
Abstract
Chronic kidney disease (CKD) results in a delay in wound healing because of its complications such as uremia, anemia, and fluid overload. Mesenchymal stem cells (MSCs) are considered to be a candidate for wound healing because of the ability to recruit many types of cells. However, it is still unclear whether the CKD-adipose tissue-derived MSCs (CKD-AT-MSCs) have the same function in wound healing as healthy donor-derived normal AT-MSCs (nAT-MSCs). In this study, we found that uremic toxins induced elevated reactive oxygen species (ROS) expression in nAT-MSCs, resulting in the reduced expression of hypoxia-inducible factor-1α (HIF-1α) under hypoxic conditions. Consistent with the uremic-treated AT-MSCs, there was a definite imbalance of redox state and high expression of ROS in CKD-AT-MSCs isolated from early-stage CKD patients. In addition, a transplantation study clearly revealed that nAT-MSCs promoted the recruitment of inflammatory cells and recovery from ischemia in the mouse flap model, whereas CKD-AT-MSCs had defective functions and the wound healing process was delayed. Of note, the expression of prolyl hydroxylase domain 2 (PHD2) is selectively increased in CKD-AT-MSCs and its inhibition can restore the expression of HIF-1α and the wound healing function of CKD-AT-MSCs. These results indicate that more studies about the functions of MSCs from CKD patients are required before they can be applied in the clinical setting.
Introduction
C
Functional renal failure also causes delay of wound healing in CKD patients [4]. In addition to the uremic toxin accumulation, systemic anemia caused by erythropoietin deficiency, fluid overloading, and impaired angiogenesis are considered as underlying causes of the impairment of wound healing [4]. Wound healing is a complex process that requires the combined activity of growth factors and extracellular matrix proteins, and involves many cell types such as inflammatory, endothelial, epithelial, and fibroblast cells [5]. Central to this process are mesenchymal stem cells (MSCs) that serve a keystone role in recruitment of many other cells in the body to the wound site [6]. MSCs are known to secrete a mixture of growth factors and matrix proteins that are essential for wound healing [7]. Therefore, MSCs are considered to be a promising therapeutic candidate to enhance poor wound healing caused by metabolic disease [6,7].
MSCs were firstly isolated from bone marrow (BM); therefore, the characteristics of BM-derived MSCs (BM-MSCs) have been extensively studied [8]. However, many other sources of MSCs have been reported and it is shown that compared to BM, adipose tissue is an abundant source of MSCs from which the isolation of MSCs is easier, safer, and larger amounts of MSCs can be obtained [8]. Adipose tissue-derived MSCs (AT-MSCs) are reported to be more genetically and morphologically stable with a lower senescence, higher proliferative capacity, and differentiation potential in a long-term culture. In clinical application, AT-MSCs are as effective as BM-MSCs, and in some cases maybe more effective than BM-MSCs [9].
Recently, several studies have shown that transplantation of MSCs into rat models of CKD resulted in the improvement of renal function [10 –13]. Klinkhammer et al. reported that transplantation of BM-MSCs prepared from healthy rat can help to accelerate the healing of glomerular lesions whereas those from CKD rat failed to exhibit such activity [14]. Roemeling-van et al. reported that MSCs derived from adipose tissues (AT-MSCs) of human CKD patients showed no differences in differentiation capacity and stability than those from healthy donors, hinting at autologous use of these cells in CKD therapy [15]. However, Yamanaka et al. reported that adipose-derived MSCs from patients with ESKD, who have suffered long-term uremia, exhibit downregulation of hypoxic response factors and poor angiogenesis activation [16]. Given that availability of AT-MSCs are superior to other stem cell sources, functional evaluation of patient-derived AT-MSCs is important for autologous cell therapy.
Hypoxia-inducible factor-1α (HIF-1α) is a central transcription factor that regulates the expression of a number of genes related to wound healing [17]. Hypoxic stimuli result in the stabilization of HIF-1α protein, which is regulated by oxygen-dependent hydroxylase enzymes [18]. Several studies have shown that the hypoxia-induced activation of HIF-1α is attenuated in CKD-derived MSCs, although the underlying molecular mechanisms have not been fully elucidated [16,19,20]. In addition, recent studies showed that HIF-1α activation is dynamically regulated by ROS production [21 –23]. Because the accumulation of ROS is a hallmark of CKD [24,25], understanding the role of ROS in the regulation of HIF-1α is important for the use of CKD-AT-MSCs in regenerative medicine.
In this study we have isolated AT-MSCs from early stage CKD patients and examined their ability to repair tissue damage. Our study provides a tight relationship between the increase of intracellular ROS levels and the impaired activation of HIF-1α. In particular, the expression of prolyl hydroxylase domain 2 (PHD2) is selectively increased in CKD-AT-MSCs and its inhibition successfully restores HIF-1α activation and the wound healing ability in these cells. Hence, further studies related to the functions and modification of CKD-AT-MSCs would be necessary for the use in stem cell therapy.
Materials and Methods
The isolation and culture of AT-MSCs
All human cell experiments were approved by the Ethics Committee of the University of Tsukuba. Human adipose tissues were obtained from stage 2 to stage 3, nondiabetic CKD patients (n = 5, creatinine level = 1.71 ± 0.3, GFR = 45.5 ± 9.5 mL/min per 1.73 m2, male, age = 75 ± 10 years) and nondiabetic non-CKD donors (n = 5, creatinine level = 0.82 ± 0.1, GFR = 95.5 ± 5.5 mL/min per 1.73 m2, male, age = 75 ± 10 years) from the Department of Cardiovascular Surgery, University of Tsukuba Hospital, Tsukuba, Japan. AT-MSCs were isolated from adipose tissues according to a previously described method [26]. Briefly, the adipose tissues were cut into small pieces and incubated in phosphate-buffered saline (PBS) containing 0.1% collagenase (Invitrogen, CA), separated by centrifugation and resuspended in culture medium, Iscove's modified Dulbecco medium (Invitrogen), supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 2 mg/mL
FACS analysis of AT-MSCs
To purify AT-MSCs, CD45+ and CD31+ cells were eliminated from adherent cells in tissue culture dishes by fluorescence activated cell sorting (FACS) (MoFlo XDP; Beckman Coulter), as described previously [26]. The following antibodies were used for the analysis of AT-MSC markers: Fluorescein isothiocyanate (FITC)-labeled anti-HLA-ABC (BioLegend, CA), FITC-labeled anti-CD90 (BioLegend), FITC-labeled anti-CD34 (BD Biosciences, CA), phycoerythrin (PE)-labeled anti-CD13 (BioLegend), PE-labeled antti-CD166 (BD Biosciences), PE-labeled anti-CD105 (BioLegend), PE-labeled anti-CD73 (BD Biosciences), PE-labeled anti-HLA-DR (BioLegend), PE-labeled anti-CD31 (BioLegend), PE-labeled anti-CD14 (BioLegend), and allophycocyanine (APC)-labeled anti-CD45 (BD Biosciences). FITC-labeled anti-IgG1 (BD Biosciences), APC-labeled anti-IgG1 (BD Biosciences), and PE-labeled anti-IgG1 (BD Biosciences) were used as the isotype controls.
Reverse transcription–polymerase chain reaction and quantitative real-time polymerase chain reaction
AT-MSC total RNA was prepared using Sepasol-RNA I Super G (Nacalai, Japan) according to the manufacturer's instructions. One microgram of total RNA was reverse transcribed to cDNA using a reverse transcriptase (RT)-polymerase chain reaction (PCR) kit (Toyobo, Japan). The resulting cDNAs were analyzed using a GeneAmp PCR System (Life Technologies, CA) with 25–35 cycles of denaturation at 95°C for 5 s followed by anneal and extension at 68°C for 30 s and then performed a fluorescence read step. The reaction mixtures for the quantitative PCR were prepared using SYBRGreen Realtime PCR mastermix (Toyobo) and analyzed using a GeneAmp 7500 Fast Realtime PCR System (Life technologies). The sequences of primers used for the PCRs are listed in Table 1. The experiments were performed in triplicate and data were calculated by the double delta cycle number of threshold (ddCt) method.
HIF-1α, hypoxia-inducible factor-1α.
