
Abstract
Skewed X chromosome inactivation (XCI) is a rare reason for hemophilia B in females. It is indefinite whether X chromosome reactivation (XCR) would occur when cells of hemophilia B patients with skewed XCI were reprogrammed into induced pluripotent stem cells (iPSCs). In this study, we investigated a female hemophilia B patient with a known F9 gene mutation: c.676C>T, p.Arg226Trp. We demonstrated that skewed XCI was the pathogenesis of the patient, and we successfully generated numerous iPSC colonies of the patient from peripheral blood mononuclear cells (PBMNCs), which was the first time for generating hemophilia-specific iPSCs from PBMNCs. Then we detected the XCI state of these iPSCs. Ninety-two iPSC lines were picked for XCI analysis. All of them retained an inactive X chromosome, which could be proved by amplification of the androgen receptor gene and XIST (X inactivation-specific transcript), expression of H3K27me3, and existence of XIST clouds in XIST RNA fluorescence in situ hybridization (FISH) analysis. We attempted to obtain iPSC lines with the wild-type F9 gene on the active X chromosome for further disease treatment. But it turned out that the patient's iPSCs were still skewed such as the somatic cells with 92 iPSC lines having mutant F9 on the active X chromosome. In conclusion, skewed XCI is one reason for hemophilia in females. PBMNCs are excellent somatic cell resources for hemophilia patients to do reprogramming. More attentions should be paid to generate naive iPSCs with two active X chromosomes for further clinical disease treatment. The state of skewed XCI is retained in the iPSCs from a female with hemophilia B.
Introduction
U
Hemophilia B is a hereditary disorder caused by mutations of coagulation factor IX (F9) gene, affecting ∼1 in 25,000 males worldwide [8]. Being an X-linked recessive disorder, hemophilia B is inherited by heterozygous females who are defined as carriers, and generally does not affect females. However, female hemophilia does exist. There are two reasons for this: first, homozygous mutations and compound heterozygous mutations in females [9 –13]; second, skewed XCI, defined as the preferential inactivation of one of the two X chromosomes in female cells [14,15]. In the cells of normal females, there are two X chromosomes with wild-type F9 (X F9-WT ); while in the cells of female carriers of hemophilia B, there is an X F9-WT chromosome and an X chromosome with mutant F9 (X F9-mut ). In mammals, one of the two X chromosomes is randomly inactivated in female cells to maintain a balance in X-linked gene dosage with males during early embryonic development [16]. The preferential inactivation of X F9-WT chromosome induces the onset of hemophilia B in female carriers. The inactivation of X chromosome depends on the noncoding RNA XIST (X inactivation-specific transcript), which coats and silences the chromosome in which it is expressed [17 –19]. Skewed XCI can be caused by mutations of genes involved in XCI, a reduced number of precursor cells being present at the time of inactivation, or the selection of a small number of normal cells to contribute to the inner cell mass of the blastocyst [20].
In majority of studies, human iPSCs are generated from dermal fibroblasts [21]. Being a hemorrhagic disorder, hemophilia is not suitable for this invasive approach. Usually, iPSCs of hemophilia patients are generated from urine cells [22 –24]. However, peripheral blood mononuclear cells (PBMNCs) have been widely accepted as the best cell source for cell reprogramming due to their easy accessibility and better quality and quantity [25,26].
In this study, we investigated the pathogenesis of a female hemophilia B patient. We generated numerous iPSC colonies of the patient from PBMNCs, which was the first time for generating hemophilia-specific iPSCs from PBMNCs. Then, we detected the XCI state of these iPSCs. We attempted to obtain iPSC lines with X F9-WT chromosome activated for further disease treatment.
