
Abstract
Mesenchymal stem cells (MSCs) have shown promise for use in cell therapy, and due to their tumor tropism can serve as vehicles for delivering therapeutic agents to tumor sites. Because interleukin-8 (IL-8) is known to mediate the protumor effect of MSCs, elimination of IL-8 secretion by MSCs may enhance their safety for use in cancer gene therapy. However, little is known concerning the effect of endogenously secreted IL-8 on MSCs. We performed studies using placenta-derived MSCs (PMSCs) to determine whether knockdown of IL-8 would influence their biological activity. We first verified that IL-8 and its membrane receptor CXCR2, but not CXCR1, were highly expressed in PMSCs. We then employed lentivirus-mediated small hairpin RNA interference to generate stable IL-8-silenced PMSCs, which displayed a variety of characteristic senescent phenotypes. We observed that at day 9 post-transfection, IL-8-silenced PMSCs had become larger and displayed a more flattened appearance when compared with their controls. Moreover, their proliferation, colony forming unit-fibroblast formation, adipogenic and osteogenic differentiation, and immunosuppressive potentials were significantly impaired. Enhanced senescence-associated β-galactosidase (SA-β-gal) activity and specific global gene expression profiles confirmed that IL-8 silencing evoked the senescence process in PMSCs. Increased levels of p-Akt and decreased levels of FOXO3a protein expression suggested that reactive oxygen species played a role in the initiation and maintenance of senescence in IL-8-silenced PMSCs. Notably, the majority of CXCR2 ligands were downregulated in presenescent IL-8-silenced PMSCs but upregulated in senescent cells, indicating an antagonistic pleiotropy of the IL-8/CXCR2 signaling pathway in PMSCs. This effect may promote the proliferation of young cells and accelerate senescence of old cells.
Introduction
M
Previous studies identified IL-8 as a major component of the senescence-associated secretory phenotype (SASP); furthermore, overexpression of IL-8 has been observed in senescent cells. Acosta et al. [10] reported that IL-8 could reinforce replicative and oncogene-induced senescence (OIS) by binding with its G protein-coupled receptor (GPCR) CXCR2. Medina et al. [11] showed that IL-8 could facilitate the replicative senescence of outgrowth endothelial cells (OECs), and that knockdown of IL-8 expression prolonged the life span and enhanced the function of ex vivo-expanded OECs. Accordingly, reduction of IL-8 expression appears to be an effective method for expanding stem cell numbers without diminishing their functionality.
Currently, placenta-derived MSCs (PMSCs) have received greater attention because they can be easily obtained without ethical problems. Furthermore, PMSCs possess more potent proliferative and immunomodulatory capacities when compared with their adult control cells [12,13]. While PMSCs are known to express IL-8, it has remained unclear whether knockdown of IL-8 expression improves their replication potential and therapeutic efficacy. In this study, we demonstrated that stable knockdown of IL-8 expression in PMSCs induced a variety of senescent phenotypes, which included enlarged cell morphology, decreased cell proliferation, increased cell death, enhanced senescence-associated β-galactosidase (SA-β-gal) activity, impaired functions, and significant changes in the gene expression profile. After analyzing the expression levels or subcellular locations of p53, p21, p16, AKT, p-AKT, and FOXO3a proteins, our results suggest participation of the AKT-FOXO3a signaling pathway in initiation and maintenance of the senescence response of IL-8-silenced PMSCs. We also found that CXCR2 ligand expression was downregulated in presenescent IL-8-silenced PMSCs, but upregulated when the cells became senescent, indicating antagonistic pleiotropic regulation of the IL-8-CXCR2 pathway; for example, promoting the growth of young cells, while accelerating senescence in older cells.
Materials and Methods
Reagents
SB225002, a selective CXCR2 antagonist, was from Millipore. Protamine sulfate salt and phytohemagglutinin (PHA) were purchased from Sigma Aldrich. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was purchased from Invitrogen. Recombinant human IL-8 was from R&D Systems. Recombinant human IL-2 was from Peprotech. Anti-hCXCR1-APC, anti-hCXCR1-APC, and +antibodies were purchased from Miltenyi Biotec. Anti-hCD45-PE-Cy7, anti-hCD4-PE, and +antibodies were purchased from eBioscience. Antibodies against BCL-2, BAX, p21, p-AKT, FOXO3a, and GAPDH were purchased from Cell Signaling Technology. Puromycin dihydrochloride and p53 (DO-1) antibody were purchased from Santa Cruz Biotechnology. p16 antibody was purchased from Abcam.
Cell culture
Human tissue collection for research was approved by the Institutional Review Board of the Chinese Academy of Medical Science and Peking Union Medical College. Term placenta, was obtained from local maternity hospitals with donors' informed consents. Bone marrow aspirates, adipose tissues, and peripheral blood were obtained from healthy donors with written consents. PMSCs, umbilical cord-derived MSCs (UMSCs), adipose-derived MSCs (AMSCs), and bone BMSCs were isolated as previously described [7,12,14]. All human MSCs were maintained in complete culture medium: Dulbecco's modified Eagle's medium (DMEM): nutrient mixture F-12 (Ham) (1:1) supplemented with 10% fetal bovine serum (FBS; Gibco), 2 mM L-glutamine (Sigma Aldrich), 100 IU/mL penicillin, and 100 mg/mL streptomycin (Gibco), in 37°C humidified incubator with 5% CO2. Each tissue was from three donors.
Lentiviral shRNA transduction
Target sequence of lentiviral shRNA vectors (GeneCopoeia) were as follows: negative control shRNA: 5′-TGGCTGCATGCTATGTTGA-3′; two shRNAs targeting different regions of human IL-8 transcript (NM_000584.2): IL-8 shRNA-1, 5′-GCACGGGAGAATATACAAATA-3′: IL-8 shRNA-2, 5′-GGATCCACAAGTCCTTGTTCC-3′. The third generation replication-incompetent lentivirus was produced in HEK293T cells using the Lenti-Pac™ HIV Expression Packaging Kit (Genecopoeia) according to the manufacturer's instruction. In brief, shRNA expression plasmids and HIV-based packaging plasmids (containing genes: Gag/Pol, Rev, VSV-G) were mixed with EndoFectin Lenti reagent to form DNA-EndoFectin complex in Opti-MEM I (Invitrogen) to transfect HEK293T cells. The culture medium was replaced with fresh DMEM supplemented with 3% fetal calf serum (FCS) within 16 h post-transfection. The lentivirus-containing culture medium was collected, aliquoted, and stored at −80°C. PMSCs were transduced with lentiviral media at a multiplicity of infection of 5, in the presence of 100 μg/mL prostamine sulfate (Sigma Aldrich) for 24 h. The percentage of enhanced green fluorescent protein (EGFP) positive cells was determinded by flow cytometry analysis. Three days after transfection, stable transduced cells were selected with 2.5 μg/mL puromycin (Santa Cruz Biotechnology) for 48 h or sorted by FACS Aria Cell Sorter. Knockdown of IL-8 was confirmed by real-time reverse transcription–polymerase chain reaction (RT-PCR) and enzyme-linked immunosorbent assay (ELISA).
Cell proliferation assay by Cell Counting Kit-8
Cell Counting Kit-8 (CCK-8; Dojindo) was used to detect cell proliferation according to the manufacturer's instruction. To observe the effect of SB225002 on PMSC proliferation, PMSCs were seeded at a density of 2 × 103 cells/well in plastic 96-well plates. About 4 h later, old medium was replaced by the fresh with addition of different concentrations of SB225002 (0.055, 0.11, 0.22, 0.45, 0.9, and 1.8 μM), and the OD value was determined by microplate reader under 450 nm wavelength after 48 h incubation. To examine the growth ability of PMSCs after IL-8 silence, IL-8-silenced PMSCs and their controls (non-transfected PMSCs or control shRNA-transfected PMSCs) were seeded at a density of 1 × 103 or 2 × 103 cells/well in plastic 96-well plates. At the indicated time points, CCK-8 assay solution was added, and then the OD value per well was read after incubation for 2 h at 37°C and 5% CO2. The absorbance at wavelength 450 nm (A450) was measured every day for 7 days. Each group was performed in six parallel wells, and more than three indepedent experiments were carried out.
Flow cytometry analysis
Cell surface molecules were detected by relevant monoclonal antibodies. Briefly, cells were incubated with corresponding antibodies in 4°C for 30 min in staining buffer [phosphate-buffered saline (PBS) supplemented with 1% FBS], washed with staining buffer, and analyzed by flow cytometry (FACS Canto II; BD Biosciences). Nonspecific isotype-matched antibodies served as controls.
Cell cycle and cell death assay
After puromycin selection, cells were seeded at a density of 5 × 104 cells/well for another 4 days culture. Then, cells were collected by trypsin digestion and fixed in 70% ice-cold ethanol and maintained overnight at −20°C. DNA content was analyzed with Cell Cycle Analysis Kit (Multiscience) according to the protocol provided by the manufacturer. Briefly, after washed with PBS, cells were incubated with propidium iodide (PI) staining buffer for 30 min at room temperature, and then examined by BD LSRII (BD Pharmingen). At the same time, cell death level was examined by Annexin V/PI double staining. In short, cells were incubated with APC-Annexin V and PI in the binding buffer according to the manufacturer's protocol (BD Biosciences) for 30 min. The labeled samples were detected within 1 h by BD LSRII (BD Pharmingen). Data were analyzed by FlowJo 7.6.1 software.
Luminescent cell viability assay
To detect the anti-inflammatory ability of PMSCs after IL-8 knockdown, cells were seeded into 96-well plates at a density of 3 × 103 cells/well and treated with combined tumor necrosis factor-α (TNF-α; 50 ng/mL) and interferon-gamma (IFN-γ) (100 ng/mL) for 48 h. Cell viability was measured using the CellTiter-Glo®Luminescent Cell Viability Assay (Promega) according to manufacturer's protocol. Luminescent signal was recorded using a Synergy H4 Hybrid Microplate Reader (BioTek).
Colony forming unit-fibroblastic assay
Colony forming unit-fibroblastic (CFU-F) assay was performed as follows. Cells were counted and seeded in 3.5 cm diameter dishes at a density of 3 × 103 cells/dish. After 14 days incubation, the culture dishes were washed with PBS, fixed with 4% paraformaldehyde (PFA), and stained with 0.1% crystal violet for 30 min. A cell cluster that had more than 50 cells was counted as a colony under microscopy. The relative CFU-F number was calculated.