Western blotting
Nuclear protein was extracted according to the manufacturer's protocol (ThermoFisher Scientific, MA). The extracted protein was then mixed with sodium dodecyl sulfate (SDS) loading buffer (Wako) and heated at 95°C for 3 min. Equal amounts of protein were separated through SDS–polyacrylamide gels and transferred onto polyvinylidene fluoride (PVDF) membranes (EMD Millipore, Germany) for western blotting. The membranes were immunoblotted with primary antibodies, including rabbit anti-HIF-1α (Novus Biologicals, CO), goat anti-Superoxide dimutase-1 (SOD1; R&D System, MN), goat anti-Glutathione peroxidase-1 (GPX1; R&D System), goat anti-Catalase (R&D System), and rabbit anti-PHD2 (Cell Signaling Technology, MA). Goat anti-Lamin B antibody (Santa Cruz Biotechnology, CA) was used to monitor protein loading and transfer. Horseradish peroxidase (HRP)-conjugated rabbit anti-goat IgG (Invitrogen), and HRP-conjugated goat anti-rabbit IgG (Invitrogen) were used as secondary antibodies. The signals were detected by incubating the membrane with an enhanced chemiluminescence HRP substrate (EMD Millipore) for 1 min and visualized using an Image Quant LAS 4000 System (GE Healthcare Life Sciences, United Kingdom).
Measurement of the intracellular ROS level
The intracellular ROS level was measured using 2′,7′-dichlorodihydrofluorescein diacetate (H2-DCFDA; Invitrogen) according to the manufacturer's instructions. Briefly, after reaching 80% confluency, the cells were washed with PBS and incubated with PBS containing 10 μM DCFDA at 37°C for 30 min. The fluorescence intensity was analyzed by absorbance at 495 nm and 525 nm wavelengths using a photoemission spectrophotometer (Corona Electric, Japan).
The skin flap mouse model
Male C57BL/6 mice were purchased from Charles River Japan, Inc. (Japan), given food and water ad libitum, and maintained in a 12-h-light/dark cycle in the Animal Research Center of the University of Tsukuba. All animal experiments were approved by the Animal Care Committee of the University of Tsukuba.
The skin flap mouse models were performed as described previously [27]. Male 10-week-old C57BL/6 mice were anesthetized and the skin on the dorsal surface was cut into a peninsular-shaped incision (3 × 2 cm) to generate an ischemic gradient by blood flow restriction. The mice were divided into four groups: no operation (n = 5); sham injection with PBS (n = 5); healthy donor normal adipose tissue-derived MSCs (nAT-MSCs) injection (n = 15); and CKD-AT-MSCs injection (n = 15). To detect the existence of AT-MSCs at the wound site, both nAT-MSCs and CKD-AT-MSCs were labeled with PKH26 (Sigma-Aldrich, MO) according to the manufacturer's instructions. On the first day after the surgical process, a total of 5 × 105 cells were intramuscularly injected into four divided sites of the flap skin at 1.25 × 105 cells/50 μL/site. The wound healing of the flap tissues was analyzed at two time points (3- and 7-day post-transplantation) based on two criteria (the inflammatory effect after 3 days of transplantation and the necrotic area after 7 days of transplantation.) After 3 days, mice were sacrificed, the flap tissues were sectioned and stained with inflammatory cell markers to analyze the inflammatory activity. After 7 days, images of the ischemic flaps were captured and the necrotic areas were analyzed using Image J software (NIH, MD). The flap tissues then were sectioned and stained with endothelial markers.
Histological analysis
The frozen flap tissue sections were mounted, stained with hematoxylin and eosin (Wako), and observed under a microscope (Olympus, Japan). The inflammatory cells recruited in the ischemic area were visualized by immunohistochemical staining with PE-labeled anti-CD45, PE-labeled anti-Mac1, and PE-labeled anti-31 (BD Pharmigen, CA). The numbers of positive cells were counted in 10 fields and the average was calculated.
Establishment of PHD2 knockdown MSCs
OmicsLink shRNA Expression Clone targeted to PHD2 (GeneCopoeia, United States) were transfected into HEK293T cells using the Lenti-Pac HIV Expression Packaging Kit (GeneCopoeia). Then, harvested viral particles were infected to target MSCs and selection of the infected cells was performed with puromycin (Wako, Japan). The PHD2 expression was confirmed by quantitative realtime PCR (qRT-PCR).
Statistical analysis
The data were analyzed by ANOVA. SPSS software (IBM Corp., NY) was used to perform the statistical analyses. P values of <0.05 were considered significant. The data are presented as the mean ± standard deviation (SD).
Results
A temporary treatment of uremic toxins causes the reduced expression of antioxidant-related genes and ROS accumulation in AT-MSCs
It is reported that uremic toxins such as p-cresol (an end-product of protein metabolism and greatly increased in CKD) increase the expression of intracellular ROS in many types of cells [28,29]. First, we treated the AT-MSCs from non-CKD donors (nAT-MSCs) with p-cresol and evaluated the intracellular ROS level. We found that treatment with p-cresol at low concentration (20 μM) for short periods (24 h) had no effect on cell viability and morphology under normoxic and hypoxic conditions (Fig. 1A). The intracellular ROS levels were examined by using CM-H2-DCFDA, a ROS indicator (Fig. 1B). We found that the intracellular ROS level was greatly increased in the presence of p-cresol under both normoxic and hypoxic conditions (2.3-fold increase under normoxic conditions; 3-fold increase under hypoxic conditions, n = 3, P < 0.05). MSCs are known to constitutively express high levels of antioxidant enzymes, such as SOD1, GPX1, and catalase, which can regulate the balance of intracellular ROS [30]. As expected, the treatment with p-cresol resulted in a significant reduction of the mRNA and protein expression levels of antioxidant enzyme genes in nAT-MSCs under both normoxic and hypoxic conditions (SOD1 expression: 3.4-fold decrease, GPX1: 5.6-fold decrease, catalase: 3.1-fold decrease in the presence of p-cresol under normoxic conditions, n = 3, P < 0.05) (Fig. 1C, D). Furthermore, the intracellular ROS accumulation was also observed in nAT-MSCs treated with other uremic toxins, hippuric acid (HA), and indoxyl sulfate (IS) (Fig. 1E). To examine whether the cells were able to reduce ROS levels following 24-h period of p-cresol treatment, the nAT-MSCs were continued to culture in the absence of p-cresol and passaged for several times. We found that mRNA levels of antioxidant enzyme genes were restored and the elevated ROS levels were decreased to baseline after six passages of cells (Fig. 1F, G). Collectively, these results indicate that uremic toxins attenuate the expression of antioxidant enzyme genes, which causes ROS accumulation in nAT-MSCs.
