
Abstract
Adult-derived human liver stem/progenitor cells (ADHLSCs) have the potential to alleviate liver injury. However, the optimal delivery route and long-term biodistribution of ADHLSCs remain unclear. In this article, we used a triple fusion reporter system to determine the kinetic differences in the biodistribution of ADHLSCs following intrasplenic (IS) and intrahepatic (IH) administration in severe combined immunodeficiency/beige mice. ADHLSCs were transduced with a lentiviral vector expressing a triple fusion reporter comprising renilla luciferase, monomeric red fluorescent protein, and truncated HSV-1 thymidine kinase. The stability and duration of the transgenes, and the effects of transduction on the cell properties were evaluated in vitro. The acute retention and long-term engraftment in vivo were revealed by positron emission tomography and bioluminescence imaging (BLI), respectively, followed by histochemical analysis. We showed that ADHLSCs can be safely transduced with the triple fusion reporter. Radiolabeled ADHLSCs showed acute cell retention at the sites of injection. The IH group showed a confined BLI signal at the injection site, while the IS group displayed a dispersed distribution at the upper abdominal liver area, and a more intense signal. In conclusion, ADHLSCs could be monitored by BLI for up to 4 weeks with a spread out biodistribution following IS injection.
Introduction
A
Orthotopic liver transplantation (OLT) has been demonstrated to extend life expectancy and enhance quality of life. However, the procedure is invasive, irreversible, and expensive and limited by organ shortage. Liver cell transplantation, an alternative to ease the requirement for OLT, is less invasive, less expensive, and fully reversible [2]. Liver cells have a mature metabolic capacity and a proliferative advantage over diseased host hepatocytes when transplanted into an individual with liver disease. In the clinic, hepatocyte transplantation successfully alleviated inborn metabolic liver diseases [2], acute liver failure, and chronic liver disease [3]. However, the shortage of cadaveric donor livers, poor resistance of hepatocytes against cryopreservation, and loss of metabolic capability in vitro limit the clinical use of hepatocytes.
Adult-derived human liver stem/progenitor cells (ADHLSCs), which were first characterized by our laboratory, possess a higher resistance to cryopreservation and a high proliferative potential; they display a hepato-mesenchymal phenotype and possess the ability to proliferate and to differentiate into hepatocyte-like cells in vitro and in vivo and allow for longer in vitro amplification [4]. These features make ADHLSCs a good candidate for the treatment of liver disease. In addition, ADHLSCs integrate and repopulate the mouse liver following hepatectomy or chronic disease [4], respectively, and enhance cell proliferation [5]. Preclinical studies have shown that ADHLSCs maintain appropriate gatekeeper mechanisms against transformation following in vitro culture [6].
Clinically, a “first in man” infusion of ADHLSCs to a 3-year-old girl harboring ornithine carbamoyltransferase deficiency not only led to the repopulation of donor cells up to 100 days but also to a decrease in ammonia and orotic acid levels [7]. Finally, a recent international multicenter trial has clearly documented the safety and preliminary efficacy of ADHLSCs in pediatric patients suffering from urea cycle disorders or Crigler–Najjar syndrome (Smets et al., manuscript in preparation).
The delivery route may influence the safety, engraftment, and function of the infused cells. The optimal cell injection route for hepatic targeting needs to be balanced between invasiveness of the procedure, donor cell viability, and engraftment. In the context of mesenchymal stem cells, intravenous (IV) injection, which is the least invasive route, has been shown to ameliorate liver fibrosis, enhance mouse survival, cell proliferation, and decrease collagen deposition [7]. However, IV-injected donor cells commonly lead to pulmonary entrapment [8]. Moreover, mice receiving 2 and 3 million mesenchymal stem cells (MSCs) by IV suffered lethal pulmonary embolism accounting for 10% and 80% mortality, respectively, as MSCs tend to aggregate in high concentration [9], and the volume for injection must be kept low [10].
On the contrary, intrahepatic (IH) injection restricts the distribution of injected cells locally [11] and may cause minimal loss of transplanted cells; therefore, more cells can be expected to settle in damaged liver tissues [12]. However, when the liver is too injured to tolerate further surgical intervention, the delivery of therapeutic donor cells should avoid direct damage to the liver.
Injection of donor cells via the superior mesenteric vein (SMV) enables the dispersed distribution of donor cells in the host liver up to 7 days posttransplantation [11]. However, this route does not support the delivery of large numbers of cells in a mouse model. Doses of 5 × 105 and 106 cells per infusion carry a high risk of lethal portal vein embolization (33.3% and 50% mortality within 24 h posttransplantation, respectively) [11]. Injection of MSCs following the intraportal (IP) route, downstream of the SMV, can be performed with minimal invasiveness in humans by percutaneous transhepatic catheterization [13] or use of an implantable catheter [14]. Moreover, it shows a superior homing and engraftment in animals with fibrotic livers. Unfortunately, it is a more traumatic procedure that may cause hemorrhage and death (22.2% death) in small animals [15].