Materials and Methods
Study participants
The female patient, admitted to our center in 2014, 46 years old, had suffered from bleeding episodes in the lower and upper limb muscles, elbow, knee, and hip joints, 4 to 5 times per year for 35 years. Her activated partial thromboplastin time was 57.3 s (normal value 23–33 s); prothrombin time was 14.4 s (normal value 10–14 s); FIX:C was 6% (normal value 50%–120%), FII:C (112.1%), FV:C (113.8%), FVII:C (103.6%), FVIII:C (106.9%), FX:C (102.8%), FXI:C (60%), FXII:C (91.7%), FXIII:C (blood clot did not dissolve after 24 h in urea), and von Willebrand factor antigen (Ag) (93.1%) were normal. Both the qualitative and quantitative detection of coagulation factor inhibitor were negative using the Bethesda assay. Based on these test results, the patient was clearly diagnosed as hemophilia B. As shown in the family pedigree (Fig. 1), there was no history of hemophilia B in her family. The patient's five relatives, including her mother (II8), two sisters (III1 and III3), and two brothers (III5 and III6), were enrolled in our study. The five relatives had no muscular or articulatory hemorrhage. The study was approved by the Ethics Committee of the Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences, and written informed consent was obtained from each study subject.

The pedigree of the female patient family. There was no hemophilia B history in this family. III4 was the female patient we investigated in the study (as the black arrow points). Her mother (II8), two sisters (III1 and III3), and two brothers (III5 and III6) were enrolled in the study.
High-throughput sequencing of the F9 gene
High-throughput sequencing (including library construction, capture, and sequencing) was carried out at BGI-Tianjin (Tianjin, China). The F9 mutation found by high-throughput sequencing was then verified by Sanger sequencing. The primer sequences were as follows: forward 5′-GAACACCTATTCTATTTCCGT-3′ and reverse 5′-TAGTGCCCTGAACCTGAG-3′.
XCI assay
The XCI state was determined by PCR analysis of a polymorphic trinucleotide repeat in the first exon of the androgen receptor (AR) gene [27]. Genomic DNA was digested with HapII enzyme, and complete digestion was assessed by amplification of the 5′ region of the MIC2 gene [28,29]. Then, both digested and undigested samples were amplified for AR gene using primers: forward 5′-FAM-AAGTGCAGTTAGGGCTGGGAAGG-3′ and reverse 5′-GTAGCCTGTGGGGCCTCTACGAT-3′. If XCR did not occur after reprogramming, AR gene would be amplified. Then PCR products were sized using capillary electrophoresis on an ABI Prism 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA) [30]. In our study, the patient's alleles at the AR gene were 3 bp apart, and shadow bands of the first allele overlap with bands of the second allele. Shadow bands were calculated to represent 30% of the amplification from one allele, and peak area values were adjusted accordingly [30]. As determined by the previous study [29], the degree of skewing was determined as mild skewing (≥75%) and extreme skewing (≥90%).
Whole-genome sequencing
Whole-genome sequencing (WGS) was carried out at BGI-Shenzhen (Shenzhen, China), yielding 30× coverage. Sequencing was performed on HiSeq X Ten platform (Illumina, Santiago, CA). Raw image files were processed by Illumina pipeline for base calling with default parameters, and the sequences of each individual were generated as 150-bp paired-end reads.
Generation of the patient's iPSCs
The patient's iPSCs were generated from PBMNCs of the patient using four episomal vectors, as previously described [31]. The reprogramming process was completed under hypoxic conditions (4% O2). The episomal vectors were gifted by Xiaobing Zhang from the Department of Medicine at Loma Linda University.
Karyotype analysis
For karyotype analysis, iPSCs were harvested after 10 passages. It was done according to a general protocol. The slides were visualized under a microscope, and 20 split cells were selected randomly.
Alkaline phosphatase staining
The Alkaline Phosphatase (AP) Staining Reagents Detection Kit was bought from SiDanSai Biotechnology Co., Ltd. (Shanghai, China). The staining procedures were done according to the protocol provided.