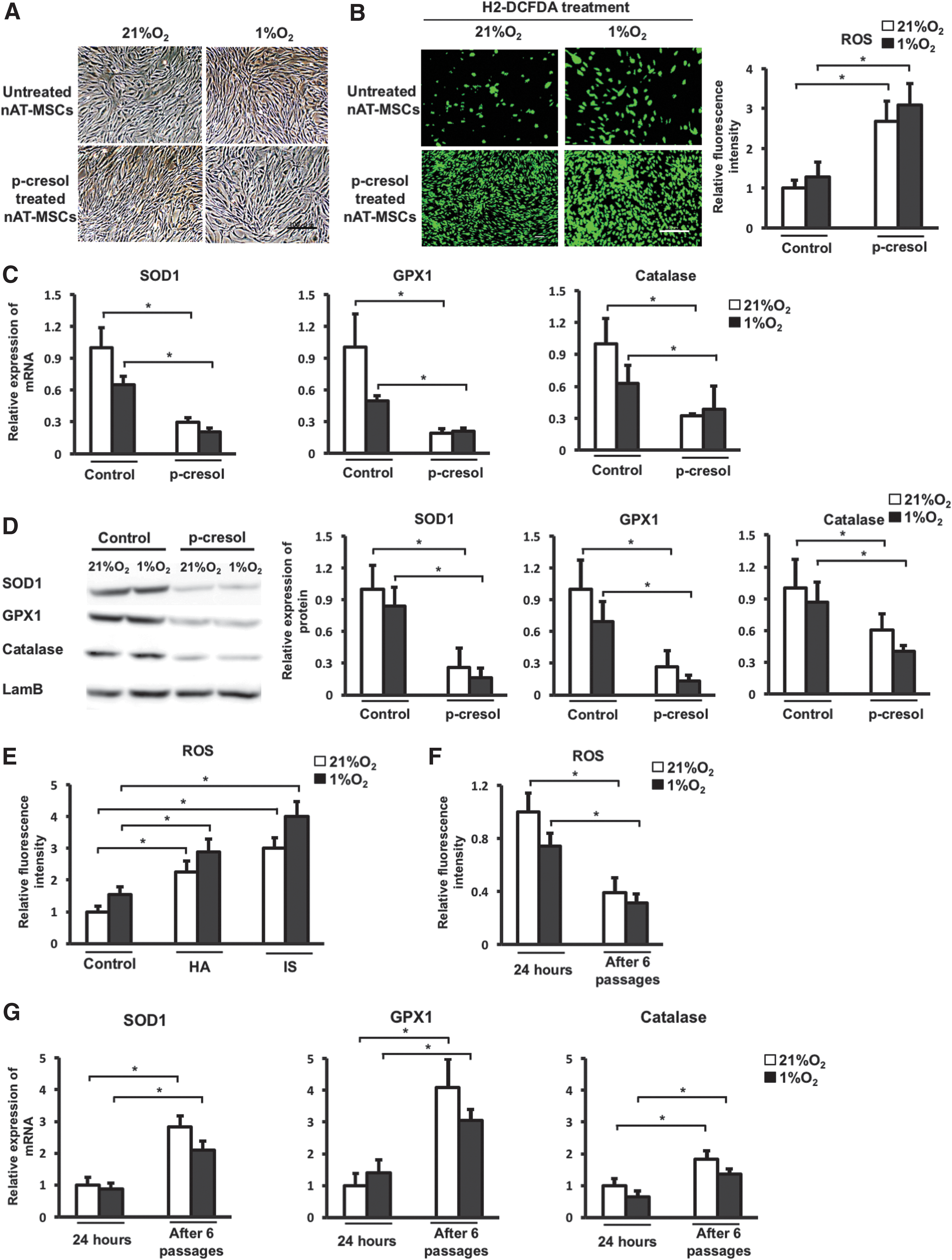
A temporary treatment of uremic toxins causes the reduced expression of antioxidant-related genes and ROS accumulation in AT-MSCs. p-cresol (20 μM), HA (25 μM), and IS (50 μM) were used as uremic toxins. nAT-MSCs were induced in the culture medium contain uremic toxins for 24 h before analysis.
A temporary treatment of uremic toxins causes the impaired hypoxic response and loses wound healing activity in AT-MSCs
HIF-1α is a key transcriptional factor in the response to hypoxic stimuli and regulates the expression of many target genes that play essential roles in the function of MSCs [31]. ROS is known to be involved in the regulation of HIF-1α but the role of ROS in the stability of HIF-1α remains controversial [21,22]. To investigate whether the induction of intracellular ROS by uremic toxin affects the stability of HIF-1α, we evaluated the mRNA and protein levels of HIF-1α in the uremic toxin-treated nAT-MSCs. The HIF-1α protein was almost undetectable under normoxic conditions and clearly detected under hypoxic conditions in the untreated nAT-MSCs (Fig. 2A). Remarkably, the HIF-1α protein level was significantly reduced under hypoxic conditions in the uremic toxin-treated cells (Fig. 2A). The HIF-1α mRNA levels were largely unaffected by the p-cresol treatment (Fig. 2A). Consistent with these observations, the mRNA levels of HIF-1α target genes VEGF and SDF-1 were significantly reduced by the p-cresol treatment under hypoxic conditions (vascular endothelial growth factor [VEGF]: 3.7-fold decreased expression; stromal cell-derived factor 1 [SDF-1]: 2.3-fold decreased expression, n = 3, P < 0.05, Fig. 2B).

A temporary treatment of uremic toxins causes the impaired hypoxic response and loses wound healing activity in AT-MSCs.
We previously showed that an injection of nAT-MSCs into the injured area facilitates healing process in a mouse skin flap wound model and HIF factors play key roles for the wound healing capability [32,33]. Because both VEGF (which is involved in angiogenesis) and SDF-1 (which is involved in migration) are major factors that promote wound healing activity, we surmised that the uremic toxin-treated nAT-MSCs may have defective wound healing capability in this model. As expected, the p-cresol treatment almost completely lost the wound-healing effect of nAT-MSCs as revealed by the size of necrotic area on day 7 post-transplantation (uremic toxin-treated nAT-MSCs: 55.08% ± 7.21% vs. nAT-MSCs: 28.23% ± 8.05%, n = 3, P < 0.05, Fig. 2C). These data indicate that uremic toxins cause the impaired hypoxic response and lose the wound healing activity of nAT-MSCs.
AT-MSCs isolated from the early stage of CKD patients exhibit high levels of intracellular ROS and impaired hypoxic response
We next examined AT-MSCs derived from CKD patients, which are supposed to be continuously exposed to uremic toxins in the patient body. Prolonged exposure of uremic toxins often results in the damage of multiple tissues and cell types resulting in secondary organ failure at the terminal stage of CKD [34,35]. Therefore, to investigate direct effects of uremic toxins, we have chosen the early stage of CKD patients as donors of AT-MSCs.
First, we briefly characterized the morphology, proliferation/differentiation abilities, and the expression of MSC cell surface markers of CKD-derived MSCs (CKD-AT-MSCs), comparing with those from non-CKD donors. Both nAT-MSCs and CKD-AT-MSCs had a fibroblast-like morphology, were spindle-shaped, and expressed the typical markers of MSCs, such as CD13, CD90, CD73, and CD105 (Fig. 3A, B). In addition, CKD-AT-MSCs showed ability similar to nAT-MSCs to differentiate into osteoblasts, adipocytes, and chondrocytes (n = 5 in each, P < 0.05).
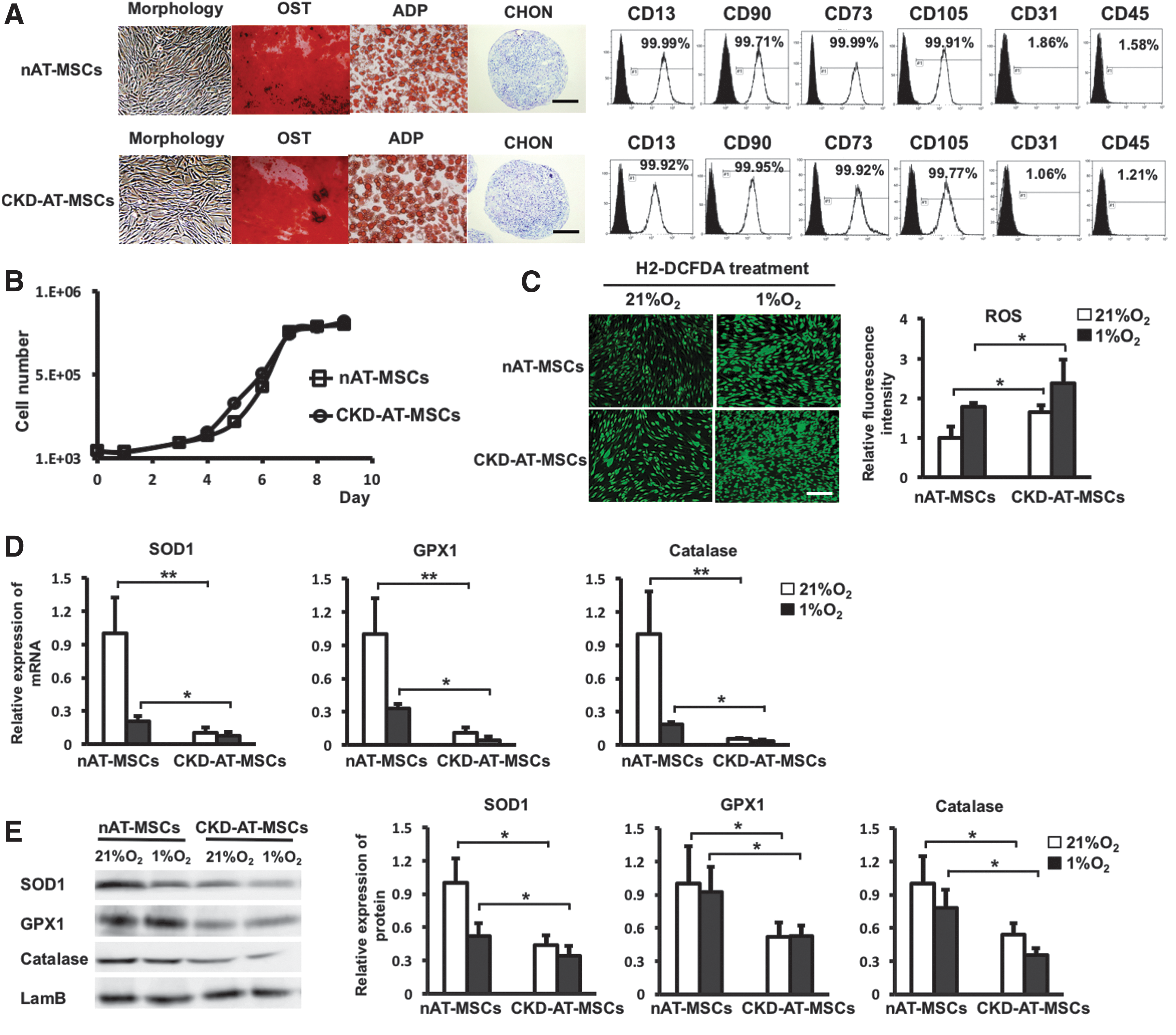
AT-MSCs isolated form the early stage of CKD patients exhibit high levels of intracellular ROS and impaired hypoxic response.