To take advantage of the portal system [16] without the risk of embolization [17], intrasplenic (IS) injection of donor cells is a promising alternative [12]. Indeed, using IS instead of IV injection shows better donor cell distribution in the liver at 1 h posttransplantation [8]. In addition, IS injections lead to higher liver regeneration than IH injections in rats with fibrotic livers [12]. Transplantations via IH and IS routes are considered to encompass a higher delivery efficacy to the liver by bypassing the lung entrapment. Nevertheless, the best delivery route for hepatic targeting and engraftment remains to be identified for ADHLSCs.
Up until now, the evaluation of ADHLSC engraftment mostly depended on postmortem analyses, such as fluorescence in situ hybridization [7], immunohistochemistry [4,5], reverse transcription-polymerase chain reaction (RT-PCR) [4], and quantitative PCR (qPCR) [18]. These methods largely depend on the site of sampling, instead of providing a whole view on in vivo biodistribution. The noninvasive monitoring of ADHSLCs was first performed by direct radiolabeling with 111In-diethylenetriaminepentaacetic acid (DTPA), an isotope that enters the cell by passive diffusion and allows tracking up to 5 days after IP infusion in the clinic [19]. This cell tracking method is limited by the half-life of the radioisotope and thus does not allow long-term tracking. Furthermore, it does not differentiate between live and dead cells.
Reporter gene-based noninvasive tracking has been established to longitudinally monitor the trafficking, viability, proliferation, and migration of injected cells [20]. The investigation of triple fusion multimodality reporter genes, composed of renilla luciferase (rluc), monomeric red fluorescent protein (mrfp), and truncated HSV-1 sr39tk mutant (ttk), further allows the detection with positron emission tomography (PET) imaging, bioluminescence imaging (BLI), fluorescence microscopy, and flow cytometry [20]. Each detection method has its pros and cons.
For in vivo studies, BLI is rather cheap and is the most sensitive among these triple fusion reporters [21]). However, it only provides signal localization in two dimensions (2D) and it is limited to a few cm in depth. The application of mRFP is also cheap and potentially allows the enrichment of the transgene-positive cell population by fluorescence-activated cell sorting (FACS) to enhance signal. However, the autofluorescence of tissue and the high absorption and scattering of photons may hinder its detection in vivo. The exact detection limit for in vivo mRFP is unclear. It was demonstrated that 2 millions of mRFP+ cells were detectable when subcutaneously implanted into a nude mouse [22]; however, this cell number is far more than the tolerance of small animal for both IV and SMV infusions.
Although PET is expensive and merely enables the detection of a cell cluster of more than 104 cells, it conveys the signal in three dimensions, providing an anatomical view when combined with computed tomography (CT) imaging [23], which eases the interpretation of cell biodistribution. In addition, it is the only modality that can be noninvasively imaged in the clinic.
The purpose of this study was to determine (1) the feasibility and duration of tracking ADHLSCs transduced with a triple fusion reporter in vivo and (2) the safety and efficacy in monitoring the biodistribution of ADHLSCs following IS and IH injection.
Materials and Methods
This study was accepted by the institution's Ethics Review Board (comité d'éthique hospitalo-facultaire) (reference JMM/sy/2010/12) for the use of human-derived tissue, with appropriate informed consent obtained from all tissue donors. In addition, the animal experiments were approved by the Ethics Committee for animal experimentation of the Université Catholique de Louvain Health Sciences Department (ref 2012/UCL/MD/009).
Cell culture
The human embryonic kidney cell line (HEK) 293T is highly transfectable and was used for lentiviral packaging. HEK293T cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM, Cat. No. 41965-039; Invitrogen, Merelbeke, Belgium) supplemented with 10% fetal bovine serum (FBS, Cat. No. A15-101; A&E Scientific, Enghien, Belgium), 1 × penicillin/streptomycin (Cat. No.15140-122; Invitrogen), and 1 × L-glutamine (Cat. No. 1529-026; Invitrogen) (complete medium).
ADHLSCs were emerged and expanded in vitro from hepatocytes of five healthy cadaveric donors (Table 1. Aged 6.00 ± 5.03 years) following protocols described by Najimi et al. [4]. ADHLSCs were grown in high-glucose DMEM containing 10% FBS and 1 × penicillin/streptomycin. Both cell types were grown on CellBIND® (Cat. No. CLS3298; Sigma Aldrich, Machelen, Belgium) surfaces for better cell attachment. ADHLSCs at passage number 2 were transduced with a lentiviral vector expressing the triple fusion reporter. ADHLSCs at passage number 4 were trypsinized and cryopreserved at a density of 0.5–10 × 106 cells/mL with the freezing media, CryoStor™ CS10 (Cat. No. 07930; STEMCELL Technologies, Grenoble, France), for later cell transplantation.