Adipogenic and osteogenic differentiation
PMSCs were plated in six-well plates at a density of 1 × 103 cells/cm2 for osteogenetic differentiation, and 1 × 104 cells/cm2 for adipogenic differentiation. The composition of differentiation medium was as follows: (1) adipogenic differentiation medium: Iscove's modified Dulbecco's medium (IMDM) supplemented with 10% FBS, 1 μM dexamethasone, 10 μg/mL insulin, 0.5 mM 3-isobutyl-1-methylxanthine, and 60 mM indomethacin; (2) osteogenic differentiation medium: IMDM supplemented with 10% FBS, 100 nM dexamethasone, 0.2 mM ascorbic acid 2-phosphate, and 10 mM glycerol 2-phosphate. All reagents were purchased from Sigma Aldrich. The medium was replaced twice a week. After 21 days of induction, cultures were stained with Oil red O solution or Alizarin red S for adipogenic or osteogenic differentiation, respectively. RNA was isolated after 14 days of differentiation induction. Gene markers for adipogenesis and osteogenesis were analyzed by real-time RT-PCR.
SA-β-gal staining
SA-β-gal was used as a biomarker of cellular senescence [15]. SA-β-gal activity was qualitatively assessed by a Histochemical Staining Kit (Beyotime Biotechnology) according to the manufacturer's instruction on day 9 post-transfection. After SA-β-gal activity staining, cells were counterstained with DAPI (Invitrogen) to calculate the total cell number. The percentage of senescent cells was represented by the number of blue stained cells in the total population.
Quantitative real-time PCR analysis
Total RNA was isolated from cultured cells using a TRIzol reagent (Invitrogen) according to the manufacturer's instruction, and was reverse-transcribed using TransScript First-Strand cDNA Synthesis SuperMix (TransGen Biotech China Co., Ltd.) according to the manufacturer's instruction. Real-time PCR was carried out with SYBR Green Master Mix (Applied Biosystems) using an ABI PRISM 7900 (Applied Biosystems) following manufacturer's instruction. Three independent experiments were performed for each sample. All primers used in the experiment were listed in Table 1. Relative mRNA expression was calculated using the 2−ΔΔCT method. GAPDH was used for normalization.
Determination of cytokine secretion by ELISA
Cell-free supernatants was collected and kept in refrigerator at −80°C. IL-8 and IFN-γ was tested using ELISA kits (Neobioscience Biotech Co., Ltd.) following the supplier's instruction.
Peripheral blood mononuclear cells proliferation assay
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by lymphocyte separation medium (TBD). After having stained with CFSE (Invitrogen) according to manufacturer's instruction, PBMCs were cocultured with irradiated PMSCs (30 Gy) at a ratio of 1:10 (PMSCs:PBMC), or cultured alone in RPMI1640 medium (Gibco) supplemented with 10% FCS, 100 IU/mL penicillin, 100 mg/mL streptomycin, and 10 ng/mL hIL-2 (Peprotech), with or without 5 μg/mL PHA (Sigma Aldrich) for 5 days. Then, PBMCs were collected and stained with antibodies (anti-hCD45-PE-Cy7, anti-hCD4-PE, and anti-hCD8-APC; Ebioscience) and analyzed by flow cytometry. Average number of cell divisions and the percentage of divided cells were calculated using the Flowjo 7.6.1 proliferation platform.
Western blot analysis
Cell total protein was extracted using RIPA lysis buffer (Beyotime Biotechnology) supplemented with phenylmethylsulfonyl fluoride (Invitrogen). The protein concentration was quantified by the BCA protein assay kit (Pierce). An equal amount of protein sample was separated by SDS-PAGE gel and transferred to PVDF membranes (Millipore). After blocked with 5% BSA for 2 h, the membrane was incubated with primary antibodies: anti-BCL-2 (1:1,000), anti-BAX (1:1,000), anti-GAPDH (1:1,000), anti-p53 (1:200), anti-p21 (1:1,000), anti-p16 (1:500), anti-AKT (1:1,000), anti-p-AKT (1:1,000), and anti-FOXO3a (1:500) overnight at 4°C. The membrane was washed and incubated with horseradish peroxidase-conjugated secondary antibody (Pierce) at 1:2,000 dilution. Protein bands were visualized by enhanced chemiluminescent (ECL; Thermo Scientific) detection reagents.
Laser confocal staining
On day 9 post-transfection, cells were fixed with 4% PFA and stained with rhodamine phalloidin to reveal actin filament (red) and counterstained with DAPI to show cell nucleus.
For p53, p21, and p16 protein staining, cells was permeabilized by Triton X-100 (Sigma Aldrich), and then incubated with primary antibodies overnight at 4°C. After washing, cells were incubated with relevant second antibody for 1 h at room temperature. Then, cells were counterstained with DAPI before observation under laser confocal microscope.
Transcriptome analysis
On day 6 post-transfection, control or IL-8 lentiviral shRNA stably transfected PMSCs were replated in 10-cm diameter cell culture plastic dishes at a density of 3 × 103/cm2 for another 4 days culture. Three pairs of samples were from three independent transfection experiments. Total RNA was extracted by Qiagen miRNeasy Micro Kit (Qiagen), and were qualified and quantified by agarose gel electrophoresis and ultraviolet spectrometry. Then, sense strand cDNAs were generated using the Ambion® WT Expression Kit (Ambion), and were fragmented and biotin-labeled using the AffymetrixGeneChip® WT Terminal Labeling Kit (PN 900671; Affymetrix). According to the protocol provided by Affymetrix, biotin-labeled cDNAs were hybridized with probes on AffymetrixGeneChip Human Transcriptome Array 2.0 that covers more than 245,000 coding transcripts, which were followed by washing and staining with R-Phycoerythrin Streptavidin (Molecular Probes; P/N S-866) using the Fluidics Station 450. After washing, the probe arrays were scanned on the Affymetrix® GeneChip® Scanner 3000. Probe cell intensity data (CEL) of different microarrays were normalized and log2 scale transformed using the RMA measure in Expression Console Software (Affymetrix). The differentially expressed genes were selected if they had fold change >1.2 at a P < 0.05 level. Gene Ontology (GO) and pathway enrichment analysis were performed using GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Statistical analysis
All data are presented as mean ± standard deviation. GraphPad Prism 5 was used for the statistical analysis. The Student's t-test was used for two group comparisons. Differences are considered significant when P < 0.05.
Results
High expression of IL-8 and its receptor (CXCR2) in PMSCs
To establish the expression status of IL-8 and its receptors CXCR1 and CXCR2 for further study, we isolated and cultured MSCs from four different types of human tissue: placental tissue, umbilical cord tissue, bone marrow aspirate, and adipose tissue. Each type of tissue was collected from three different donors. All MSCs (passage 3) displayed a typical fibroblast-like morphology under an inverted phase contrast microscope (Fig. 1A), and expressed MSC-related cell surface antigens when analyzed by flow cytometry (data not shown). Expression of IL-8 mRNA and secreted protein was detected by real-time RT-PCR and ELISA methods, respectively. IL-8 was widely expressed in all types of MSCs, and its concentration in culture supernatants ranged from 14.02 ± 0.06 ng/mL to 38.70 ± 2.45 ng/mL (Fig. 1B). The levels of CXCR1 and CXCR2 expression in MSCs remain unknown because conflicting results have been obtained from different studies [16 –19]. It is well known that IL-8 stimulates cellular responses mainly by binding and activating its cell membrane receptors (CXCR1 or CXCR2) [20]. Therefore, CXCR1 and CXCR2 proteins expressed on the surface of MSCs were measured by flow cytometric analysis after being labeled with APC-conjugated monoclonal antibodies. A subpopulation of peripheral blood neutrophils known to express both CXCR1 and CXCR2 served as a positive control (Fig. 1C) [21], and also confirmed the specificity and efficacy of the monoclonal antibodies. Based on fluorescence intensity, we confirmed that CXCR2, but not CXCR1, was expressed on MSCs from the four different types of tissue (Fig. 1C). Additionally, CXCR2 receptor activity was evaluated by treating the PMSCs with different concentrations of SB225002 (a selective CXCR2-specific antagonist). As shown in Fig. 1D, SB225002 decreased PMSC proliferation in a dose-dependent manner. In addition to IL-8, other CXCR2 ligands, including CXCL1, 2, 3, 5, 6, and 7 may also bind CXCR2 and activate downstream signaling pathways [20]. To expand our knowledge of the CXCR2 signaling pathway, we examined the expression status of all the CXCR2 ligands mentioned above. Interestingly, a RT-PCR analysis showed that all of the ligands were expressed in PMSCs (Fig. 1F); furthermore, the ligands were visualized following agarose gel electrophoresis (Fig. 1E). Taken together, our data showed that chemokine IL-8 was ubiquitously and highly expressed in MSCs of different origins. However, the importance of IL-8 for regulating activity of the CXCR2 signaling pathway remains to be clarified.

IL-8 and its receptor CXCR2 were highly expressed in placenta and other tissue-derived MSCs.
Stable knockdown of IL-8 in PMSCs by lentivirus-mediated RNA interference retarded their proliferation
To investigate the autocrine effect of IL-8 on PMSCs, we employed lentivirus-mediated RNA interference to generate PMSCs with persistent IL-8 downregulation. Two lentiviral shRNAs targeting different sequences of IL-8 transcripts (IL-8 shRNA-1 and IL-8 shRNA-2), and a scrambled shRNA that served as a negative control were constructed for use in all the following experiments to minimize any siRNA off-target effects. All lentiviral vectors co-expressed EGFPs as reporter markers and puromycin resistance genes as selection markers (Fig. 2A). Three days after transfection, the PMSCs were subjected to 48 h of puromycin selection to identify stably transfected cells. Selected cells were observed under a fluorescence microscope (Fig. 2B), and a cell population that was >90% GFP-positive was used in the following experiments (Fig. 2C). Knockdown efficiency was determined by real-time RT-PCR and ELISA (2 weeks after transfection). Both IL-8 shRNAs significantly reduced IL-8 expression at both the mRNA and protein level (Fig. 2D).