Reverse transcription PCR and quantitative real-time PCR
RNA was extracted using RNeasy Mini Kit (Qiagen, West Sussex, UK) following the manufacturer's instructions. RNA (3 μg) was reversely transcribed using a First Strand Synthesis Kit (Life Technologies, Carlsbad, CA). Quantitative real-time PCR (qRT-PCR) analysis was performed using a Step One Plus Real-Time PCR System (Applied Biosystems, Foster City, CA). The primer sequences are presented in Table 1.
F, forward; R, reverse.
Teratoma formation assay
iPSCs were injected intramuscularly into Nonobese Diabetic/Severe Combined Immunodeficiency Disease (NOD/SCID) mice. Teratomas formed within 6–8 weeks, and harvested samples were fixed in 4% paraformaldehyde overnight, the paraffin sections were stained with hematoxylin and eosin for histological determinations.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for 30 min at room temperature (RT) and blocked with blocking buffer (phosphate-buffered saline +5% goat serum +0.3% bovine serum albumin) for 1 h at RT. Primary antibodies were incubated overnight at 4°C. Cells were incubated with secondary antibodies for 1 h at 37°C. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole for 5 min. Descriptions of the primary and secondary antibodies are listed in Table 2.
RNA fluorescence in situ hybridization analysis
iPSCs (6 × 104) were spun onto slides using the cytospin. After adding probes (Empire Genomics, Buffalo, New York), slides were placed in ThermoBrite platform (Thermo Fisher Scientific, Waltham, MA) in the program: 73°C 10 min and 37°C 24 h. Finally, slides were visualized under a fluorescence microscope.
Results
F9 sequencing of the patient and her five relatives
The high-throughput sequencing results showed that the patient was a carrier with a known hemophilia B mutation: c.676C>T, p.Arg226Trp, which was verified by Sanger sequencing. Sanger sequencing showed that none of her relatives had this mutation (Supplementary Fig. S1; Supplementary Data are available online at
XCI assay of the patient, her mother, and two sisters
The results of capillary electrophoresis are shown in Figure 2. According to the formula, 100*(d1/u1)/((d1/u1) + (d2/u2)) [23], the percentage of skewing of the patient was 94.69% (Table 3), considered as extreme skewing. Because of the excessive amplification of the 306 bp fragment, we considered that the X chromosome with 306 bp fragment had excessive inactivation. As the female was a hemophilia B patient, we inferred that mutation c.676C>T, p.Arg226Trp was located at the X chromosome with 303 bp fragment. The percentages of skewing of her mother and two sisters were 64.20%, 59.12%, and 59.85%, respectively (Table 3). None of the three female relatives showed skewed XCI.

The XCI assay of the patient, her mother, and two sisters. The capillary electrophoresis results of II8 (
AUC, area under the curve.
WGS results for the patient and analysis of XIST and TSIX
According to previous studies [32], both XIST and TSIX (XIST antisense RNA) play important roles in the process of XCI. In our unpublished study, we found mutations of 1061G>A and 11947-130_11947-128delAAT in the XIST gene of this female patient by WGS, while no mutations were found in the TSIX gene. Both mutations were verified by Sanger sequencing and were also found in the XIST genes of her mother, a sister, and a brother (Supplementary Fig. S2). No copy number variations or structural variations were found in the XIST and TSIX gene.
Generation and characterization of the patient's iPSCs
More than 100 iPSC colonies were obtained from 2 × 106 PBMNCs. The colonies exhibited the characteristic human embryonic stem cell morphology with a high nucleus to cytoplasm ratio (Fig. 3Ai) and were positive for AP staining (Fig. 3Aii). Ninety-two iPSC colonies were picked and expanded for further analysis. The iPSC lines were randomly chosen for characterization of pluripotency. They had normal karyotypes (Fig. 3B), and immunofluorescence staining showed expression of pluripotent markers, including TRA-1-60, SSEA4, OCT4, and SOX2 (Fig. 3C). The pluripotency of the iPSCs was further assessed by qRT-PCR analysis (Fig. 3D). Weeks after injection into NOD/SCID mice, iPSCs produced teratomas containing multiple derivatives of the three germ layers (Fig. 3E). Collectively, these results demonstrated that the iPSCs were well reprogrammed.