Production of lentiviral vector encoding triple fusion reporter genes and lentiviral transduction
The plasmid DNA of the triple fusion multimodality reporter [20] (CellSight Technologies, San Francisco, CA) (Supplementary Fig. S1; Supplementary Data are available online at
Thirty million HEK293T cells in growth phase were plated on one T175 flask 1 day before transfection. The next day, the cells were cotransfected with a second-generation lentiviral packaging system encompassing three plasmids: (1) pHR' that contains triple fusion gene, (2) pMD.G for pseudotyping with vesicular stomatitis virus (VSV)-G, and (3) pCMV-ΔR8.9 with structural genes, gag, pol, and rev, using linear polyethylenimine (PEI, Cat. No. 23966; Polysciences, Warrington, PA) at a PEI:DNA ratio of 2:1 (w/w). Cells were replenished with growth media containing 10 mM HEPES (Cat. No. 15630-080; Invitrogen) and 1% bovine serum albumin (BSA, Cat. No. A2153; Sigma) at day 1 and 2 after transfection.
The supernatants containing lentiviruses were harvested at day 2 and 3 after transfection, with further centrifugation and filtration with a 0.45-μm syringe filter (Cat. No. 516-0428; VWR, Leuven, Belgium) to remove cell debris. Lentiviruses were titrated on HEK293T cells, and the transduction rate, in terms of mRFP expression, was analyzed on a BD FACSAria III using the BD FACS Diva software (BD Biosciences, Erembodegem, Belgium).
For lentiviral transduction of ADHLSCs, cells were treated with protamine sulfate (Cat. No. 539122; Calbiochem, Darmstadt, Germany) for 5 min at room temperature (RT), followed by addition of media containing lentiviruses. Fresh media were replenished the next day. ADHLSCs were then subjected to cell sorting at least 3-days after transduction.
Cell sorting
Transduced ADHLSCs were separated with a BD FACSAria according to their mRFP expression levels into non-, medium-, or highly transduced cells, to examine the effects of transduction on cell properties. Nontransduced ADHLSCs served as negative control.
In brief, ADHLSCs were rinsed with Dulbecco's phosphate-buffered saline (Cat. No. BE17-512F, DPBS, Lonza, Braine-l'Alleud, Belgium) and detached with 0.05% trypsin-EDTA (Cat. No. 25300-062; Invitrogen), subsequently neutralized with complete growth media. After centrifugation, cell pellets were resuspended at a concentration of 1–1.5 × 106 cells/mL in phenol red-free DMEM (Cat. No. 31053-028; Invitrogen) supplemented with 1% FBS and 1 × penicillin/streptomycin to avoid background fluorescence. Cells were passed through 100-μm cell strainers (Cat. No. 352360; BD Biosciences) and collected in FACS tubes (BD Biosciences). Cell sorting was performed with a nozzle sized 85 μm under a pressure of 45 psi. The cell viability after sorting was determined with 0.4% trypan blue (Cat. No. T8154; Sigma) dye exclusion assay.
In vitro hepatogenic differentiation of ADHLSCs
In brief, ADHLSCs (104 cells/cm2) were plated on growth surfaces coated with rat tail collagen type I (Cat. No. 354236; BD Biosciences) at a concentration of 5 μg/cm2.
Cells were either grown in control media [Iscove's modified Dulbecco's media (IMDM, Cat. No. 21980032; Invitrogen) containing 1 × penicillin/streptomycin and 2% FBS] [24] or differentiation media. Cells were replenished with fresh control or differentiation media twice per week. For the first step of differentiation, media consisted of IMDM containing 20 ng/mL epidermal growth factor (EGF; Cat. No. 100-15) for 1 week. The medium for the second step was composed of 1 × penicillin/streptomycin, 20 ng/mL hepatocyte growth factor (HGF, Cat. No. 100-39), 20 ng/mL oncostatin M (OSM, Cat. No. 300-10), 1 × insulin/transferrin/selenium (Cat. No. 41400045; Invitrogen), and 1 μM dexamethasone (Cat. No. D4902; Sigma) for 2 weeks. EGF, HGF, and OSM were purchased from Peprotech (London, United Kingdom).
Cyp3A4 activity assay
The Cyp3A4 activity was examined to check the efficacy of cell differentiation (Cat. No. V9001; Promega Leiden, the Netherlands). It was performed according to the manufacturer's instructions as previously described [25].
Population doubling time
To determine whether lentiviral transduction affected cell proliferation, cells with different expression levels of mRFP were seeded onto CellBIND® 24-well plates (Cat. No. CLS3337; Sigma) in triplicate (cell density of 5,000 cells/cm2). Media were changed once before analyses. One week after seeding, cells were trypsinized and counted. Calculation of population doubling time was performed as duration × log (2) ÷ [log(final concentration) − log(initial concentration)] [26]. The results of population doubling time were averaged from three independent experiments (donors). Results are expressed as mean ± standard error of the mean (SEM).