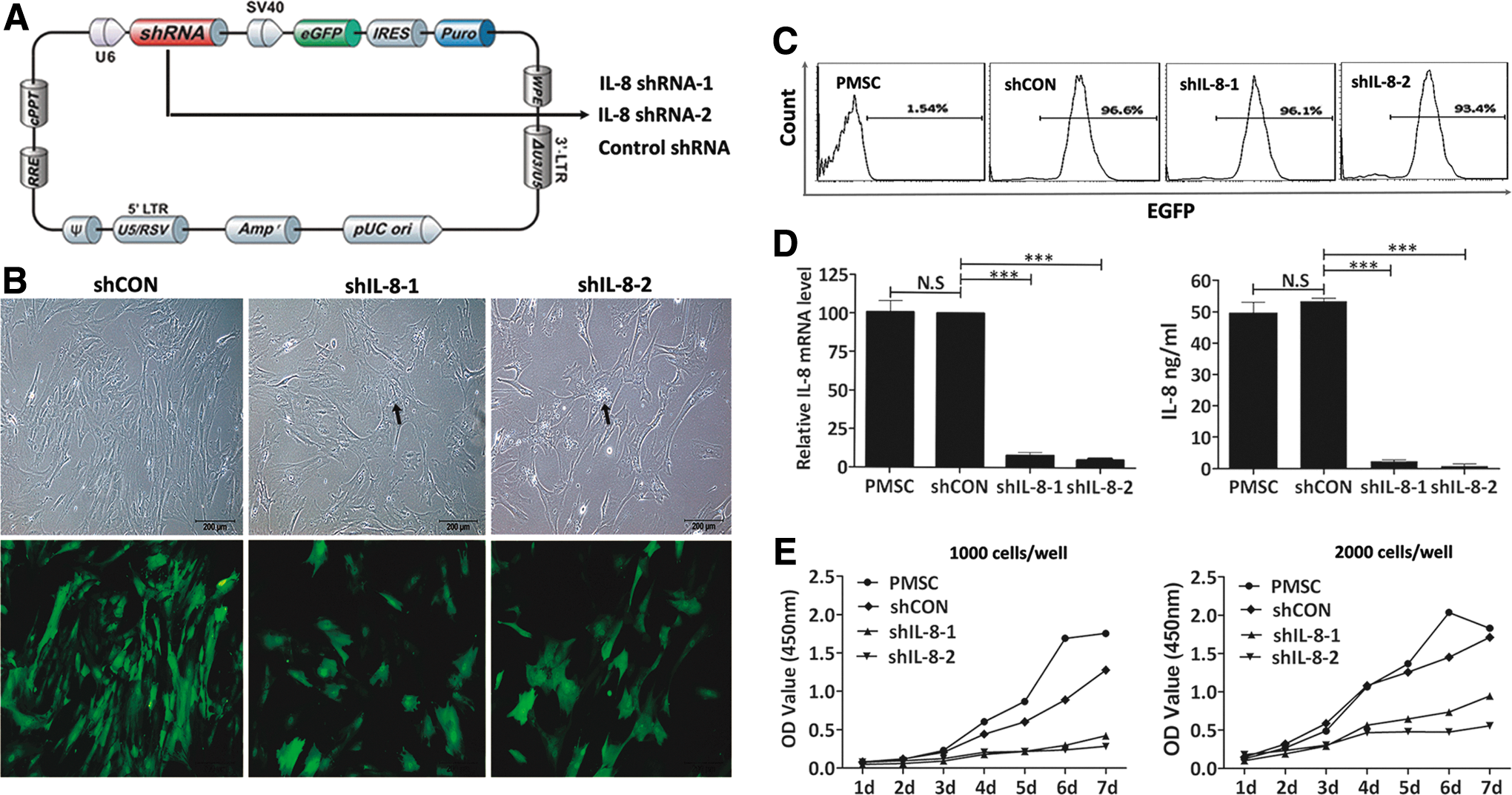
Stable knockdown of IL-8 expression by lentiviral shRNAs retarded PMSCs proliferation.
After puromycin selection, IL-8 shRNA-transfected PMSCs or their controls were reseeded at the same density in T75 culture flasks. During the following 4 days of culture, obvious morphological changes and growth retardation were observed in the IL-8-silenced PMSCs. They became hypertrophic, flattened, and heterogeneous in morphology when compared with the control cells (Fig. 2B). Furthermore, larger numbers of granules accumulated in the cytoplasm of the IL-8-silenced PMSCs (Fig. 2B), and most of the enlarged and flattened PMSCs did not adhere to the plastic culture flask after being trypsinized. After 4 days of culture as described above, the control cells reached near complete confluence, while the IL-8-silenced PMSCs reached <50% confluence (Fig. 2B), suggesting that IL-8 knockdown resulted in reduced cell proliferation. To confirm this result, cell growth curves were generated using a cell counting kit-8 assay. As shown in Fig. 2E, when grown under high-density culture conditions (2,000 cells/well), there was little difference between the growth rates of the control shRNA-transfected and nontransfected PMSCs. However, the IL-8-silenced PMSCs showed significant growth retardation, and that feature was even more prominent for cells grown in low-density cultures (1,000 cells/well).
Knockdown of IL-8 arrested PMSCs cell cycle progression and increased cell death
To clarify the cause of retarded cell growth among IL-8-silenced PMSCs, their cell cycle phase distribution was analyzed by PI staining as described in the Materials and Methods section. A flow cytometric analysis indicated that the cellular G2/M phase ratio was significantly increased (PMSC, 10.77% ± 0.39%; shCON, 13.23% ± 0.86%; shIL-8-1, 29.33% ± 0.57%; Fig. 3A), indicating that the G2-M phase was prolonged in the IL-8-silenced PMSCs when compared with their controls. We then evaluated the percentages of dead cells by Annexin V/PI double staining. As described in previous reports, Annexin V recognizes externalized phosphatidylserine, which is a suitable marker for cell death resulting from apoptosis, necrosis, or autophagy [22,23]. The percentage of Annexin V-positive cells was significantly higher among the IL-8-silenced PMSCs (PMSC, 5.14% ± 1.50%; shCON, 5.78% ± 1.81%; shIL-8-1, 10.44% ± 1.58%; shIL-8-2, 21.11% ± 2.08%; Fig. 3B), suggesting that cell death among PMSCs had increased after downregulation of IL-8. To gain further insight into the underlying mechanisms for this result, we performed western blot studies to examine the expression levels of relevant proteins involved in cell survival and apoptosis. We found that BCL-2 protein expression was reduced after IL-8 knockdown, while BAX protein expression had not increased (Fig. 3C). Additionally, real-time RT-PCR revealed that the expression of cell proliferation-related genes such as CCNA2, CCNB2, Ki-67, and PCNA, was significantly reduced in IL-8-silenced PMSCs (Fig. 3F).
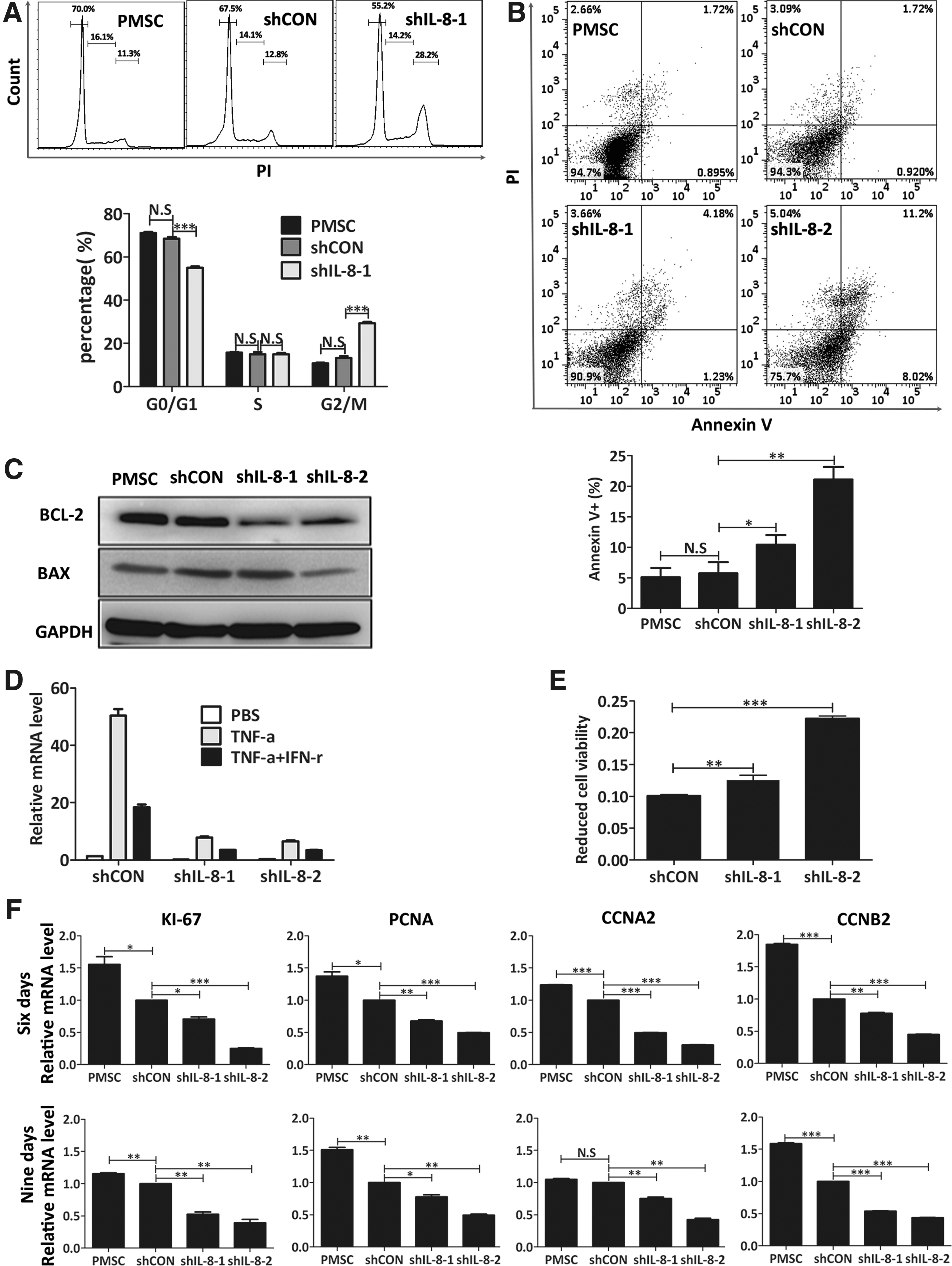
Knockdown of IL-8 arrested PMSCs cell cycle progress and increased cell death.