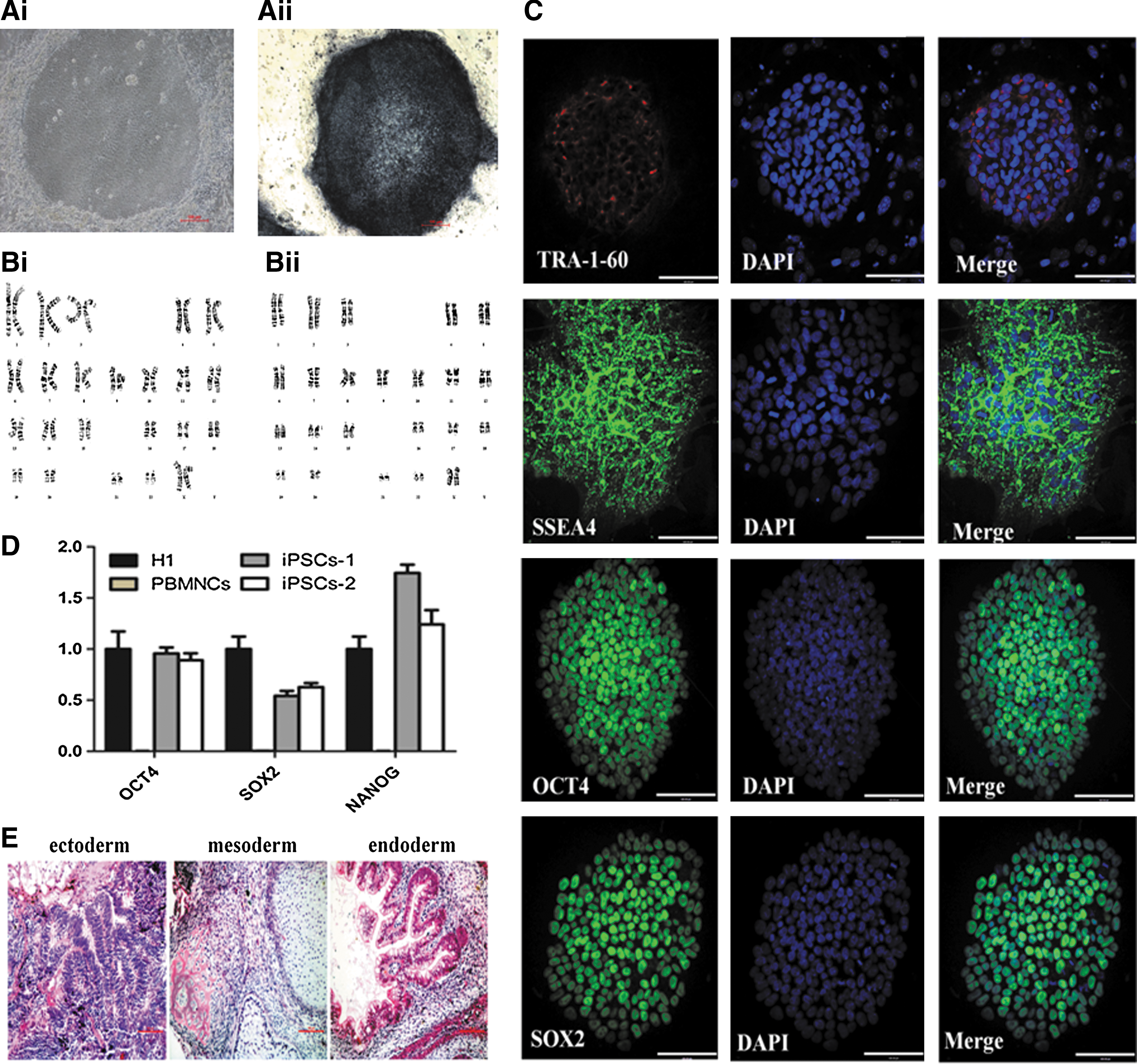
Characterization of the patient-specific iPSCs derived from PBMNCs with episomal vectors. All scale bars represent 100 μm.