Annexin-V staining
Cells were seeded onto CellBIND® 12-well plates (Cat. No. CLS3336; Sigma) (cell density of 5,000 cells/cm2). Cells were grown to confluency after 1 week. Nontransduced cells treated with 125 μM of H2O2 (Cat. No. 23613.397; VWR) for 3 h and 4% formaldehyde (Cat. No. 902405000; VWR) for 15 min served as positive control of apoptotic cells [27] and dead cells, respectively. Nontransduced cells served as blank.
Cells were harvested with trypsin, neutralized with complete culture medium, and washed with cold DPBS. All the following procedures were performed with annexin-binding buffer [10 mM HEPES (Cat. No. 15630-080; Invitrogen), 140 mM NaCl (Cat. No. 424290010; Acros Organics, Geel, Belgium), and 2.5 mM CaCl2 (Cat. No. 1.02379.1000; Merck, Overijse, Belgium), pH 7.4]. Cells were incubated with annexin-V Alexa Fluor® 488 conjugate (Cat. No. V13241; Invitrogen) for 20–30 min at RT. Nuclei were then stained with 1 μg/mL of 4′,6-diamidino-2-phenylindole (DAPI, Cat. No. D1306, Molecular Probes) for 5 min at RT. Cells were washed once and resuspended at a cell concentration of about 1–1.5 × 106 cells/mL. Fluorescence intensity was analyzed on a BD FACSAria III, using the BD FACS Diva software.
Immunofluorescence staining
Cells were fixed with 3.5% formaldehyde for 15 min at RT. The remaining free aldehyde groups were quenched by incubation with 100 mM glycine (Cat. No. 104201; Merck) for 10 min. Permeabilization of cell membranes was performed with 0.1% Triton X-100 (Cat. No. 93420; Sigma) for 10 min. After blocking with DPBS containing 1% BSA for 1 h, cells were further stained with primary antibodies (Table 2) overnight (around 16 h) at 4°C, followed by incubation with appropriate secondary antibodies conjugated with fluorochrome (Table 3) for 1 h at RT. Finally, slides were mounted with ProLong® Gold Antifade Reagent with DAPI (Cat. No. P36931; Molecular Probes, Inc., Eugene, OR) and the resulting fluorescence analyzed using an EVOS microscope (Advanced Microscopy Group, Mill Creek, WA) or a ZEISS ApoTome (Carl Zeiss MicroImaging GmbH, Jena, Germany).
IHC, immunohistochemistry staining; RFP, red fluorescent protein; WB, western blotting analysis.
In vitro rluc activity
ADHLSCs were trypsinized and subjected to rluc assay according to the manufacturer's instructions (Cat. No. E2810, Renilla Luciferase Assay System; Promega, Madison, WI).
Western blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer [150 mM sodium chloride (Cat. No. 1.06404.5000; Merck), 1.0% Triton X-100, 0.5% sodium deoxycholate (Cat. No. D6750; Sigma), 0.1% sodium dodecyl sulfate (Cat. No. 226145000; Acros organics), and 50 mM Tris base (Cat. No. T6066; Sigma), pH 8.0] supplemented with protease inhibitor cocktail (Cat. No. 5892791001, cOmplete ULTRA Tablets, Mini, EDTA-free; Roche Diagnostics, Penzberg, Germany). Mouse tissues were dissected and frozen in liquid nitrogen, followed by homogenization with FastPrep® Lysing Matrix D (Cat. No. 6913-100; MP Biomedicals, Brussels, Belgium) in RIPA buffer, followed by sonication. After centrifugation, the protein concentration of the supernatants was determined by bicinchoninic acid assay (Cat. No. 23227; Pierce biotechnology, Rockford, IL).
Protein lysates were subsequently subjected to 10% sodium dodecyl sulfate/polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride (Cat. No. IPFL00010; Merck) membranes and blocked with 5% BSA in Tris-buffered saline Tween-20 (TBST). Each membrane was incubated with primary antibodies (Table 2) overnight (around 16 h) at 4°C. The membrane was subsequently incubated with an appropriate secondary antibody conjugated with either horseradish peroxidase or fluorochrome. Target proteins were revealed with the Clarity™ Western ECL Substrate Kit (Cat. No. 170-5061; Bio-Rad Laboratories, Temse, Belgium) and exposed to X-ray film (Amersham Hyperfilm, Cat. No. 28-9068-35; GE Healthcare, Diegem, Belgium) or Odyssey CLx Infrared Imaging System (LI-COR Biosciences, Bad Homburg, Germany).
Animal model and cell transplantation
The mRFPhigh ADHLSCs were used in in vivo experiments to maximize the imaging results. Before animal experiments, cells at passage number 5 were trypsinized and resuspended in DPBS containing 4% N-acetylcysteine (NAC) (Lysomucil®, Zambon, Italy) at a cell concentration of 107/mL. The cell viability after trypsinization was 94.86% ± 1.01%.