We also examined cell viability in an inflammatory environment, which was modeled by adding of 50 ng/mL TNF-α and 100 ng/mL IFN-γ into the cell culture medium. IL-8 mRNA expression induced by TNF-α alone or in combination with IFN-γ was significantly inhibited in the IL-8-silenced PMSCs (Fig. 3D). IL-8-silenced PMSCs were more sensitive to combined TNF-α and IFN-γ-induced cell death, whose mechanism was elucidated by previous studies performed in our laboratory [24], and verified by a significant reduction in cell viability (Fig. 3E).
Downregulation of IL-8 accelerated PMSCs senescence
Cytoskeleton staining with rhodamine phalloidin revealed that actin filaments were excessively expressed and irregularly arrayed in IL-8-silenced PMSCs when compared with their controls, whose fibers were arranged in bundles. The increased sizes of the IL-8-silenced PMSCs were quantified by measuring the area of the cell skeleton (Fig. 4A), and forward scatter intensity was assayed by flow cytometry (Fig. 4B). Based on their morphologic changes, reduced proliferation capacity, and increased numbers of dead cells, we speculated that the IL-8-silenced PMSCs may have undergone a senescence process. To confirm this hypothesis, we used a cytochemical method to detect SA-β-gal activity, which is a widely used marker of cellular senescence [25]. The enlarged and flattened PMSCs stained blue, indicating they have become senescent. The percentage of SA-β-gal-positive PMSCs was significantly increased on day 9 after IL-8 knockdown (PMSC, 2.13% ± 0.18%; shCON, 3.95% ± 0.63%; shIL-8-1, 54.46% ± 16.61%; shIL-8-2, 48.25% ± 16.55%; Fig. 4C). Collectively, IL-8 downregulation reduced the growth and survival of PMSCs, and ultimately led to their senescence, which was indicated by a characteristic morphologic change, permanent cell cycle arrest, and accelerated termination of viability. These findings revealed that IL-8 plays a prosurvival role in proliferating PMSCs, by exerting an autocrine effect.
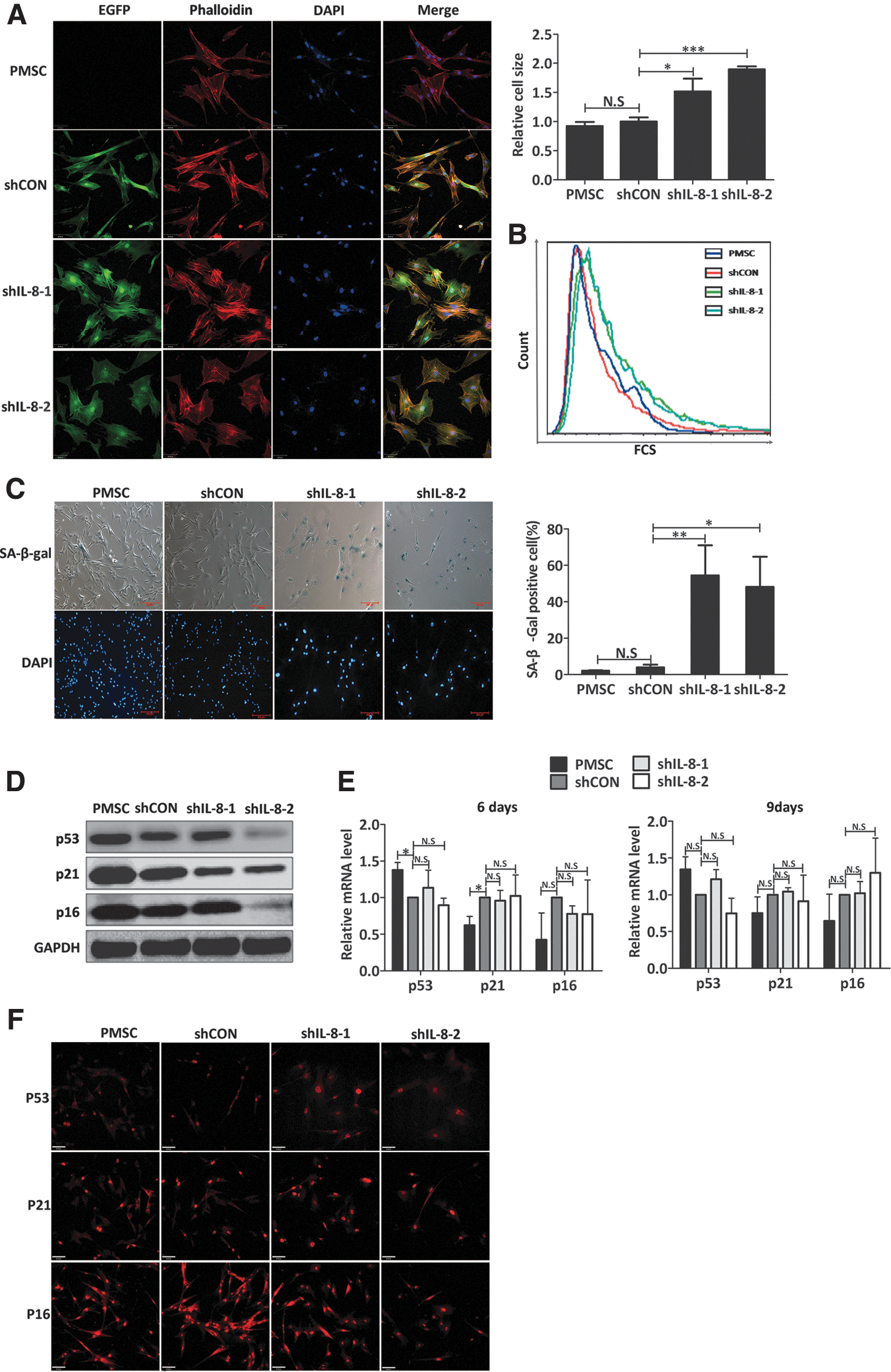
Knockdown of IL-8 induced premature senescence of PMSCs.
In most cases, cell senescence is established and maintained by the p53/p21 and p16 pathways [26]. As a transcription factor, p53 regulates numerous downstream genes involved in cell cycle checkpoints, apoptosis, and senescence. Expression of p21 is directly regulated by p53 protein, and mediates p53-induced cell senescence [27]. As a G1 phase regulator, p21 accumulates in the cell nucleus and inhibits the activity of cyclin-cdk complexes [28]. To determine whether cellular senescence induced by IL-8 downregulation was mediated by classical effectors (p53, p21, or p16), we examined the levels of p53, p21, and p16 protein expression in senescent IL-8-silenced PMSCs (day 9 post-transfection). Our data showed that the levels of p53, p21, and p16 proteins were not increased in IL-8-silenced PMSCs when compared with their controls; but rather decreased in some experiments (Fig. 4D). In addition, real-time RT-PCR showed that p53, p21, and p16 mRNA levels had not significantly changed following IL-8 downregulation at day 6 or 9 post-transfection (Fig. 4E). Furthermore, immunofluorescence staining results showed that the subcellular locations of p53, p21, and p16 had also remained unchanged (Fig. 4F). In conclusion, further studies must be conducted to determine whether the p53/p21 and p16 signaling pathways participate in the initiation and maintenance of IL-8 silence-induced cellular senescence.
Self-renewal and adipogenic and osteogenic capacities were attenuated in IL-8-silenced PMSCs
Recently, MSCs have become widely used in cell-based regenerative therapies because of their capabilities for self-renewal and multi-differentiation [29,30]. To evaluate the response of stem cells to IL-8 knockdown, the CFU-F formation and adipogenic and osteogenic potentials of IL-8-depleted PMSCs were analyzed and compared with those of shCON-PMSCs. shCON-PMSCs had a reduced clonogenic potential when compared with nontransfected PMSCs (Fig. 5A), suggesting that the process of lentivirus transfection and puromycin selection had damaged the PMSCs. However, two IL-8 shRNAs-transfected PMSCs had nearly lost their CFU-F capacities when compared with the shCON-PMSCs (Fig. 5A). Similar results were found in two IL-8 shRNAs-transduced PMSCs, suggesting that lentiviral IL-8 shRNA-mediated knockdown was specific.