XCI assay of 92 patient's iPSC lines
Considering that prolonged passaging affects XCI characteristics [2], we analyzed the 92 iPSC lines before fifth passage. The nonamplification of MIC2 indicated the complete digestion by HapII enzyme (Supplementary Fig. S3A). Then, AR gene was amplified to see whether XCR occurred, as shown in Supplementary Figure S3B, all cell lines, even iPSCs from passage one, yielded amplified products of the AR gene after HapII enzyme digestion, indicating that XCR did not occur after reprogramming, and all iPSCs had one X chromosome activated and the other inactivated (XaXi).
Further proving the XaXi state of the patient's iPSCs
To further prove the XaXi state of these iPSCs, we amplified the noncoding XIST RNA. As shown in Figure 4A, XIST was expressed in all female patient's iPSC lines. FISH is an important non-radioactive in situ hybridization technique. If the detected DNA on the chromosome is complementary with the homologous nucleic acid probe, they will form hybrids. RNA–FISH analysis indicated that 93% of the cells showed one XIST cloud per nucleus (Fig. 4B; 200 cells were analyzed per cell line). Furthermore, H3K27me3 (trimethylation of histone H3 on lysine 27), a silencing marker on chromatin that correlates with XIST expression, was also detected in the patient's iPSCs (Fig. 4C).