We then examined whether the intracellular ROS levels were increased in the CKD-derived MSCs as we observed in the p-cresol-treated nAT-MSCs. Even in the absence of the exogenous administration of uremic toxins, the ROS generation in CKD-AT-MSCs was higher than that in nAT-MSCs under both normoxic and hypoxic conditions (2.3-fold higher in normoxic condition and 2.7-fold higher in hypoxic condition, n = 4, P < 0.05) (Fig. 3C). Moreover, consistent with the case of the p-cresol-treated nAT-MSCs, the mRNA and protein expression levels of antioxidant enzyme genes (SOD1, GPX1, and catalase) were significantly lower in CKD-AT-MSCs than those in nAT-MSCs (SOD1: 9.72-fold lower; GPX1: 9.23-fold lower; Catalase: 19-fold lower, respectively, in AT-MSCs, n = 4, P < 0.05) (Fig. 3D, E). Collectively, these data indicate that the antioxidant ability was severely impaired in CKD-AT-MSCs.
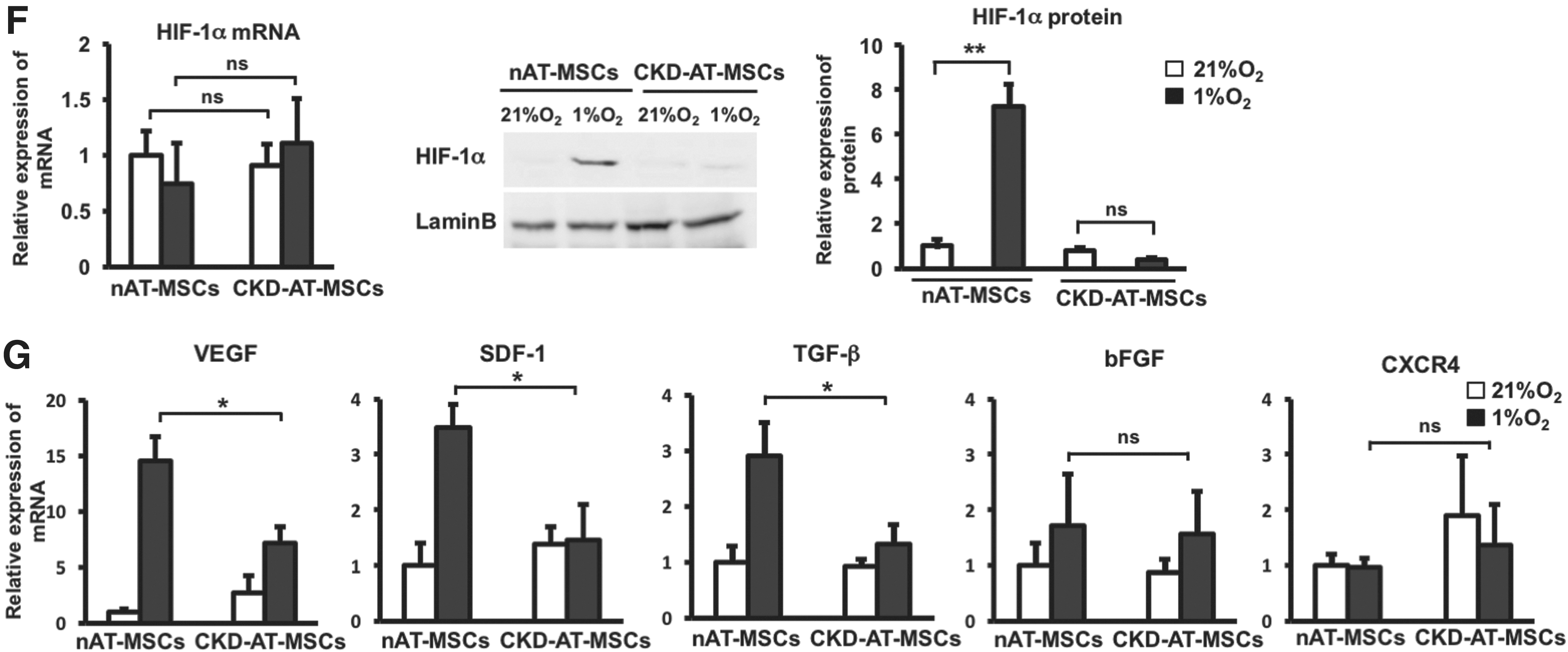
We next examined whether the hypoxic response of HIF-1α protein is altered in the CKD-derived MSCs. As is the case of the p-cresol-treated nAT-MSCs, the HIF-1α protein level was significantly lower in CKD-AT-MSCs compared to nAT-MSCs under hypoxic conditions, whereas the mRNA expression level was comparable in these cells (Fig. 3F). In association with the reduction of HIF-1α protein, the mRNA levels of VEGF, SDF-1, and transforming growth factor (TGF)-β were significantly reduced under hypoxic conditions. By contrast, the moderate increase of bFGF mRNA expression was retained in CKD-AT-MSCs under hypoxic conditions. In addition, the expression of a CXC chemokine receptor CXCR4, another HIF-1α target gene in macrophages, endothelial cells, and cancer cells [36], was not increased in nAT-MSCs and CKD-AT-MSCs under hypoxic conditions.
CKD-AT-MSCs exhibit impaired wound healing ability
We thought that the reduced expression of HIF-1α target genes might affect the wound healing capacity of CKD-AT-MSCs in vivo. To test this possibility, we examined whether an injection of CKD-MSCs into the injured area facilitates healing process as nAT-MSCs did in a mouse skin flap wound model. Compared to the mice injected with nAT-MSCs, those with CKD-AT-MSCs revealed larger unhealed wound area on day 7 post-transplantation (Fig. 4A). The area of wound site was comparable to the control mice, indicating that CKD-AT-MSCs are incapable of facilitating physiological healing process (the necrotic area: sham control (PBS): 60.02% ± 9.33%, CKD-AT-MSCs: 73.34% ± 7.02% vs. nAT-MSCs: 37.52% ± 8.13%, n = 3, P < 0.05).