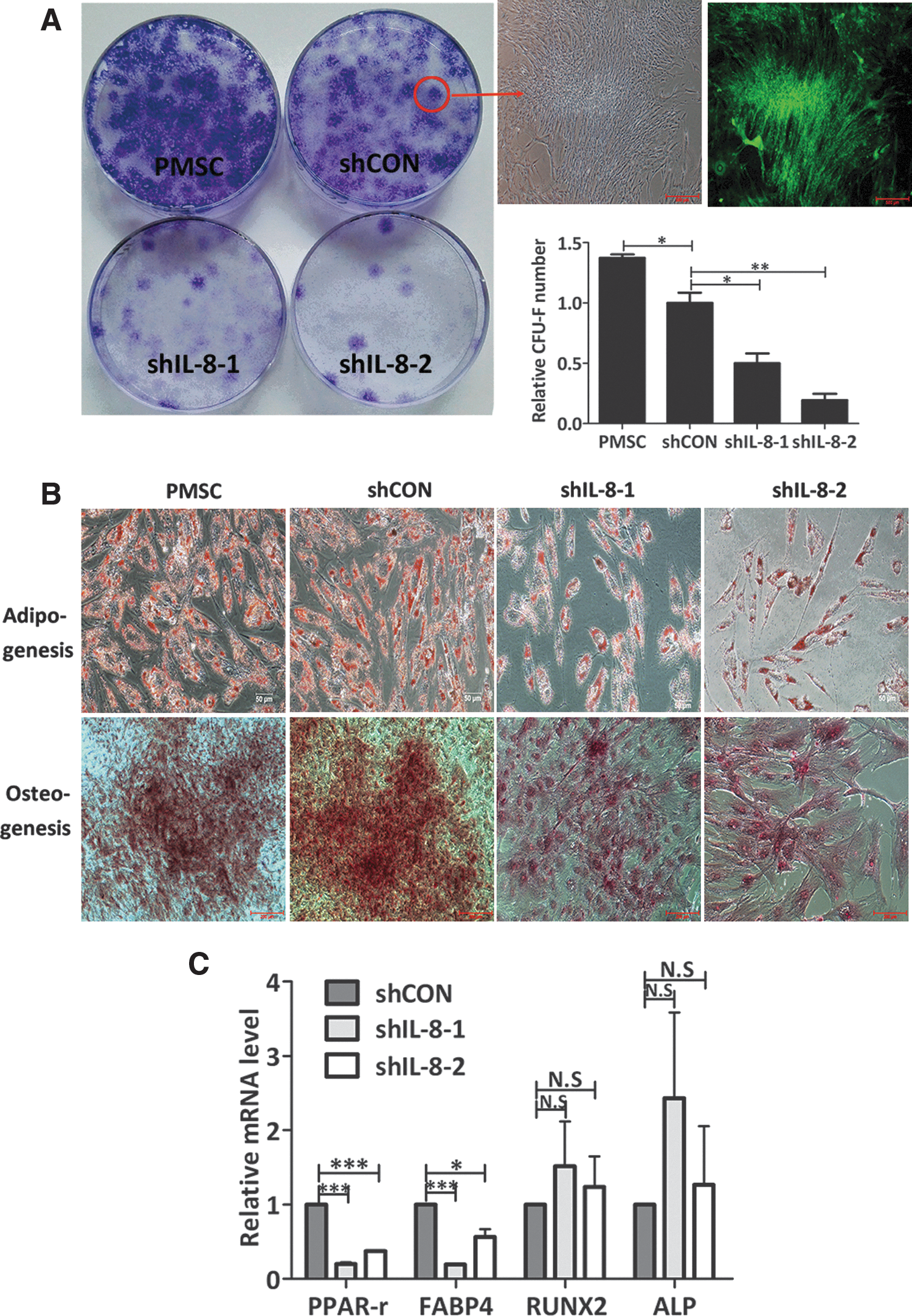
Knockdown of IL-8 expression impaired the self-renewal, adipogenic and osteogenic capacities of PMSCs.
When studying in vitro adipogenesis, more adipocyte-like cells were generated from nontransfected and control shRNA-transfected PMSCs than from IL-8-silenced PMSCs. Moreover, the nontransfected and control shRNA-transfected PMSCs were more homogeneous in shape than the IL-8-silenced PMSCs, indicating that the enlarged senescent shIL-8-PMSCs were unable to differentiate into adipocytes. Oil droplets accumulation in the cytoplasm of all PMSCs was observed by Oil red O staining performed after 21 days of adipogenic induction (Fig. 5B). Because it is difficult to compare the adipogenic capacities of different groups of cells based on the number of oil droplets, we quantified these differences by measuring the mRNA levels of adipogenic markers PPARγ and FABP4 by real-time RT-PCR analysis on day 14 of adipogenic induction. We found that although both PPARγ and FABP4 mRNA expression in all groups had increased during the process of adipogenic differentiation, those expression levels still remained lower in the IL-8-silenced PMSCs than in the control PMSCs (Fig. 5C).
When studying in vitro osteogenesis, an evident morphological change from a spindle-like shape to a cuboidal shape was observed in most of the nontransfected and control shRNA-transfected PMSCs after several days induction in osteogenic medium; however, this change was not observed in the IL-8 silenced -PMSCs. An Alizarin red staining assay showed that after 21 days induction, fewer mineralized matrix nodules had formed in the IL-8-silenced PMSCs than in their controls (Fig. 5B). However, a mineralized matrix was seen in a single PMSC, indicating that a decreased proliferative capacity of osteoprogenitor cells, rather than their decreased function, led to impaired osteogenic potential. Unchanged expression of RUNX2 and ALP mRNA may account for the results mentioned above (Fig. 5C).
In summary, downregulation of IL-8 expression in human PMSCs induced a senescence-associated phenotype and attenuated the stemness characteristics of self-renewal and differentiation capacities. Furthermore, our results are in line with previous reports suggesting that aged MSCs not only manifest a reduced proliferative capacity, but also a reduced differentiation potential [31].
Immunosuppressive function was impaired in IL-8-silenced PMSCs
The low immunogenicity and immunomodulatory capacities of MSCs make them an ideal stem cell type for use in allogeneic transplants [32]. While previous studies performed by our group confirmed that MSCs derived from placental tissue possess potent immunomodulatory capabilities [12], it remained unknown whether IL-8 knockdown would affect the immunosuppressive ability of PMSCs. Therefore, we performed a CFSE-labeled PBMC proliferation assay to test the immunomodulatory function of IL-8-silenced PMSCs. As shown in Fig. 6A, without PHA stimulation, PBMCs displayed little cell division when cultured alone or with different modified PMSCs (PBMC:PMSC = 10:1), suggesting that no immunoreaction had been stimulated by the PMSCs either before or after IL-8 downregulation. When stimulated with PHA, clusters of activated PBMCs in an IL-8-silenced PMSC coculture system were seen under a light microscope; however, these clusters were almost absent in a control PMSCs (nontransfected or control shRNA-transfected PMSCs) coculture system. Although PHA-stimulated PBMC proliferation was inhibited by both IL-8-silenced PMSCs and their controls, the inhibition effect, to some extent, was attenuated for IL-8-silenced PMSCs. The percentage of divided PBMCs in the CD45, CD4, and CD8-positive subpopulation was significantly higher in the IL-8-silenced PMSC co-culture system than in the control systems. Similar results were obtained with the BrdU cell proliferation assay, which showed that coculture with IL-8-silenced PMSCs inhibited PMSC proliferation by ∼36%, while co-culture with the control PMSCs inhibited proliferation by >80% (Fig. 6C). As an important parameter of T lymphocyte proliferation and function, IFN-γ secretion in culture supernatants was assayed. High levels of IFN-γ were detected in IL-8-silenced PMSC coculture systems when compared with those levels in their control systems (Fig. 6B). All of the above data suggest that reducing the levels of IL-8 had impaired the immunosuppressive ability of PMSCs. Furthermore, we used recombinant human IL-8 to exclude any effect that reduced IL-8 secretion may have had on the impaired immunomodulatory function of PMSCs (Fig. 6D). Previous reports proposed that the immunomodulatory function of MSCs is cell dose-dependent; in which case, having too few MSCs might reduce their immunosuppressive effect but stimulate lymphocyte proliferation [33]. In line with the opinions mentioned above, we suggest that an attenuated survival ability and increased sensitivity to inflammatory factor-induced cell death may account for the impaired immunosuppressive ability of IL-8-silenced PMSCs.
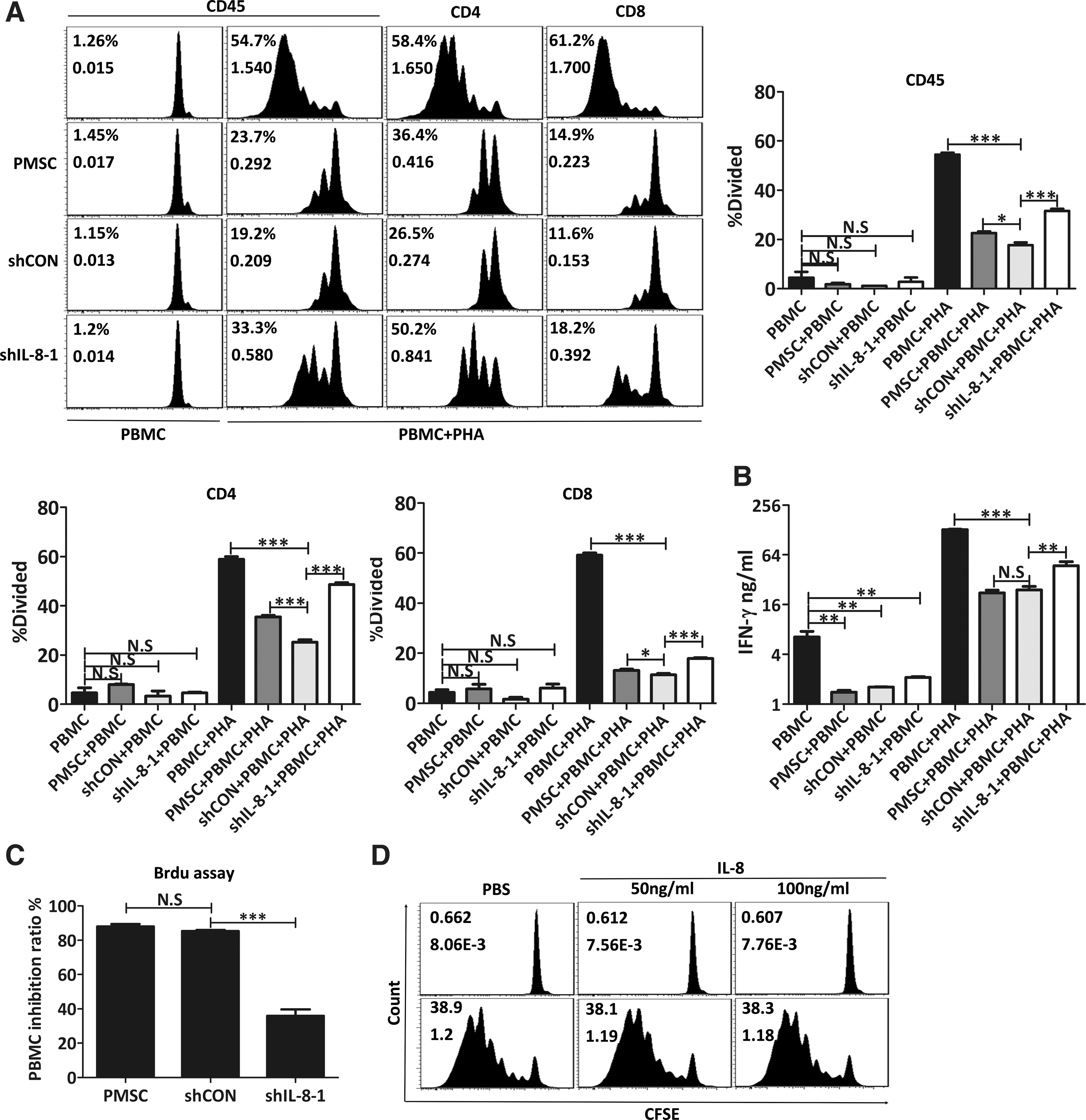
Knockdown of IL-8 impaired immunosuppressive capability of PMSCs. PMSCs, shCON-PMSCs or shIL-8-1-PMSCs were cocultured with PBMCs labeled with CFSE and stimulated with 2.5 μg/mL PHA or vehicle (PBS) for 5 days, and then proliferation responses of PBMCs were analyzed by flow cytometry according to CFSE fluorescence dilution.