Severe combined immunodeficiency (SCID)/beige mice (Charles River) at an average age of 14 weeks were used as recipients for ADHLSC xenotransplantation. Each mouse received 106 ADHLSCs, which would be equivalent to ∼2% of the host liver hepatocytes [28]. Three to four mice were included per injection group. Two percent of isoflurane (IsoFlo, Cat. No. b506; Abbott GmbH & Co., Wiesbaden, Germany) with 1% O2 was used to induce anesthesia. Isoflurane was adjusted to 1.5% during surgery.
For the IH route, a 1.5–2 cm transverse bilateral subcostal incision was performed to expose the left lateral lobe (LLL) of the liver. The cell suspension was injected at two sites (50 μL each) of the LLL. For IS injection, a 1 cm left subcostal incision was performed, followed by handling of the surrounding adipose tissues with cotton swabs to expose the entire spleen. A volume of 100 μL of cell suspension was injected directly into the lower tip of the spleen. A 27G needle (Cat. No. 3022000; BD Biosciences) on a 1-mL syringe and absorbable hemostat (SURGICEL®, Cat. No. 1901B; Johnson & Johnson, Beerse, Belgium) were used for both injections.
Immediately after surgery and the following day, saline and Temgesic were subcutaneously injected to prevent dehydration and pain. All experimental procedures were conducted under the practices described in the Ethical Project CMMI-2015-03 previously accepted by the Animal Ethics and Welfare Committee of the Center for Microscopy and Molecular Imaging (CMMI), Charleroi and Université Catholique de Louvain (UCL), Brussels, Belgium.
In vitro radiolabeling of ADHLSCs with 18F-9-[4-fluoro-3-(hydroxymethyl)butyl]guanine
To examine the donor cell trafficking, cells were prelabeled with 18F-9-[4-fluoro-3-(hydroxymethyl)butyl]guanine (18F-FHBG) or 18F-fludeoxyglucose (18F-FDG) (18F: t1/2 109.8 min). 18F-FHBG and 18F-FDG were supplied by the Center of Molecular Imaging, Radiotherapy and Oncology, Institut de Recherche Expérimentale et Clinique, UCL and PET/Biomedical Cyclotron Unit of the Nuclear Medicine Department at ULB-Hôpital Erasme, respectively. For 18F-FHBG labeling, a total of 7.9 × 106 ADHLSCs (viability 97.5%, determined using trypan blue exclusion assay) in suspension were labeled at a specific activity of 5.3 mCi (196.1 MBq) at 37°C for 1 h in 3 mL of DMEM containing 10% FBS and 1 × penicillin/streptomycin. Cells were then washed and resuspended as previously described.
In vitro radioactivity was measured with Genesys Genii™ Single Well Counter- Gamma 1 (Sheffield, United Kingdom). The labeling efficiency was calculated as (activitycell pellet)/(activitycell pellet + activitysupernatant + activitywashes out) × 100%. Leakage of 18F-FHBG at 3 h postlabeling was calculated as (activitysupernatant)/(activitycell pellet + activitysupernatant) × 100%. For 18F-FDG radiolabeling, two batches of 107 ADHLSCs (viability 98.9%) in suspension were labeled at a specific activity of 9.5 mCi (351.5 MBq) and 4.2 mCi (155.4 MBq), respectively, at 37°C for 1 h. The resulting labeling efficiency was 31% and 35%, respectively.
PET and CT imaging
To understand cell trafficking, whole-body PET imaging was performed on a dedicated small-animal PET scanner (Mosaic, Philips Medical Systems, Cleveland, OH) with mice transplanted with 18F-FHBG-labeled ADHLSCs immediately postsurgery. The PET scans were followed by whole-body acquisitions using a helical CT scanner (NanoSPECT/CT Small Animal Imager, Bioscan Inc., DC).
Mice were injected intravenously with a contrast agent, ExiTron NanoScan 12000 (Cat. No. 130-095-698, Viscover; Miltenyi Biotec, Bergisch Gladbach, Germany) (50 μL/20 g mouse), 1 day before PET imaging to enhance the CT contrast of the liver and spleen up to around 200% [29]. Fiducial markers were prepared to allow alignment of PET and CT images, which consisted of polymerase chain reaction tubes filled with 2 μL of iohexol (Omnipaque; GE Healthcare) and 2 μCi of 18F-FHBG [30].
The mice were anesthetized with 3% of isoflurane. PET imaging was performed before CT imaging. Superimposition of the PET and CT images was performed with the VivoQuant software (inviCRO LLC, Boston, MA). For the follow-up of the biodistribution by PET imaging, mice fasted overnight were injected with 18F-FHBG via the tail vein. PET images were acquired from 3 h postinjection. In addition, PET-CT nuclear imaging for 18F-FDG was recorded using a NanoPET-CT imager (NanoPET-CT; Mediso, Hungary). The results were converted to kBq/mL and cell retention (in percent) in different regions was calculated as a ratio of activity in that region divided by the activity in the whole mouse [31].