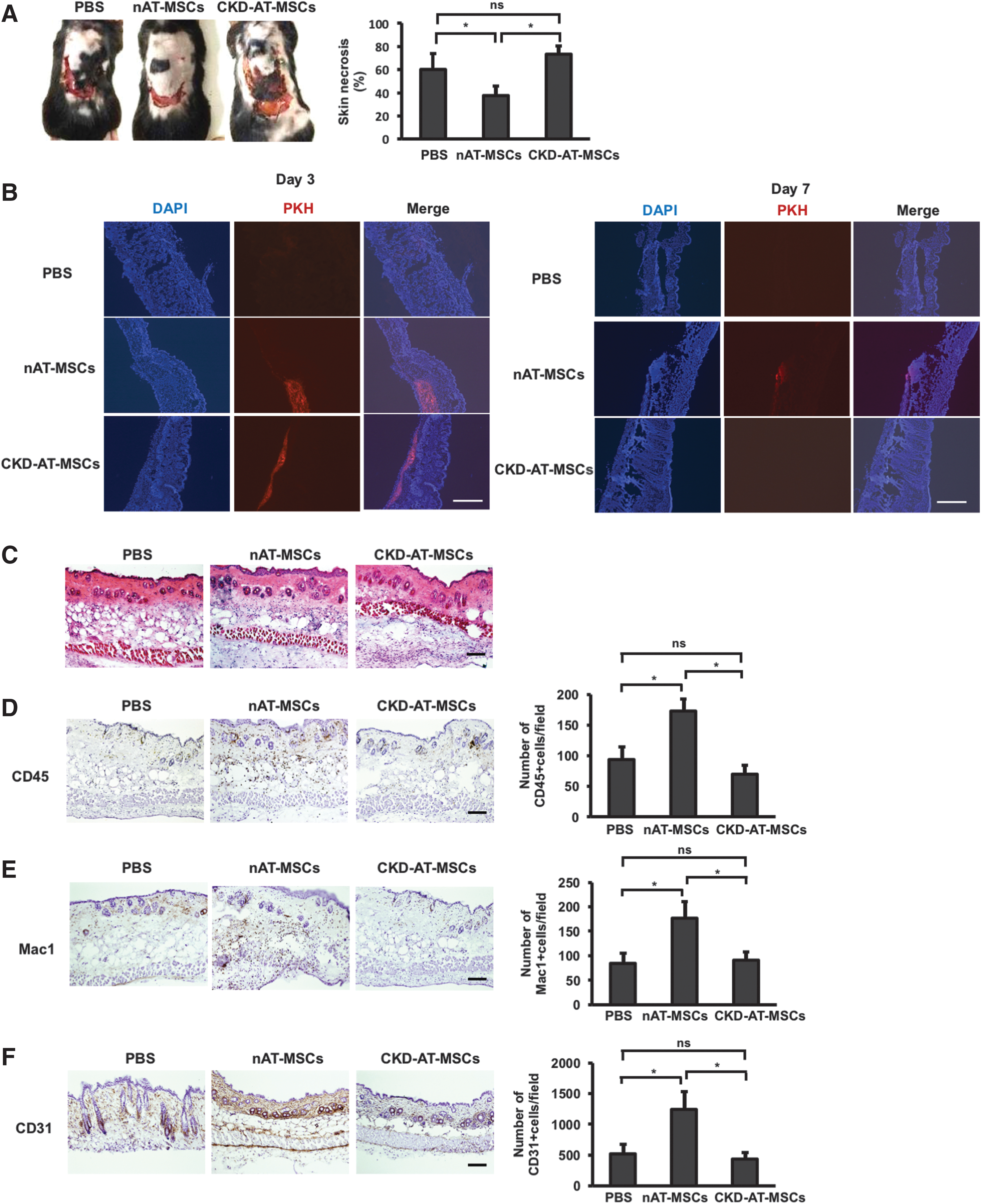
CKD-AT-MSCs exhibit impaired wound healing ability.
Next, we checked the survival of nAT-MSCs and CKD-AT-MSCs at the wound sites. Interestingly, on day 3 post-transplantation, both nAT-MSCs and CKD-AT-MSCs survived at the wound sites but on day 7, only nAT-MSCs were detected at low frequency, indicating that nAT-MSCs might have the ability to survive longer in the transplanted tissues than CKD-AT-MSCs (Fig. 4B). The histological analysis of wound healing process showed that the numbers of CD45-positive cells and Mac1-positive monocytes/macrophages were significantly increased in the ischemic area of nAT-MSC injected mice on day 3 post-transplantation, indicating that nAT-MSCs possessed the ability to recruit inflammatory cells to the site of tissue damage. In contrast to these observations, lesser numbers of migrated inflammatory cells were observed in CKD-AT-MSC injected mice (CD45-positive cells, CKD-AT-MSCs: 70 ± 14/field vs. nAT-MSCs: 173 ± 19, n = 3, p < 0.05. Macrophages, CKD-AT-MSCs: 66 ± 9/field vs. nAT-MSCs: 158 ± 14, n = 3, P < 0.05, Fig. 4D, E). On the seventh day of transplantation, the greater number of CD31-positive vascular endothelial cells was appeared in the subcutaneous region of nAT-MSC injected flap compared with that of PBS injected controls. In contrast, when CKD-AT-MSCs were transplanted at the flap site, the number of CD31-positive cells was comparable to the controls (CD31-posstive cells, CKD-AT-MSCs: 438 ± 105/field vs. nAT-MSCs: 1,292 ± 291, n = 3, P < 0.05) (Fig. 4F). Taken together, unlike nAT-MSCs, CKD-AT-MSCs failed to promote the wound healing process throughout the stage in the mouse skin flap wound model.
Treatment of CKD-AT-MSCs with an ROS inhibitor restores the impaired HIF-1α expression under hypoxic conditions
To explore a direct link between the ROS accumulation and the impaired hypoxic response in CKD-AT-MSCs, we next examined whether treatment with an ROS inhibitor [N-acetyl-
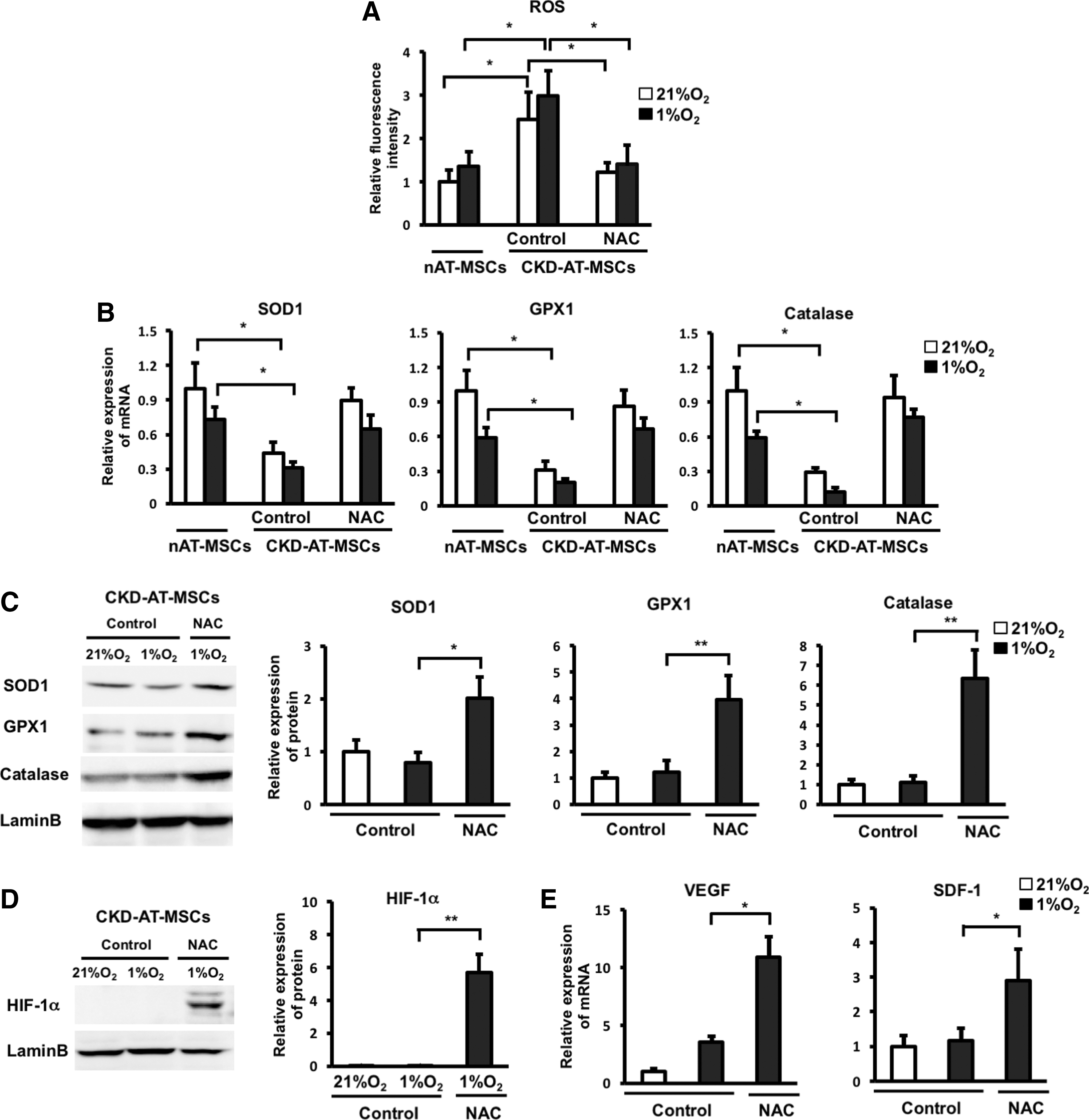
Treatment of CKD-AT-MSCs with an ROS inhibitor restores the impaired HIF-1α expression under hypoxic conditions.