Dynamic changes in expression of CXCR2 and its ligands in IL-8-silenced PMSCs
In addition to chemokine IL-8, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL7 can also bind to CXCR2 and thereby activate downstream signaling pathways regulating different cell responses, including cell growth, survival, and migration [20]. To evaluate the overall activity of the CXCR2 signaling pathway, we measured the expression levels of CXCR2 and its ligands in PMSCs after IL-8 downregulation. After 6 days post-transfection, IL-8-silenced PMSCs began to become enlarged and flattened and adopt a presenescent appearance; after which, they became senescent on day 9 post-transfection. Flow cytometric analysis of CXCR2 protein expression on the cell surface revealed no significant differences between PMSCs, shCON-PMSCs, and shIL-8-PMSCs on day 6 (Fig. 7A); however, we could not get consistent results for CXCR2 expression because of increased autofluorescence in the APC channel used to detect senescent shIL-8-PMSCs on day 9 (data not shown). Interestingly, we found that in IL-8-silenced PMSCs, the expression levels of CXCR2 ligands were time-dependent: most of the CXCR2 ligands were present at reduced levels in presenescent shIL-8 PMSCs (Fig. 7B); while their expression later increased along with the cellular senescence process (Fig. 7C). Additionally, the senescent shIL-8-PMSCs secreted higher levels of cytokines, which included IL-6, galectin-3, SDF-1α, and VEGF-α (Fig. 7D). Collectively, we speculated that downregulation of IL-8 expression in young proliferative PMSCs resulted in reduced activity of the CXCR2 signaling pathway, which subsequently led to decreased signaling for maintenance of cell growth, and induced the cellular senescence process.
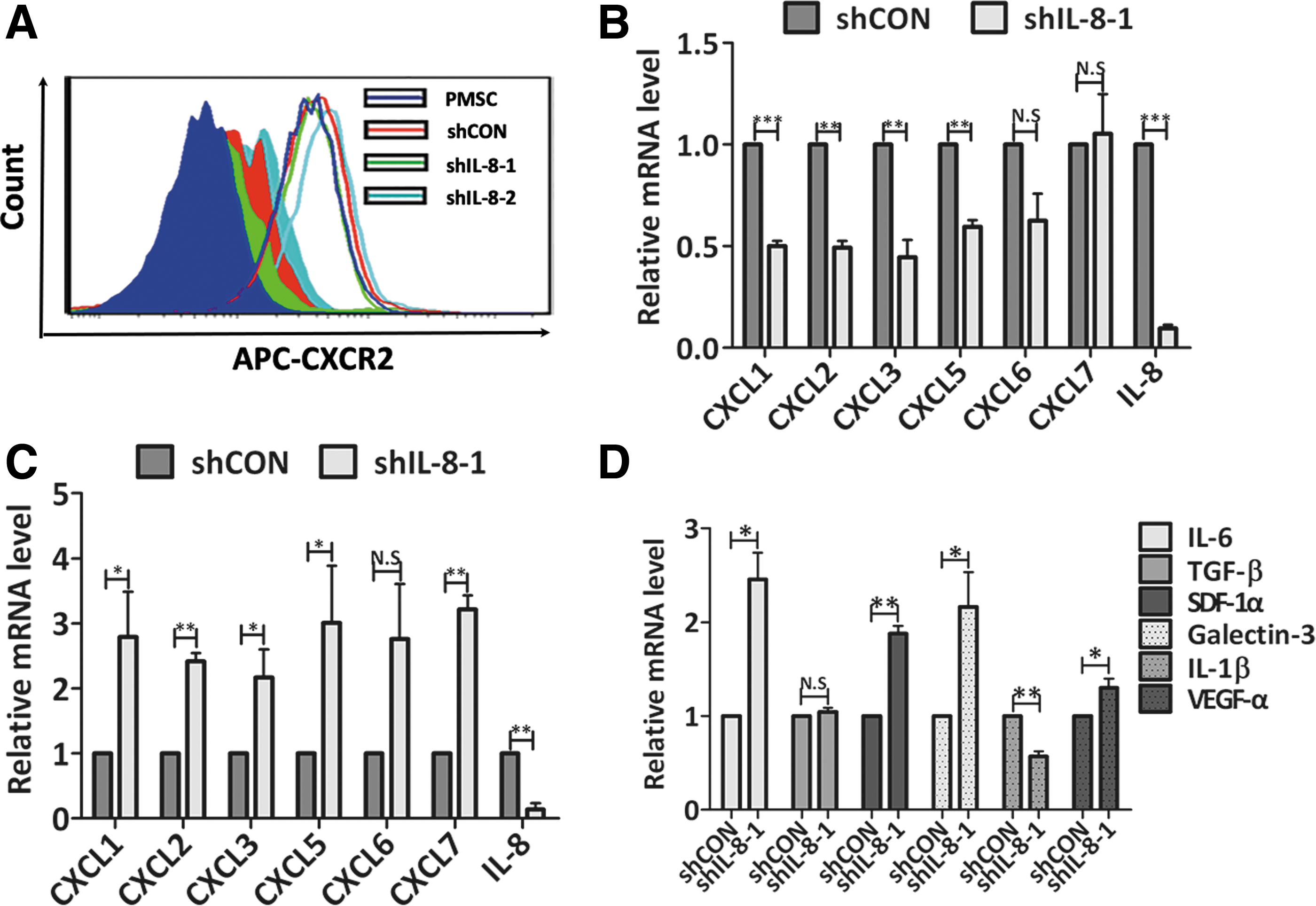
Dynamic expression of CXCLs and CXCR2 in IL-8-depleted PMSCs.
Transcriptome analysis revealed a senescence-associated gene expression profile in IL-8-silenced PMSCs
The above studies demonstrated that downregulation of IL-8 expression mainly caused premature senescence of PMSCs. To confirm the senescence phenotype caused by IL-8 silencing and gaining sights into the underlying molecular mechanisms, we employed the human transcriptome array 2.0 assay to identify differences between shIL-8-PMSCs and shCON-PMSCs regarding their global gene expression profiles. After 9 days post-transfection, when senescence-associated morphological changes could be easily observed, total mRNA was extracted and analyzed. Our analysis showed that 1,895 genes were differentially expressed with a fold change >1.2 (P < 0.05). Among these differently expressed genes, 1,153 genes were upregulated and 742 genes were downregulated in IL-8-silenced PMSCs when compared with shCON-PMSCs. A hierarchical clustering analysis of all these differentially expressed genes revealed that shCON-PMSCs and shIL-8-1-PMSCs could be easily separated into two different groups based on their systematic difference in gene expression, as visualized with a heat map (Fig. 8).
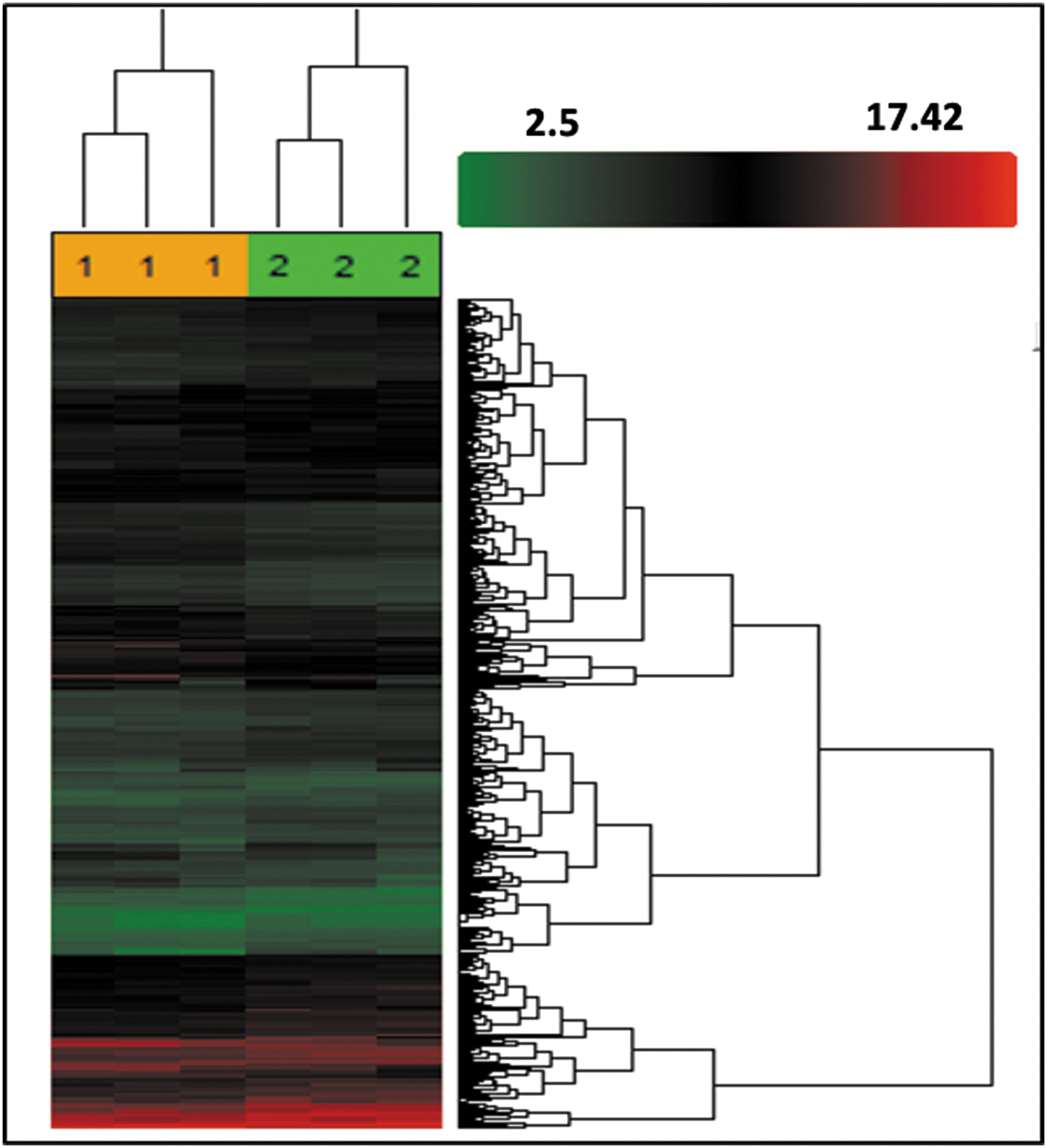
Unsupervised clustering of shCON-PMSCs and shIL-8-PMSCs based on gene expression profiling. shCON-PMSCs and shIL-8-PMSCs were classified into two clusters using differential probe sets. Expression data were depicted in a heat map, where each row represented a probe and each column represented a sample. Expression levels were depicted according to the color scale shown at the right. Red, above the median; green, below the median. The color saturation represents the magnitude of deviation from the median.