Measurement of radioactivity in isolated organs
Ex vivo radioactivity was measured with a Genesys Genii Single Well Counter-Gamma 1.
Bioluminescence imaging
Before imaging, mice were shaved to decrease the light scattering of animal fur [32]. The substrate for Renilla luciferase, coelenterazine, ViviRen (Cat. No. P1231; Promega), was diluted with PBS/medium (pH 7) containing 0.1% BSA before use. Each animal received ViviRen at 1 μg/g body weight via the tail vein. Mice were anesthetized with 2% of isoflurane and imaged with Photon Imager RT (Biospace Lab, France). The luminescent signal was captured 2.5 min after administration for 5 min. Image analysis was performed with M3Vision (Biospace Lab) software. Results are expressed as mean ± SEM.
Ex vivo fluorescence imaging
The ex vivo fluorescence (emitted from mRFP; excitation at 530 nm and emission above 570 nm, with subtraction of background at 430 nm) was collected for 4 s using a Photon Imager RT.
Immunohistochemistry
Tissue samples were placed in 4% formaldehyde immediately after sacrifice. After overnight fixation at RT, samples were trimmed and processed for paraffin or optimal cutting temperature compound (Tissue-Tek®, Cat. No. 4583; Sakura, Leiden, the Netherlands) embedding.
Five micrometers of paraffin-embedded sections were deparaffinized and rehydrated in successive baths of xylene (Cat. No. 28973-363; VWR), isopropanol (Cat. No. 149320050; Acros Organics), and H2O. Native monomeric red fluorescent protein (RFP) in transduced ADHLSCs was detected in frozen sections after a nuclear counterstain with DAPI. Before rluc and Ku80 staining, heat-induced epitope retrieval was performed in DAKO target retrieval buffer (Cat. No. S1699; DAKO, Glostrup, Denmark) for 1 h, followed by blocking with TBST containing 5% normal goat serum (Cat. No. G9023; Sigma). Sections were then incubated with primary antibodies overnight at 4°C. Target protein was visualized with secondary antibody conjugated with fluorochrome listed in Table 3 using an EVOS microscope at a 10 × magnification.
Statistical analysis
Assays performed independently at least three times were averaged and expressed as mean ± SEM. Statistical comparisons were made using one-way ANOVA post-hoc Tukey's test and paired t test when there were ≥3 or = 2 groups, respectively. The differences between groups were considered significant if the p-value was ≤0.05 (denoted *; whereas p ≤ 0.01 denoted **). Graphs were drawn with GraphPad Prism 5.00 software (GraphPad Prism Software, Inc., San Diego, CA).
Results
Susceptibility to lentiviral transduction was dose and donor dependent
