be used as potential biomarkers of neurogenesis, but requires further mechanistic studies for validation.
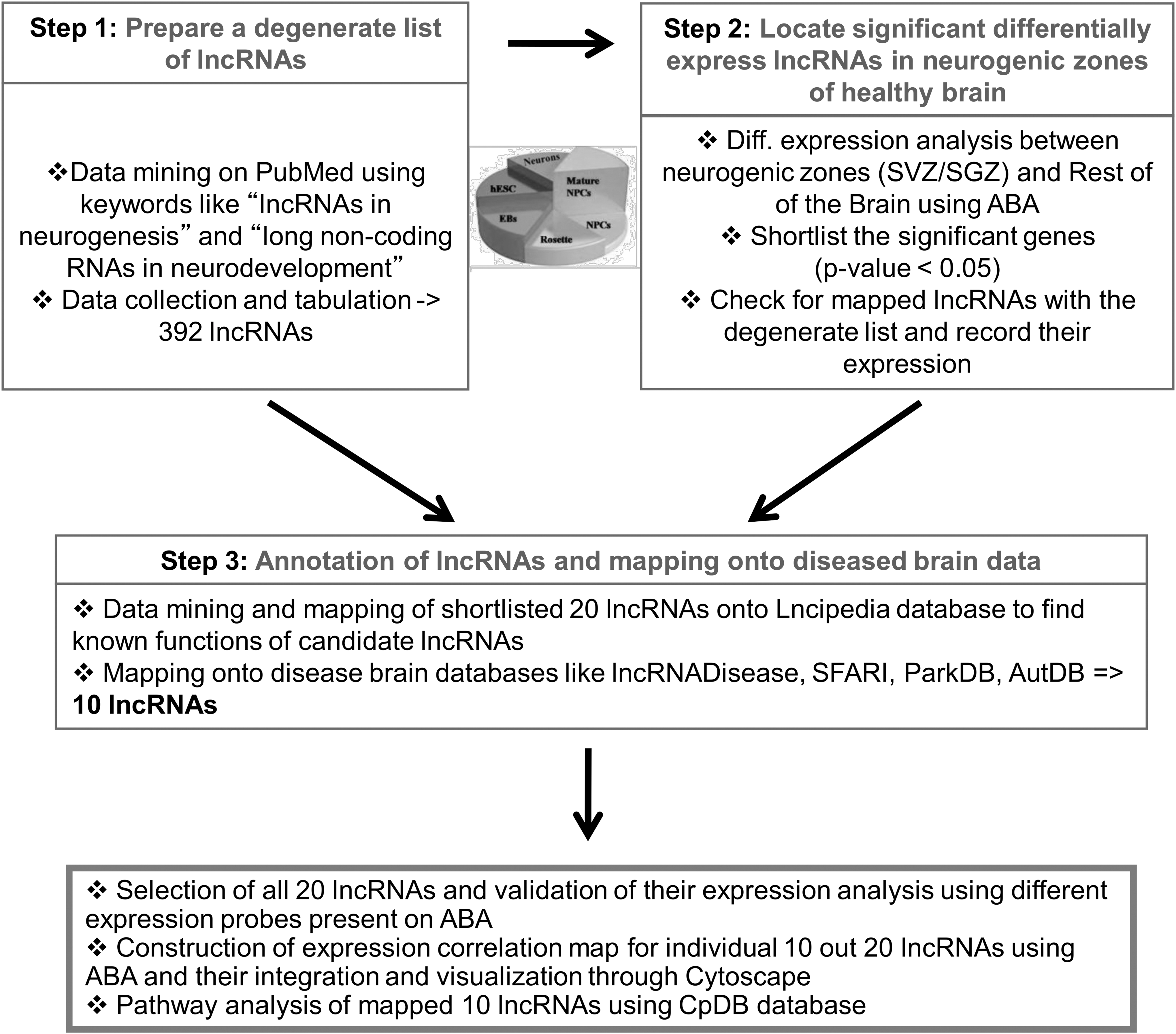
Conceptual framework for identifying potential lncRNA biomarkers implicated in neurogenesis and/or neurodegeneration.
Perspectives
One of the largely unsolved problems in human genome biology had been to calculate the exact proportion of functional RNome [87]. In the recent past, studies have resolved this problem partially and shown how non-coding RNAs are no longer nonfunctional. The ∼98% non-coding RNome has displayed substantial gene regulatory functions, depicting both cis- and trans-acting regulatory mechanisms. With the development of lncRNAs as new diagnostics, they are even seen to be promising biomarkers. For example, PCA3 (prostate cancer antigen 3) can be detected in urine samples and has shown to improve the diagnosis of prostate cancer [88]. However, only a few lncRNAs have been reported in brain development that could be used as prognostic/diagnostic markers, and given the higher fraction of non-coding genome, it is only imperative to discover more such lncRNAs. To clearly understand the mechanistic link between lncRNAs and neurological diseases, there is also a serious demand to develop proper genetic tools and generate animal models with altered lncRNA-mRNA expressions.
High-throughput technologies like RNA-Seq, ChIP-Seq, and integrative biology-based analyses have been employed to derive in-depth understanding of expression and mechanisms of action of lncRNAs [49]. LncRNA detection using RNA-sequencing has been more conclusive, and is being utilized to frame some of the preliminary studies linked to neurodevelopment [89]. In our study, we have provided a comprehensive view of lncRNA-to-disease/pathway/gene associations by employing brain disease databases/interactome information/coexpression data, respectively containing annotated lists of genetic fingerprints.
It is interesting to note that the expression of lncRNAs gets altered during pre- and postnatal brain development, leading to evident changes in the neural molecular circuitry. These changes include alteration in the associated coding gene expression and/or increasing the translation rate, regulation of the epigenome, and further affecting the downstream targets of the associated genes, thus, modulating several signaling pathways. Our in-silico data analysis has ascertained a novel set of lncRNAs, which play dual roles in neurogenesis and neuropsychiatric disorders. Further experimental studies would be essential to ascertain the feasibility of these putative candidates. To develop effective prognostic/diagnostic lncRNA biomarkers for both syndromic/nonsyndromic diseases, an integrative systems biology approach is essential, which in turn will bridge the gap in knowledge regarding altered lncRNA-based molecular mechanisms in brain diseases.
Footnotes
Acknowledgments
R.A. is grateful to Shiv Nadar University for the fellowship. The Center for Informatics located inside Shiv Nadar University is also duly acknowledged. S.S. would like to acknowledge Innovative Young Biotechnologist Award (IYBA) from the Department of Biotechnology. S.P. would like to sincerely thank Shiv Nadar Foundation for the ongoing work.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.