Increased expression of prolyl hydrolase domain 2 is responsible for HIF-1α dysregulation in CKD-AT-MSCs
As shown above, the expression of HIF-1α protein, but not transcript, was decreased both in the p-cresol-treated nAT-MSCs (Fig. 2A) and in CKD-AT-MSCs (Fig. 3F) under hypoxic conditions. To define the molecular basis of these findings, we examined the contribution of the oxygen-responsive prolyl hydroxylases (PHDs) that are known to play key roles in the regulation of HIF-1α protein [37]. PHDs hydroxylate specific proline residues of HIF-1α in the oxygen-dependent degradation domain of this protein [37]. This domain is recognized by the von Hippel-Lindau tumor-suppressor protein (VHL), a component of an E3 ubiquitin ligase complex that results in the rapid proteasomal degradation of HIF-1α [37]. Hypoxia reduces the activity of PHDs, thereby stabilizing HIF-1α protein. On the basis of these previous findings, we hypothesized that the intracellular accumulation of ROS might keep PHDs in active state, leading to continuous degradation of HIF-1α protein even under hypoxic conditions.
To test this possibility, we first examined the mRNA levels of PHD genes (PHD1, PHD2, and PHD3) by quantitative RT-PCR. As previously reported [38], the expression of all PHD genes is induced by hypoxia (Fig. 6A). Interestingly, the expression level of PHD2 was more than doubled in CKD-AT-MSCs compared to that in nAT-MSCs under hypoxic conditions (2.4-fold higher than nAT-MSCs, n = 4, P < 0.05). On the other hand, those of PHD1 and PHD3 were comparable between CKD-AT-MSCs and nAT-MSCs (Fig. 6A). Consistent with the increased mRNA expression, the PHD2 protein level was increased in CKD-AT-MSCs compared to that in nAT-MSCs under hypoxic conditions (Fig. 6A, B).
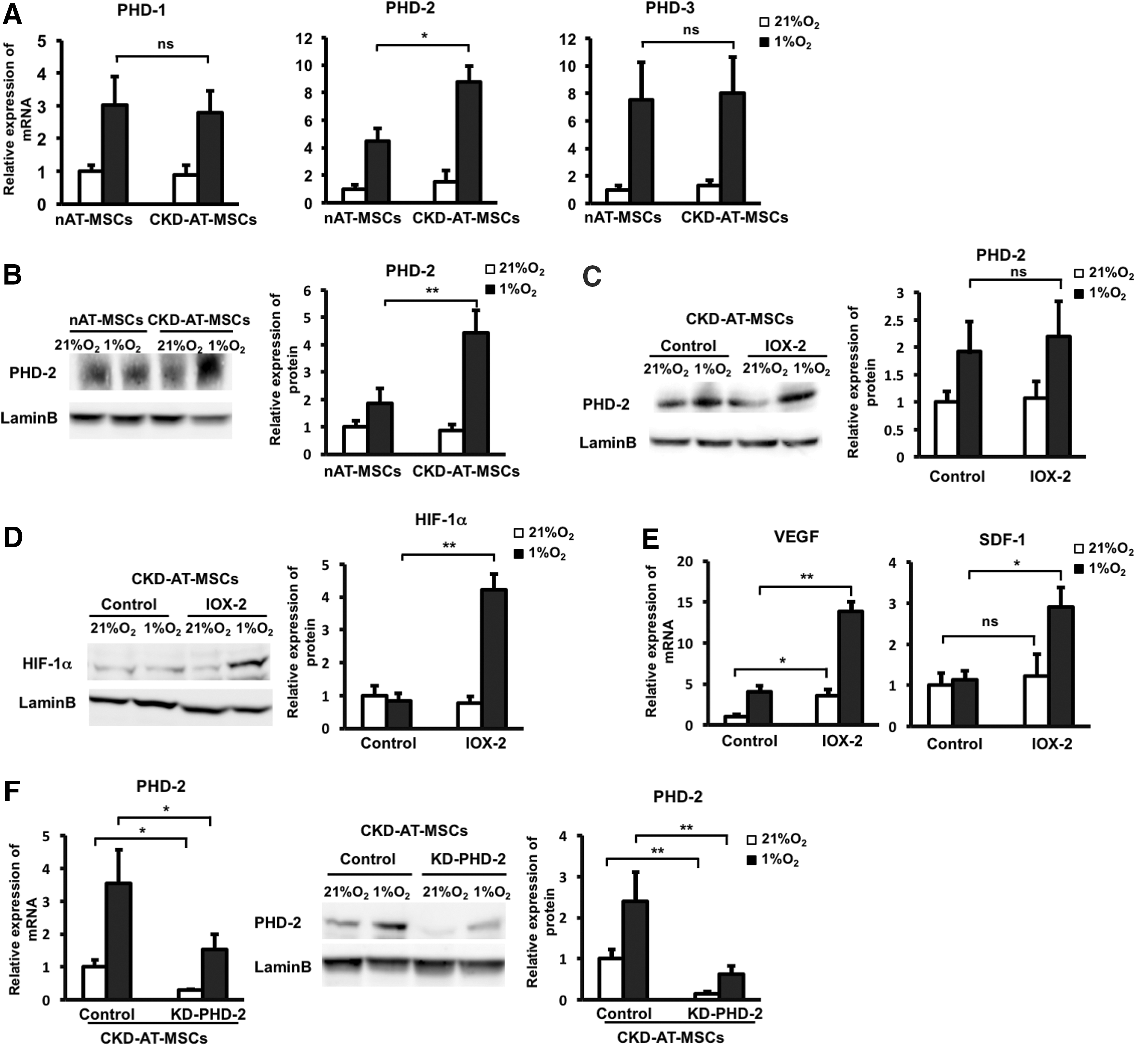
Increased expression of PHD2 is responsible for HIF-1α dysregulation in CKD-AT-MSCs.