A GO analysis showed that cell growth-related biological processes such as the mitotic cell cycle and DNA replication were inhibited in IL-8-silenced PMSCs (Fig. 9B). Accordingly, we found that 33 histone-related genes and almost all mini-chromosome maintenance (MCM) complex component-related genes, which are crucial constituents controlling DAN replication, were downregulated in IL-8-silenced PMSCs (Table 3). This finding was concordant with what we observed when studying vitro cultures: IL-8-silenced PMSCs displayed almost no propagation. Notably, numerous ribosome-related transcripts were upregulated in IL-8-silenced PMSCs (Table 2). Ribosomal proteins are important components of translational machinery, and enhancement of the translation process can accelerate the senescence process [34,35]. However, it remains difficult to confirm whether overproduction of ribosomal proteins is a cause for cellular senescence, or merely a byproduct. Interestingly, transcription factors NFKB1 and CEBPB were upregulated in senescent IL-8-PMSCs (Table 2), which might explain the dynamically changed expression of chemokines (CXCLs).
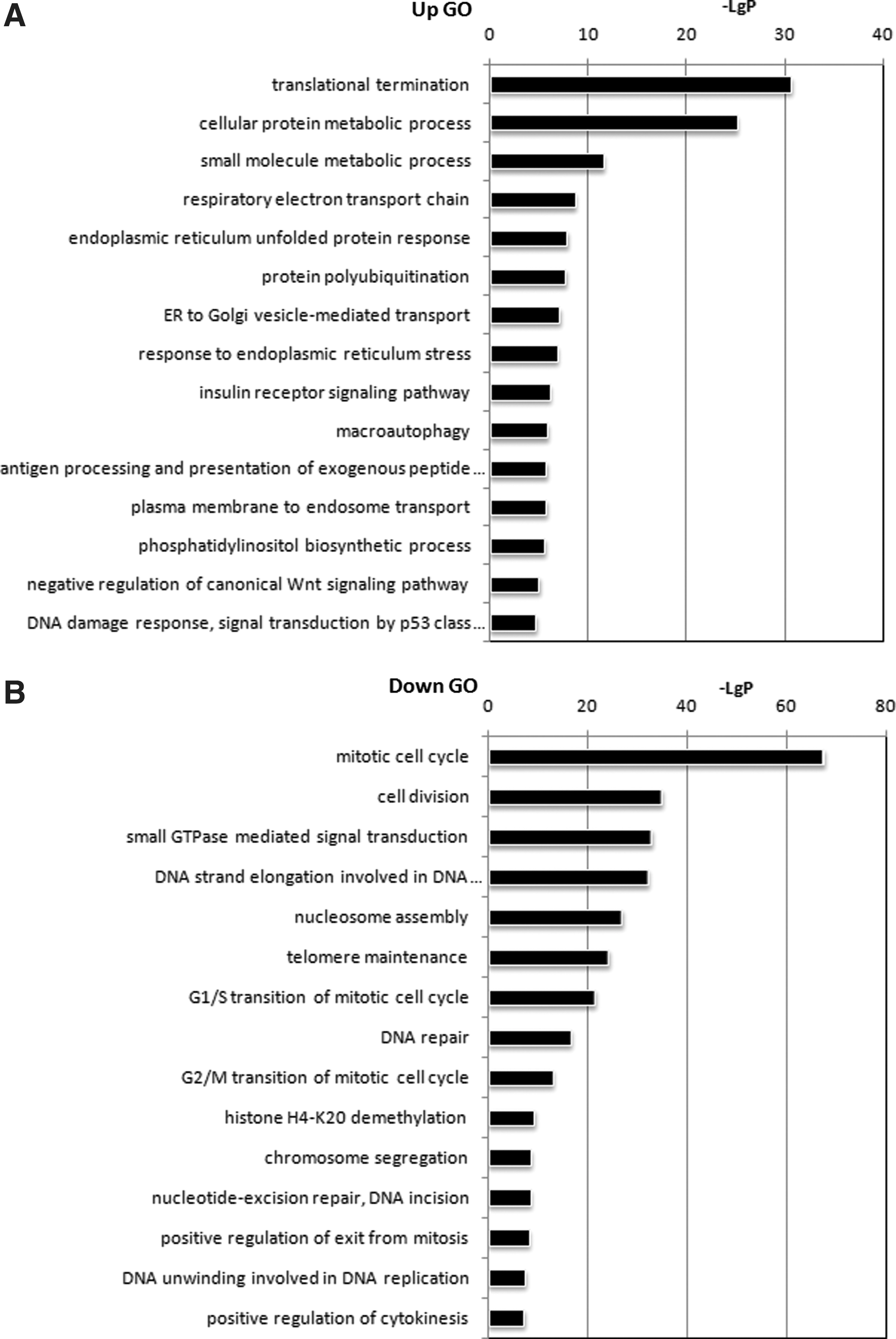
GO analysis of differential genes.
Intriguingly, our data revealed that certain biological processes such as oxidative phosphorylation, endoplasmic reticulum unfold protein responses, and protein polyubiquitination were enhanced in IL-8-silenced PMSCs (Fig. 9A). Furthermore, we found that autophagy-related genes ATG12 and ATG13 and the lysosome pathway were upregulated. It is well known that autophagy plays an important role in clearing proteins damaged by reactive oxygen species (ROS) and mediating the cellular senescence process [36,37]. Thus, autophagy might be activated by an excess of misfolded or unfolded proteins caused by ROS. A global signal transduction network analysis indicated that PIK3CA, AKT3, FOXO3, MAPK1, and NFKB1 might be the core genes that controlled changes in the whole gene expression profile of IL-8-silenced PMSCs (Fig. 10A). FOXO3a is a member of the FOXO family of forkhead transcription factors, whose expression can be repressed via the PI3K/AKT pathway. This repression has been suggested to accelerate cell aging [38,39]. Increased p-AKT expression and decreased FOXO3a protein expression in IL-8-silenced PMSCs were verified by western blot analysis (Fig. 10B). Dysfunction of the AKT-FOXO3a pathway might account for the reduced antioxidant capacity of IL-8-silenced PMSCs.
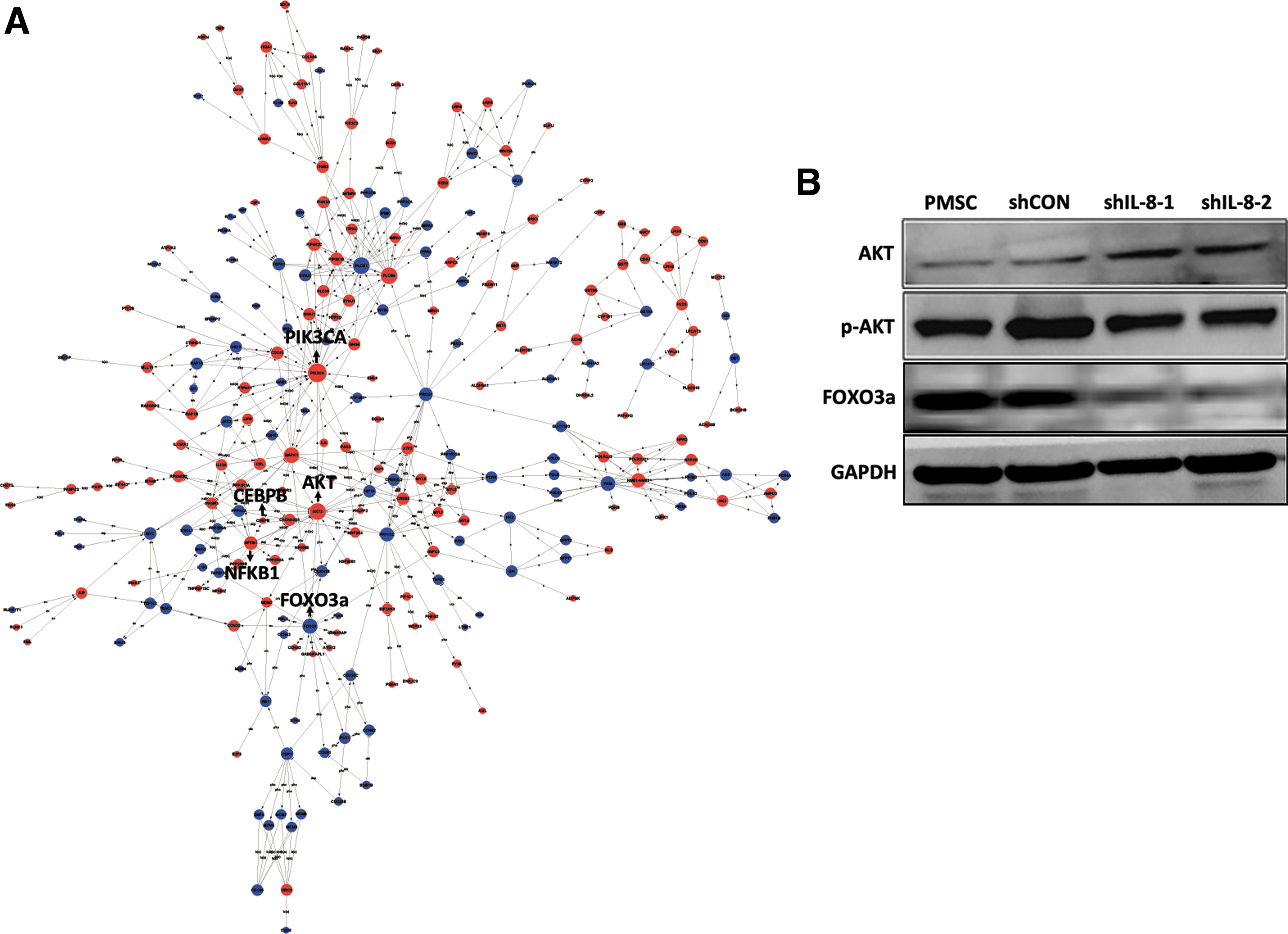
AKT-FOXO3a pathway may participate in IL-8 silence-induced premature senescence of PMSCs.