Recombinant lentiviruses are able to transduce both dividing and nondividing cells [33], leading to a high transduction rate. In lentivirally transduced ADHLSCs, upregulation of mRFP expression was observed microscopically and became stable from day 3 after transduction (data not shown). The expression of mRFP was donor and dose dependent, in terms of the percentage of mRFP-expressing cells detected by fluorescence microscopy (Fig. 1A) and protein expression evaluated by western blot analysis (Fig. 1B, C) (n = 2). These results suggest that the optimal multiplicity of infection (MOI) for lentiviral transduction needs to be optimized for each donor.
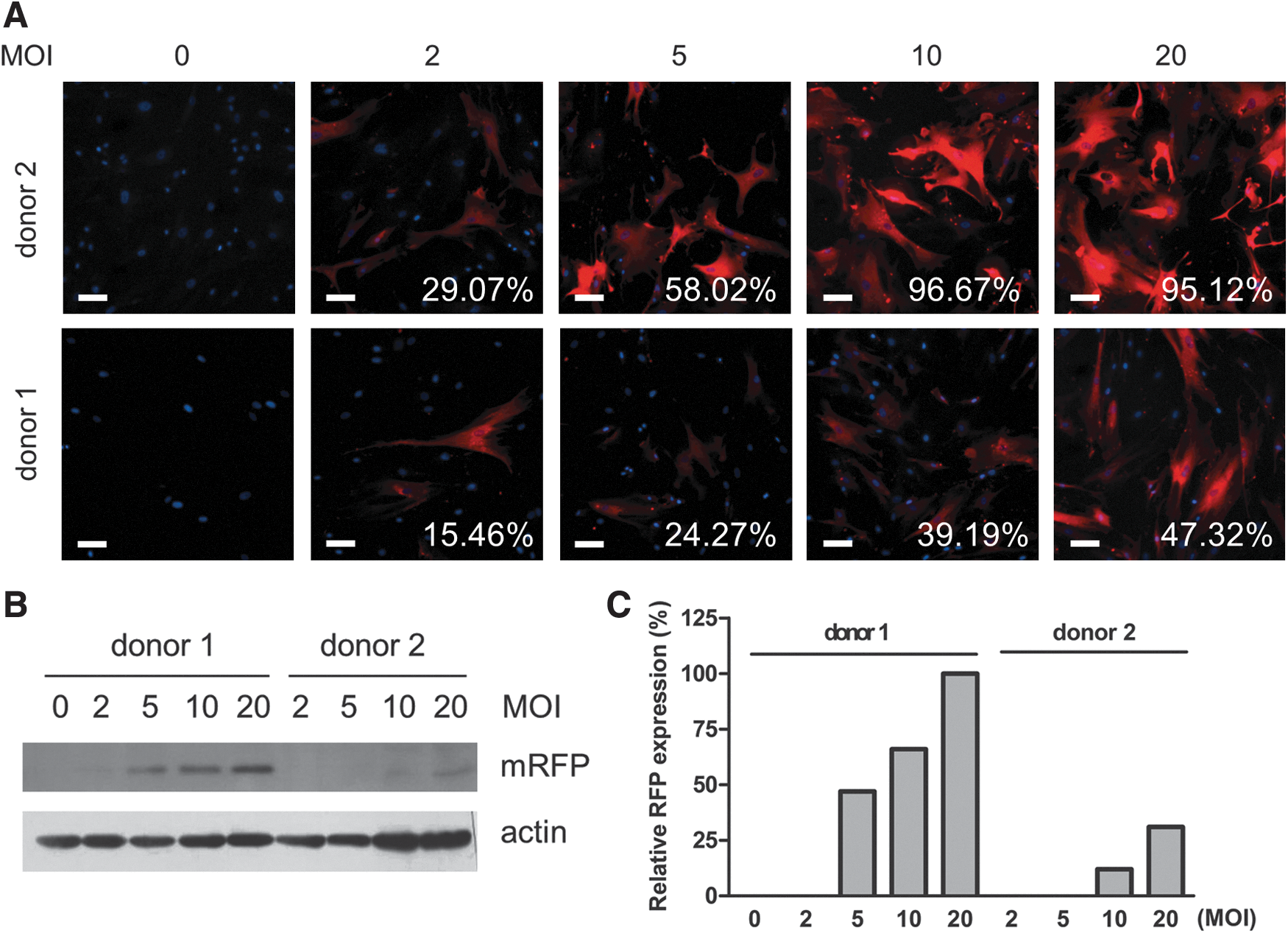
Expression of mRFP in ADHLSCs was dose and donor dependent. Adult-derived human liver stem/progenitor cells (ADHLSCs) from two healthy donors were exposed to lentivirus expressing the triple fusion reporter. Expression of monomeric red fluorescent protein (mRFP) was examined by
Positive correlation between three reporter proteins in lentivirally transduced ADHLSCs
To confirm the correlation between the expression of mRFP and the activity of rluc and ttk, ADHLSCs from two donors were transduced at different MOI, and the cells were harvested and subjected to flow cytometry, ttk and rluc activity assays with their substrates, 18F-FHBG and coelenterate luciferin (coelenterazine), respectively. Median mRFP fluorescence intensity showed positive correlation with HSV1 ttk (R 2 = 0.7294; Fig. 2A) and rluc (R 2 = 0.7178; Fig. 2B) activity in both donors. These results confirmed that enrichment of mRFP+ cells by FACS could potentially enhance the bioluminescence and PET signals.
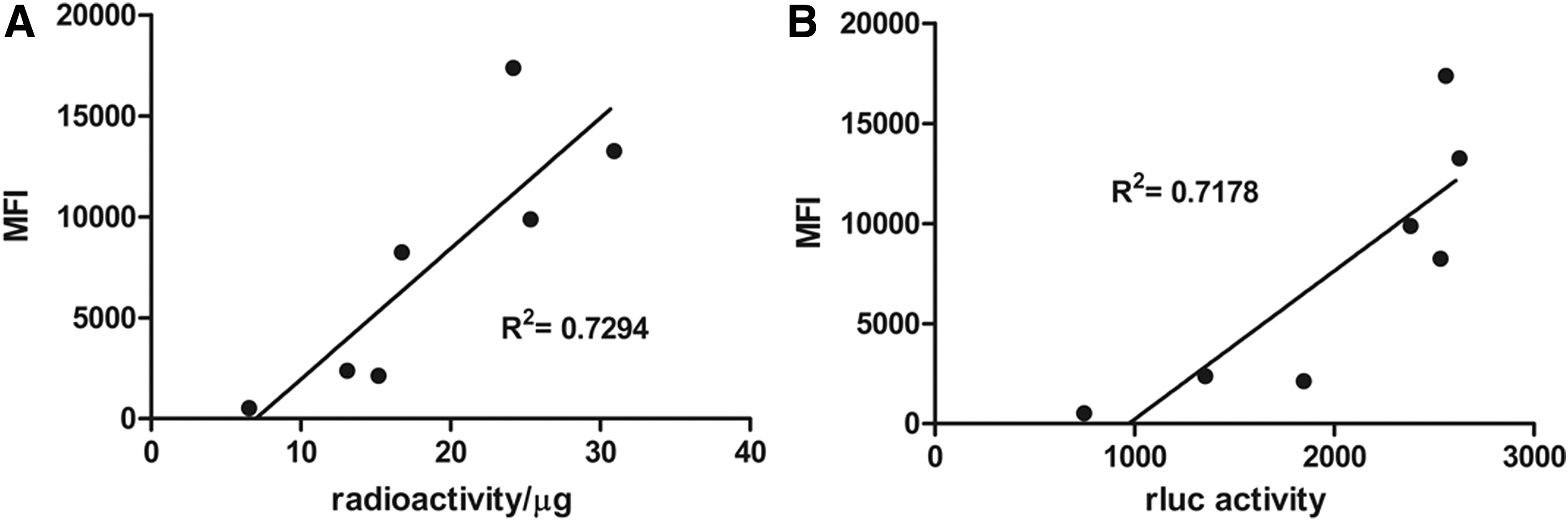
In vitro correlation between renilla luciferase (rluc) activity, thymidine kinase activity, and red fluorescence protein in lentivirally transduced ADHLSCs. ADHLSCs were transduced with lentivirus expressing the triple fusion reporter at different MOIs. The activity assays for
Expression of triple fusion reporter genes had no significant effect on the phenotype of ADHLSCs
The genomic manipulation by lentivector may lead to potential safety issues by random insertion into an undesirable site in the host genome, leading to cell transformation, and even cancer development. To ensure the safety of this triple fusion lentiviral vector, the morphology, cell proliferation, viability, and stemness of the transduced ADHLSCs (sorted into mRFPmed and mRFPhigh) were monitored and compared with nontransduced cells. Generally speaking, nontransduced ADHLSCs and RFPmed cells displayed a mesenchymal shape with a high cytoplasm to nucleus ratio (Fig. 3A), as described earlier by our laboratory [4]. However, RFPhigh cells from most donors were bigger in size and showed more round and bright cells (Fig. 3A), suggesting that higher expression of transgenes may affect the cell physiology and perhaps cell death.