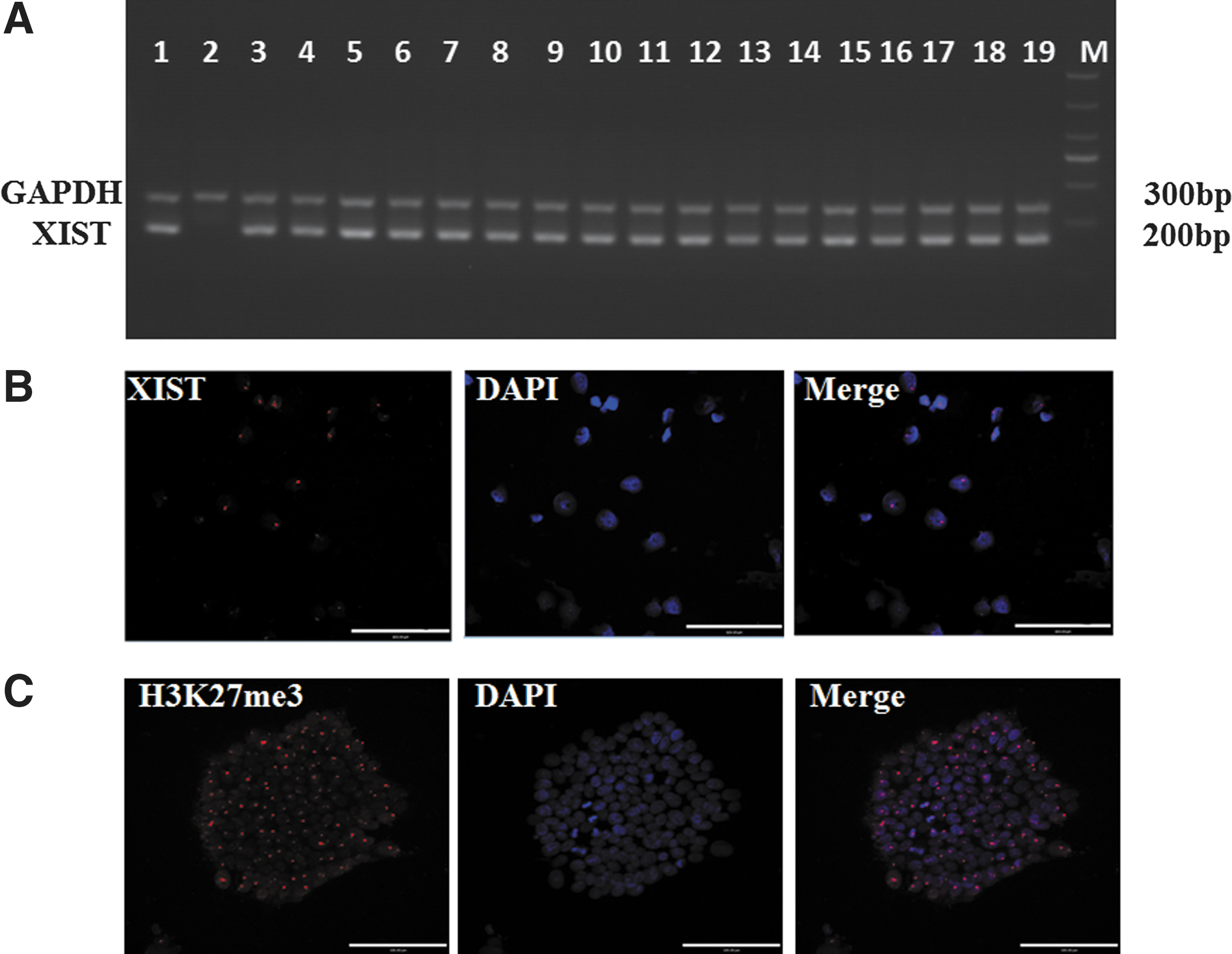
The proving of the one X chromosome activated and the other inactivated state of the patient's iPSCs.
The skewed XCI state of the patient's 92 iPSC lines
After we had evidence for the amplification of the AR gene, capillary electrophoresis was performed to assess which X chromosome in these iPSC lines was activated. Notably, all 92 iPSC lines had the same result, wherein the 306 bp fragment appeared in the electrophoresis graph (Fig. 5). Because 303 and 306 bp were 3 bp apart, the small peak at 303 bp in Figure 5B actually was the shadow of the peak of 306 bp. So the X chromosome with 303 bp fragment was activated; implying that all 92 iPSC lines had the X F9-mut chromosome activated. The XCI state of the patient's iPSCs was still skewed such as the somatic cells, reprogramming did not change the skewed state of the patient.
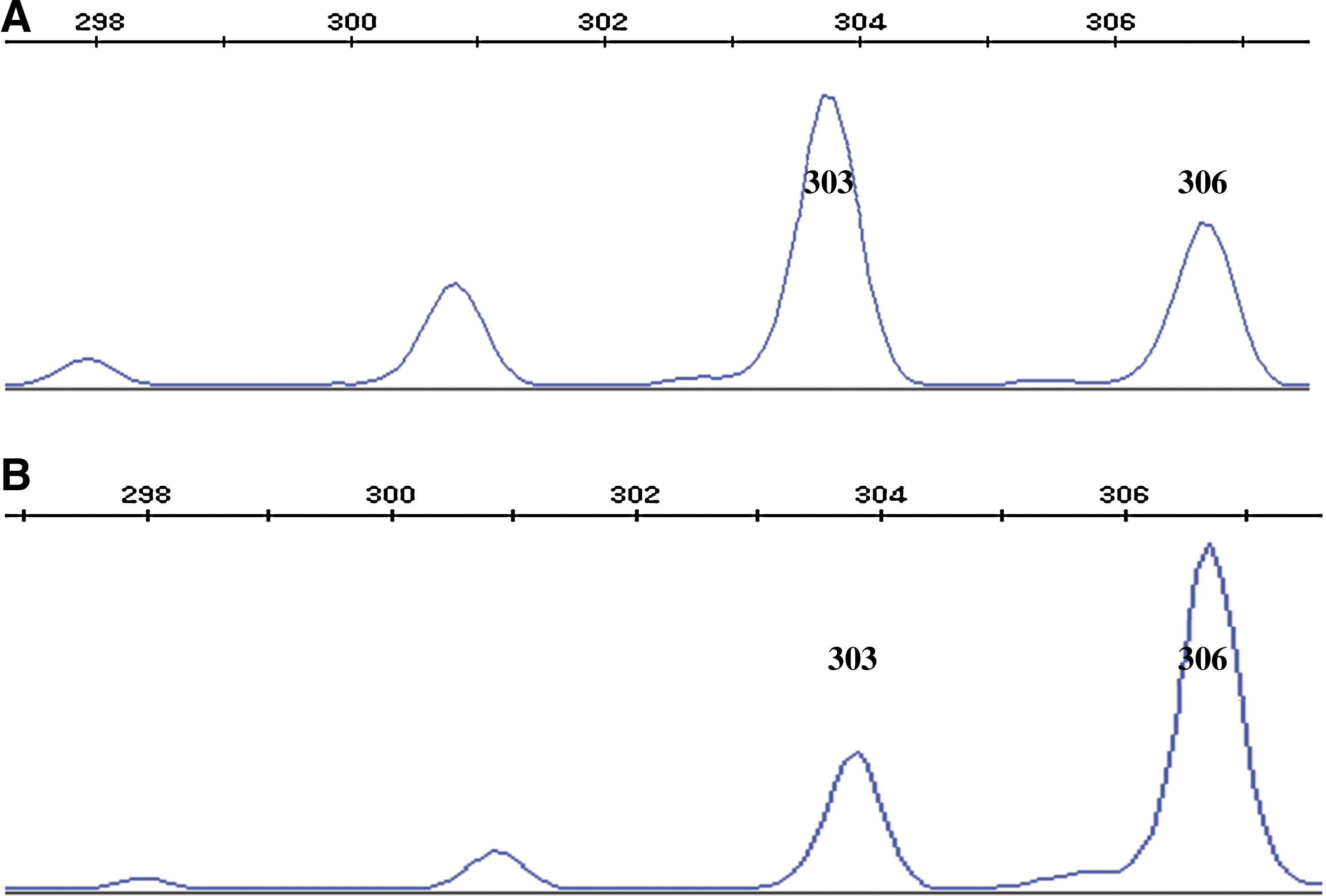
The capillary electrophoresis results of iPSCs. All 92 iPSC lines had the 306 bp fragment appeared in the electrophoresis graph after HapII digestion, so one representative capillary electrophoresis image was shown to represent all.
Discussion
Hemophilia mostly affects males and is transmitted by female carriers. Female carriers have normal phenotypes due to the random inactivation of one of the two X chromosomes in all the somatic cells. Therefore, in asymptomatic female carriers of hemophilia, the ratio of somatic cells with X F9-WT chromosome activated to somatic cells with X F9-mut activated is 50%–50% [33]. However, in our study, we diagnosed a female as hemophilia B and demonstrated that skewed XCI was the reason for the disease. Among the genetic mechanisms leading to skewed XCI [20,34], chromosomal abnormalities and parental origin effects could be ruled out because the karyotype of the patient was normal, and her mother had random XCI. The possible mechanism behind this female patient may be mutations associated with XCI-related genes such as XIST promoter mutations [35,36] and large deletions in XIST [37], or a genetically influenced choice over which X chromosome is inactivated as reported in mice [38]. TSIX noncoding RNA also participates in the process of XCI [32]. In our study, no mutations were found in the TSIX gene of this patient, while two new mutations (1061G>A and 11947-130_11947-128delAAT) were found in her XIST gene. But these mutations also existed in the patient's mother, a sister, and a brother. Therefore, the patient's skewed XCI is not caused by XIST mutations. Definite mechanisms that result in skewed XCI in the patient still need to be investigated.