Discussion
In this study, we first verified that IL-8 was highly expressed in PMSCs, and its biological activity was mainly mediated by receptor CXCR2. We next analyzed the biological activity and functionality of PMSCs after they were transfected with lentivirus expressing IL-8 sRNA. Our results showed that IL-8 silencing conveyed no benefits to PMSCs, but rather induced their senescence, which was confirmed by reduced cell growth and CFU-F formation, enhanced SA-β-gal activity, and senescence-related transcriptome changes. Additionally, the multilineage differentiation properties and immunosuppressive capacity of the IL-8 silenced PMSCs were attenuated.
The broad biological activity of IL-8 is mediated by its binding to two highly related receptors, CXCR1 and CXCR2, both of which belong to the seven-transmembrane domain rhodopsin-like GPCR superfamily, and share 78% sequence identity [40,41]. These IL-8 receptors are differentially expressed by various cell types in different metabolic states, and their expression can be induced by several cytokines, such as TNF-α. However, conflicting results have been obtained concerning their expression at the mRNA or protein level when using different experimental methods [41 –44]. Therefore, we examined the expression of CXCR1 and CXCR2 proteins on the MSC surface by flow cytometric analysis. Our results showed that the MSCs derived from all four tissues highly expressed CXCR2, but not CXCR1, which was inconsistent with the report by Abumaree et al. [45], who showed that PMSCs expressed low levels of CXCR1 but did not express any CXCR2. The PMSCs used in our studies were obtained from passage three during culture, while the cells examined by Abumaree et al. were from passage one, and thus may have been contaminated with some other cell types, such as epithelioid cells and endothelial cells. Some other reports stated that RT-PCR and immunoassay analyses detected both CXCR1 and CXCR2 in populations of UMSCs and BMSCs [17,46]. After considering all the results above, we believe that CXCR2 protein is highly expressed on the MSC surface and apparently involved in IL-8-induced intracellular signaling, as blockage of CXCR2 by its inhibitor (SB225002) reduced cell proliferation in a concentration-dependent manner.
The murine analog of human IL-8 has not yet been identified. As a result, the functionsof IL-8 and its related signaling pathways have been mainly explored in human cells [20]. As an autocrine/paracrine growth factor, IL-8 has a mitogenic effect on normal and malignant cells [18,47,48]. Multiple intracellular signaling pathways downstream of CXCR1 and CXCR2, including the Erk1/2, PI3K, Rac, and Rho signaling pathways, might be activated by IL-8 binding, and then facilitate tumor proliferation, neovascularization, and metastasis [49]. Recently, Jung et al. [50] demonstrated that IL-8 and CXCL1 secreted by PMSCs play an important role in maintaining the pluripotency and proliferation of human pluripotent stem cells by binding to their receptor, CXCR2. In this study, our data show that knockdown of IL-8 expression reduced the proliferative ability of PMSCs and increased their sensitivity to inflammatory factor-induced cell death, further supporting the growth and survival-promoting activity of the IL-8/CXCR2 signaling pathway.
Senescence is believed to be an irreversible arrest of cell division, unless some genetic modification is introduced. Normal somatic cells have a replicative limit, even when cultured under optimal conditions; namely, when they reach the Hayflick limit or replicative senescence [51]. Cell senescence can also be induced by extrinsic stress factors such as serum deprivation, oxidative stress, DNA damage, and oncogene activation; this type of senescence is termed stress-induced premature senescence [52]. Although senescent cells no longer proliferate, they are metabolically active and capable of secreting numerous proteins, which can include inflammatory factors, growth factors, and extracellular matrix (ECM)-associated factors. This type of protein secretion is termed the SASP [53]. Chemokine IL-8 is a member of the SASP and plays an important role in senescence. CXCR2 and its ligands are known to be upregulated and reinforce senescence in near-senescent fibroblast cells. Furthermore, Medina et al. [11] showed that depletion of IL-8 expression in OECs delayed the aging process in those cells and prolonged their lifespan [11]. While all of the data mentioned above indicate a role for IL-8 in old cells that are near senescence, the role played by IL-8 in young cells remains unknown. Therefore, in this study, we used young MSCs isolated from term placentas (three to six passages) and possessing a high proliferative capacity. We discovered that depletion of IL-8 in young PMSCs caused changes typical of cellular senescence: increased cell size, decreased cell propagation, cell cycle arrest, enhanced SA-β-gal activity, decreased CFU-F numbers, impaired adipogenic and osteogenic capabilities, and attenuated immunosuppressive function. However, senescent stem cells can be easily confused with quiescent stem cells, which are reversibly arrested and capable of reentering their cell cycle after a stress factor is removed [54]. Thus, caution should be taken when attempting to identify senescent stem cells. Some reports have mentioned that in most cases, senescent MSCs display an enlarged, flattened morphology, and express high levels of SA-β-gal [55]. The Rho GTPases Rac1 and Cdc42 were found to be highly active in senescent cells, and to participate in producing senescence-related morphological changes [56]. Moreover, senescent human MSCs have impaired self-renewal and differentiation potentials [31]. Our findings are concordant with the specific signs of senescent MSCs mentioned in previous reports, and suggest that downregulation of IL-8 in young PMSCs provokes a premature senescence process.
CXCR2-binding chemokines are expressed in a manner dependent on NF-kB and C/EBPβ transcription factors, whose activity can be further enhanced by secreted chemokines, resulting in a self-amplifying secretory system [10,57]. For instance, in aged fibroblasts, NF-kB activity is elevated and induces the expression of inflammatory genes [58]. Our data showed that the mRNA levels of most CXCR2 ligands were downregulated in presenescent IL-8-silenced PMSCs, but were upregulated when the cells become aged. Therefore, we believe that the survival-promoting signal is attenuated in IL-8-silenced PMSCs, and this attenuation probably initiates the senescence process. If so, NF-kB activity was enhanced in aged IL-8-silenced PMSCs, and promoted the expression of senescence-associated chemokines, resulting in reinforced cellular senescence. In a word, the IL-8/CXCR2 pathway may play different roles in young and old cells: promoting the proliferation of young cells but accelerating the aging of old cells.
It is classically believed that the p53/p21 and p16/Rb pathways are involved in the initiation and maintenance of senescence in human cells [26]. As a transcription factor, p53 regulates numerous downstream genes involved in cell cycle checkpoints, apoptosis, and senescence. Expression of p21 protein is directly regulated by p53 protein, and mediates p53-induced cell senescence [27]. Even so, p53/p21 and p16-independent senescences have been reported under some experimental conditions [59]. Furthermore, some studies revealed that a loss of p53 expression could increase genomic instability and provoke premature aging [60]. In this study, reduced expression of p53 and p21 proteins was observed in IL-8-silenced PMSCs, thus we cannot exclude the possibility that reduced p53 and p21 protein expression may have contributed to the initiation of cellular senescence in IL-8-silenced PMSCs.
Apoptosis and senescence are two well-recognized outcomes of cellular stress responses [61,62]. Although whether cells undergo apoptosis or senescence is determined by many factors, the widely accepted fundamental determinants are stress severity and cell type predisposition: severe damage induces apoptosis, whereas mild injury induces premature senescence. When exposed to the same stress, one type cell may initiate apoptosis, while others will become senescent. In a previous study, low levels of H2O2 induced senescence in human endometrium-derived mesenchymal stem cells (MESCs), while higher levels of H2O2 induced apoptosis in a proportion of human MESCs [63]. Senescence is a collective phenotype of multiple effector mechanisms; a number of which have been identified and characterized. However, the underlying mechanisms that cause cell death following senescence still need to be identified [64]. In this study, we observed that both apoptosis and senescence cell responses occurred in IL-8-silenced PMSCs, suggesting that apoptotic cell death is the end fate of senescent cells.
Mammalian cells contain four FOXO genes: FOXO1, FOXO3, FOXO4, and FOXO6. The activity of FOXO proteins is negatively controlled by the serine/threonine kinase AKT signaling pathway. FOXO factors help regulate the expression of key detoxification enzymes such as MnSOD and catalase. Inactivation of FOXO factors leads to an intracellular accumulation of ROS, and resultant oxidative damage that leads to cell aging [39]. In this study, decreased FOXO3a protein expression and increased p-AKT levels in IL-8-silenced PMSCs were confirmed by western blot analysis. A gene transcription profile assay revealed that AKT and FOXO3a were core genes regulating the global signaling network. Therefore, we hypothesize that increased ROS levels may play a role in the senescence of IL-8-silenced PMSCs.
In conclusion, we demonstrated that IL-8 is essential for maintaining the activity and functions of proliferating PMSCs during a long-term ex vivo expansion. Persistent IL-8 downregulation in vigorously proliferating PMSCs attenuates the activity of CXCR2-related prosurvival signaling pathways, resulting in reduced cell viability and persistent cell cycle arrest, which ultimately enables premature senescence. Genes that exert their beneficial effects in young cells may have deleterious effects on aging cells; a phenomenon termed antagonistic pleiotropy [65,66]. The IL-8/CXCR2 signaling pathway in PMSCs might be an example of antagonistic pleiotropy, by promoting growth in young cells while accelerating senescence in old cells.
Footnotes
Acknowledgments
This study was supported by the Key Program of the National Natural Science Foundation of China (grant no. 81330015), CAMS initiative for innovative medicine (grant no. 2016-IM-1-017), and the National Natural Science Foundation of China (grant no. 81500098).
Author Disclosure Statement
No competing financial interests exist.