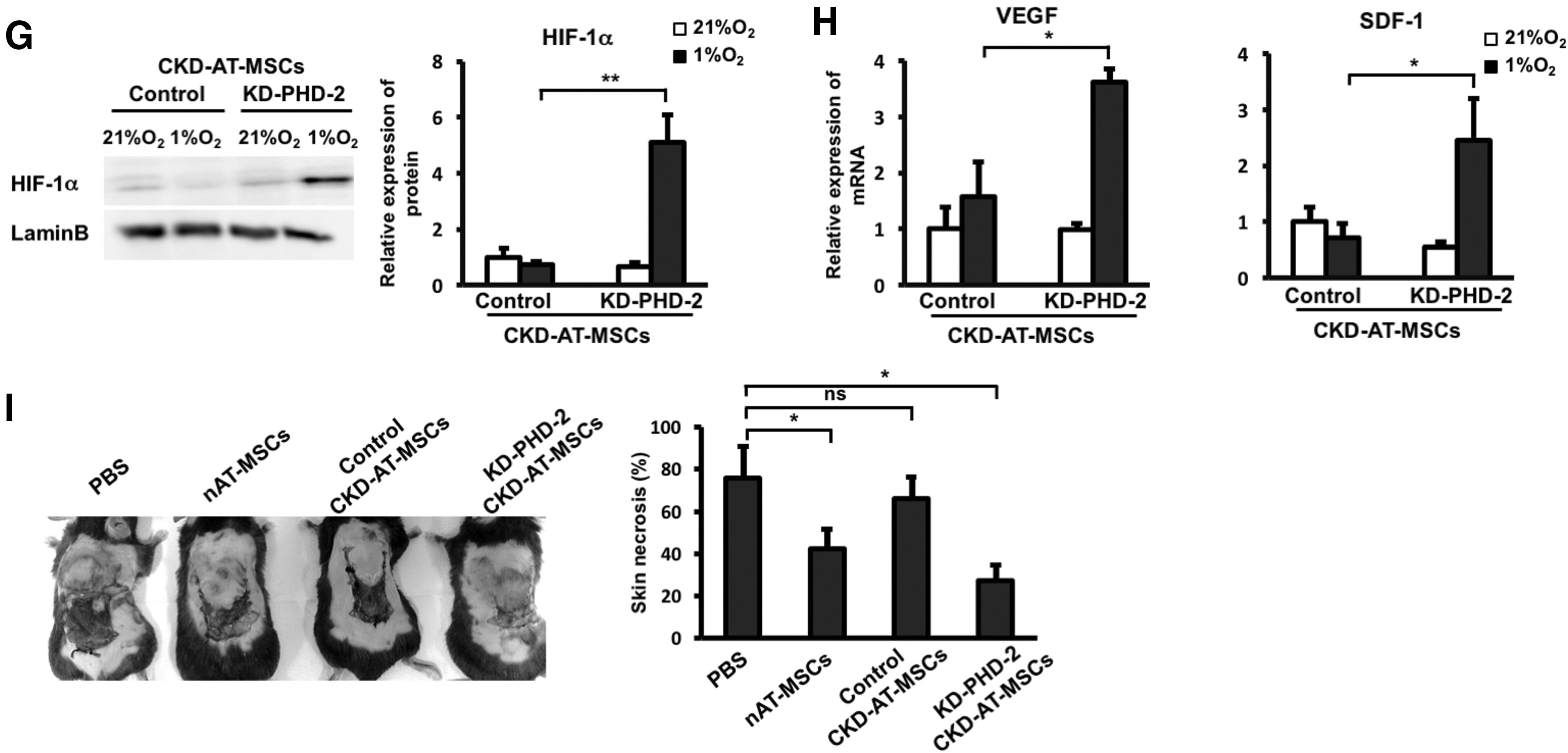
To examine whether the inhibition of PHD2 activity is capable of restoring the hypoxia-induced stabilization of HIF-1α protein, CKD-AT-MSCs were pretreated with a PHD2 inhibitor (N-[[1,2-dihydro-4-hydroxy-2-oxo-1-(phenylmethyl)-3-quinolinyl]carbonyl]-glycine, IOX2, which was reported as a selective inhibitor of PHD2 activity) and were cultured under hypoxic conditions. Treatment with IOX2 showed no significant change in the protein expression of PHD2, but there was the inhibition of PHD2 activity. As expected, the HIF-1α protein level was greater in the IOX2 treated CKD-AT-MSCs (Fig. 6C, D). Accordingly, the mRNA levels of VEGF and SDF-1 in CKD-AT-MSCs were significantly greater in these cells (threefold higher in IOX2-treated CKD-AT-MSCs, n = 4, P < 0.05, Fig. 6E).
We further examined whether similar results were observed in CKD-AT-MSCs transduced with small hairprin RNA (shRNA) against PHD2 gene (KD-PHD2-CKD-AT-MSCs). The mRNA level of PHD2 in these cells was reduced to a similar level of that in nAT-MSCs (3.39-fold decrease under normoxic conditions; 2.3-fold decrease under hypoxic conditions, n-3, P < 0.05, Fig. 6F). In addition, the impaired protein expression of PHD2 in KD-PHD2-CKD-AT-MSCs was also confirmed (Fig. 6F). Consistent with the results from IOX2 treated CKD-AT-MSCs, HIF-1α protein was clearly detectable in knockdown PHD2 (KD-PHD2), but not control siRNA transduced CKD-AT-MSCs (Fig. 6G). The mRNA levels of VEGF and SDF-1 in KD-PHD2-AT-MSCs were comparable to those in nAT-MSCs under hypoxic conditions (Fig. 6H). Lastly, we examined whether KD-PHD2-CKD-AT-MSCs can facilitate wound healing process as nAT-MSCs do in a mouse skin flap wound model. Indeed, KD-PHD2-CKD-AT-MSCs showed the recovery of wound healing similar to the control nAT-MSCs (Fig. 6I).
Taken altogether, these data indicate that hypoxic stabilization of HIF-1α protein is impaired in CKD-AT-MSCs accumulating high level of ROS derived from uremic toxins. Our data suggest that the impaired hypoxic induction of HIF-1α and functional failure in wound healing in CKD-AT-MSCs can be restored by inhibition of PHD2, which is selectively upregulated in these cells.
Discussion
This study demonstrated that AT-MSCs isolated from early-stage CKD patients (CKD-AT-MSCs) had an impaired wound healing capacity due to the absence of hypoxic induction of HIF-1α protein. The hypoxia responsible genes related to angiogenesis and recruitment of inflammatory cells, such as VEGF and SDF, were downregulated in these cells. Importantly, our data showed that the inability of HIF-1α activation is largely attributed to the increased ROS and PHD2 expression in CKD-AT-MSCs under hypoxic conditions (Fig. 7). Moreover, correcting the PHD2 expression in CKD-AT-MSCs restores the hypoxic induction of HIF-1α in these cells resulting in the recovery of wound healing activity.