After the verification of skewed XCI of the patient, we generated iPSCs using integration-free episomal vectors from PBMNCs of the patient, which was the first time for generating hemophilia-specific iPSCs from PBMNCs. In our study, more than 100 iPSC colonies occurred after reprogramming, and the iPSCs had normal karyotypes and good pluripotency. So PBMNCs are excellent somatic cell resources for hemophilia patients to do reprogramming.
Then, we analyzed the XCI state of 92 iPSC lines to see whether XCR occurred after reprogramming. As Tchieu et al. [2] and Pomp et al. [5] reported, all 92 iPSC lines retained an inactive X chromosome, which was proved by the amplification of the AR gene and the XIST gene, the existence of XIST cloud in XIST RNA-FISH analysis, and the expression of H3K27me3. Reasons that XCR does not occur might be due to different reprogramming techniques and the growth conditions, in which human iPSCs are generated and maintained, which generate “primed” rather than “naive” iPSCs [39,40]. In a recent study, it was found that culture of human iPSCs in defined conditions (naive human stem cell medium [NHSM]) resulted in more naive iPSCs and lead to efficient loss of XCI-specific markers, including XIST RNA and XCI-specific histone modifications [41]. We tried this method, domed-shaped colonies resembling murine naive cells could be seen after cultured in NHSM for three passages (Supplementary Fig. S4A), but we could still detect XIST expression by reverse transcription-PCR (Supplementary Fig. S4B). Maybe we did not get some detail points of culturing. We will improve our methods in further study.
Although XCR did not occur, considering that each iPSC colony comes from one somatic cell, we attempted to obtain two kinds of iPSC colonies after reprogramming, the ones with X F9-WT chromosome activated and the ones with X F9-mut chromosome activated. However, our actual results did not support our expected results. All 92 iPSC lines had the X F9-mut chromosome activated. According to Pomp et al. [5], using balanced populations of female Rett Syndrome patient and control fibroblasts, all colonies made from the same donor fibroblasts contained the same inactive X chromosome. They found that the mosaic nature of female fibroblasts was lost during extended passaging in vitro as a consequence of the proliferative advantage of one type of cells to its counterparts. In our study, the ratio of somatic cells with X F9-mut chromosome activated to somatic cells with X F9-WT chromosome activated was 94.69%–5.31%, the former had absolute advantage to the latter. So maybe this was the reason we could not obtain iPSC lines with X F9-WT chromosome activated. It suggests that iPSCs derived from PBMNCs have imprints of PBMNCs, which indicates that iPSCs have imprints of the original somatic cells. Thus, when using iPSCs to treat diseases limited to a specific organ, it might be a better choice to use somatic cells from the same organ to generate iPSCs.
In conclusion, skewed XCI is one reason for hemophilia in females, which should be kept in mind to avoid misdiagnosis and missed diagnosis. PBMNCs are excellent somatic cell resources for hemophilia patients to do reprogramming. And more attentions should be paid to generate naive iPSCs with two active X chromosomes for further clinical disease treatment, we will explore novel reprogramming techniques and growth conditions in further research. The state of skewed XCI is retained in the iPSCs from a female with hemophilia B, it indicates that iPSCs might have imprints of the original somatic cells, which is instructive for clinical application.
Footnotes
Acknowledgments
This study was supported by National Key Research and Development Program of China (2016ZY05002341), Ministry of Science and Technology of China (2016YFA0100600), National Natural Science Foundation of China (81470302 and 81421002), Tianjin Municipal Science and Technology Commission (15JCZDJC35800), CAMS Initiative for Innovative Medicine (2016-I2M-1-018, 2016-I2M-1-017, 2016-I2M-1-002, 2016-I2M-1-001, 2015PT310006), PUMC Youth Fund (332015129), and Novo Nordisk Hemophilia Research Fund in China.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.