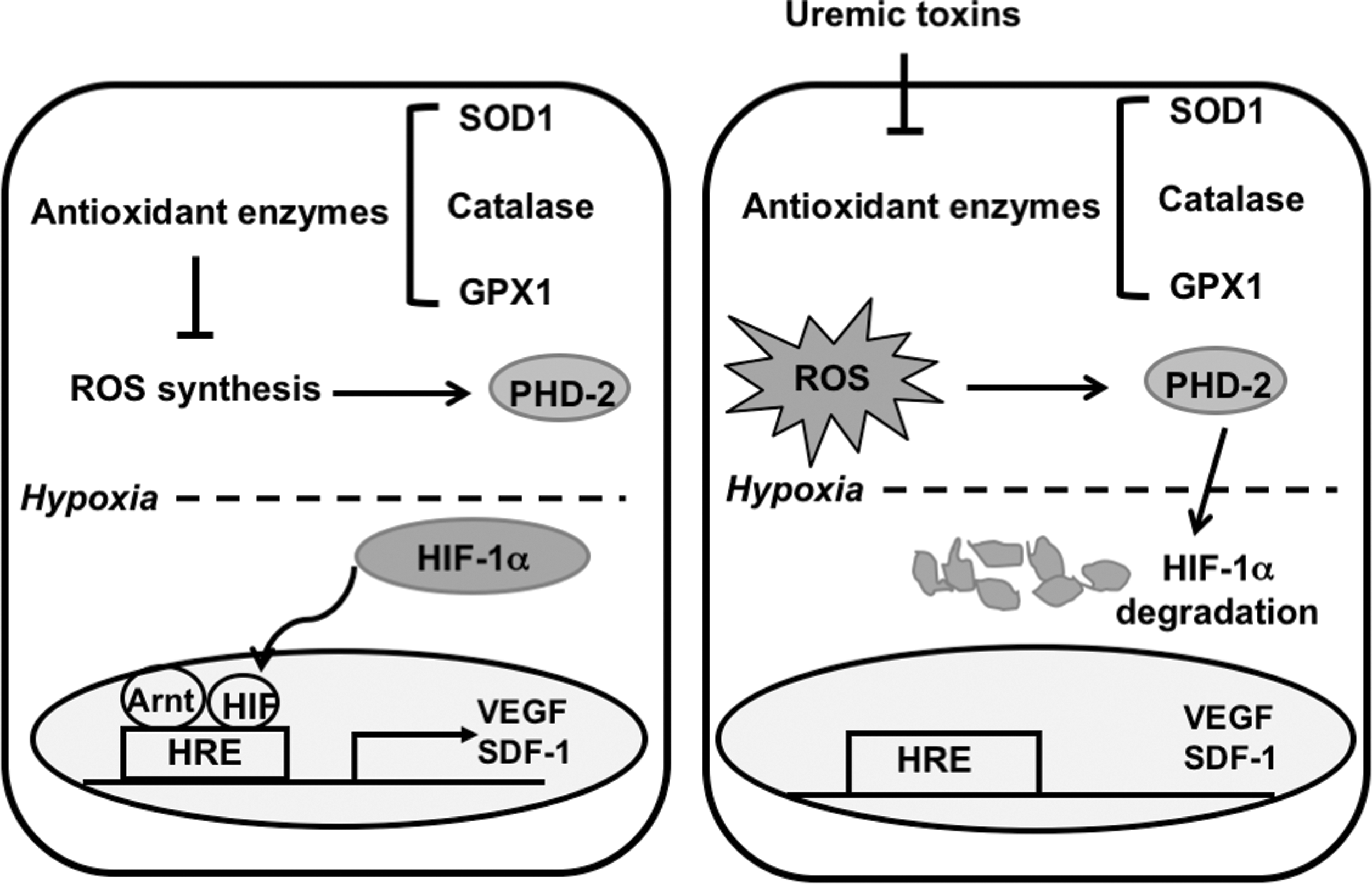
The mechanism underlying the defective function in wound healing of AT-MSCs from early-stage CKD patients. In AT-MSCs, there is a balance of the expression of antioxidant enzymes and ROS. Under normoxic condition, HIF-1α is hydroxylated by PHD2 and degraded by the ubiquitin-proteasome system while under hypoxic conditions there is no hydroxylation of HIF-1α and HIF-1α can induce the expression of its target genes involved in wound healing. In contrast, in CKD-AT-MSCs from early stage, uremic toxins induced the impairment of antioxidant enzymes, including SOD1, catalase, and GPX1, resulting in a high level of intracellular ROS. ROS can enhance the activity of PHD2 to hydroxylate HIF-1α and the degradation of HIF-1α occurs even under hypoxic conditions. Consequently, the expression of HIF-1α target genes involved in wound healing are impaired, which results in a loss of wound healing function in CKD-AT-MSCs. GPX1, glutathione peroxidase-1; SOD1, superoxide dimutase-1.
CKD results in progressive impairment of kidney function that leads to the accumulation of uremic toxins [39]. The stages of CKD are therefore associated with uremic toxin accumulation, with low levels of toxic load in early stages and high levels in the end stage [39]. In this study, we focused on nAT-MSCs treated with a low concentration of uremic toxin and AT-MSCs prepared from early stage CKD patients. Our data showed that a significant, but reversible, dysfunction of MSCs was observed, while cell proliferation, survival, and the ability of differentiation were largely unaffected. Recently, Yamanaka et al. reported that AT-MSCs from ESKD patients who receive long-term dialysis show poor angiogenesis activation [16]. Consistent with our observations, HIF-1α and VEGF expression were not upregulated under hypoxic conditions. In the case of ESKD patients, the expression of P300/CBP-associated factor (PCAF), a histone acetyltransferase, was significantly downregulated in AT-MSCs [16]. The expression of PCAF is induced by hypoxia in AT-MSCs from healthy controls, but not ESKD patients. Interestingly, we could not find any difference in the expression of PCAF between the CKD-AT-MSCs and nAT-MSCs (data not shown), while both studies showed an impairment of promoting in vivo angiogenesis in CKD-AT-MSCs. Furthermore, HIF-1α mRNA levels were comparable between these cells under both normoxic and hypoxic conditions. Collectively, these data suggest that mechanisms underlying the impaired hypoxic response in CKD-AT-MSCs might change depending on the pathological stages of CKD. There was an imbalance of redox state in CKD-AT-MSCs expressing high level of ROS. We found that there was an activation of phosphorylation of ERK1/2 in CKD-AT-MSCs compared to nAT-MSCs (data not shown), suggesting that the ROS-Raf/MEK/ERK might be involved in the dysfunction of CKD-AT-MSCs. In addition, functional failure of AT-MSCs in wound healing activity was fully restored by inhibiting PHD2 in early stage of CKD. In contrast, the persistent exposure of uremic toxin might affect the functions of AT-MSCs in multiple ways, including the PCAF-mediated epigenetic gene regulation.
Our data demonstrated that the imbalance of ROS level causes the impaired function and survival of CKD-AT-MSCs. ROS was reported to intensify the anoikids signals that lead to tranplanted cell death [40]. Although treatment with ROS inhibitor (NAC) can get the recovery of the CKD-AT-MSCs hypoxic induction in vitro, the recovered function seemed reversible and the NAC-treated CKD-AT-MSCs could not cause the wound healing in vivo (data not shown). Therefore, it might require the genetic modification of CKD-AT-MSCs for the wound healing function. In addition, the expression of PHD2 is specifically increased in CKD-AT-MSCs. Since the impaired activation of HIF-1α is closely associated with an accumulation of intracellular ROS, it is conceivable that ROS is a positive regulator of PHD2. In support of this notion, Hellfritsch et al. reported that deletion of mitochondrial thioredoxin reductase (Txnrd2) in mouse embryonic fibroblasts and in tumor cells led to the increased PHD2 protein expression and the absence of hypoxic induction of HIF-1α [41]. In Txnrd2-deficient cells, ROS levels were elevated and PHD2 protein is stabilized via c-Jun NH2-terminal kinase-dependent mechanism [41]. Another pathway that is considered to regulate PHD2 expression is the TGFβ1-Smad pathway [42]. TGFβ1 has been shown to induce HIF-1α stabilization through the selective inhibition of PHD2 expression [42,43]. As shown in Fig. 3G, the expression of TGFβ1 was significantly repressed in CKD-AT-MSCs, although the effects of TGFβ1 reduction have not been examined. Although these pathways might explain the increased PHD2 protein level in CKD-AT-MSCs, it remained unsolved that PHD2 mRNA level is also upregulated in CKD-AT-MSCs under hypoxic conditions. Compared to the post-transcriptional regulation, much less is known about the regulation of PHD2 gene transcription. It will be important to address this issue in further studies.
PHD2 is known to act as an oxygen sensor whose catalytic activity is oxygen-dependent [44]. Our data demonstrated that HIF-1α protein failed to be stabilized even under hypoxic conditions, suggesting that the enzymatic activity of PHD2 is persisted, at least in part, under hypoxic conditions in CKD-AT-MSCs. Recent studies revealed that the catalytic activity of PHD2 is regulated by a complex mechanism involving redox reductions and energy metabolism [45,46]. Since intracellular ROS is known to oxidize ferrous iron, which is an essential cofactor for PHD2 [45], the PHD2 activity of might be persisted with ROS accumulation in cells. Taking another point of view, the enzyme activity of PHD2 is also known to be dependent on 2-oxoglutarate (2-OG), an intermediate of tricarboxylic acid cycle [46]. Recently, two mitochondrial genes, oxoglutarate dehydrogenase and lipoic acid synthase, were identified as novel HIF-1α regulators [47]. Mutation of these genes results in 2-OG accumulation and L-2-hydroxyglutarate formation, which inhibits the PHD2 activity. The evaluation of oxygen metabolism in the cells with high interacellular ROS level might provide helpful information for understanding the regulation of PHD2 activity.
In summary, our study shows that uremic toxin impairs the antioxidant system and wound healing function of AT-MSCs through the ROS-PHD2 pathway. AT-MSCs isolated from CKD patients had an imbalanced redox state and lost their ability to repair wounds. In a way, we provide a therapeutic strategy in AT-MSC-based cell therapy in CKD patients. Further studies should be paid carefully to the pathological each stage of CKD, since the wound healing activity of AT-MSCs might be progressively and irreversibly lost at the end stage of CKD.
Footnotes
Acknowledgment
We thank the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) for their support.
Author Disclosure Statement
No competing financial interests exist.