Lentiviral transduction of ADHLSCs had little effects on cell properties. ADHLSCs were either left untreated or transduced with lentivirus expressing the triple fusion reporter, followed by sorting based on the level of mRFP expression.
To characterize whether lentiviral transduction affected cell proliferation and cell death, the cell population doubling time was monitored at passage number 4 until 9. RFPmed cells displayed a similar proliferative rate as nontransduced ones (Fig. 3B). RFPhigh cell proliferation appeared slower along with passaging (Fig. 3B), but the differences were not statistically significant.
To understand whether the seemingly slower growth rate of RFPhigh cells was related to cell death, annexin-V, which binds to the phosphoserine group exposed on the external leaflet of plasma membrane, and a membrane semipermeable dye (DAPI) for DNA were used to stain ADHLSCs in expansion phase and test for early apoptotic event and cell death. Of note, DAPI staining was performed after cell fixation in the positive control H2O2 group and nontransduced cells (Fig. 3C) to ensure a precise focus under the microscope. On the contrary, the intactness of plasma membrane, a feature of healthy cells, was determined by DAPI staining without prior cell fixation (Fig. 3D and transduced cells in Fig. 3C). The plasma membrane of ADHLSCs was annexin-V positive when treated with H2O2, a potent oxidizer that elicits apoptosis (Fig. 3C). ADHLSCs in all groups showed a comparably low extent of annexin-V staining (Fig. 3C).
To get a more precise idea of the proportion of dying cells in each group, annexin-V/DAPI staining was also performed on ADHLSCs in suspension and analyzed by flow cytometry. Nontransduced and RFPmed cells showed comparable results of annexin-V+ and DAPI+ cells (annexin-V: 3.63% ± 1.11% and 4.07% ± 1.54%, respectively; DAPI: 2.35% ± 0.79% and 2.78% ± 1.19%, respectively). Cells expressing a higher level of mRFP showed a seemingly higher extent of apoptotic and dead cells, however, not significantly (Fig. 3D). These results suggested that the expression of transgenes at different levels did not affect cell viability.
As a sufficient amount of ADHLSCs cannot be obtained from cadaveric donor right after liver isolation, the cells are large-scale cultivated and cryopreserved for clinical [19] use. Thus, the effects of lentiviral transduction on cell viability after thawing were investigated. Figure 3E reveals that the apoptosis and death of RFPmed and RFPhigh cells were comparable to nontransduced cells.
Together, these data suggest that the expression of the triple fusion reporter genes had no significant effect on ADHLSC characteristics.
Lentiviral transduction did not affect hepatogenic differentiation of ADHLSCs
ADHLSCs possess stemness and hepatogenic potential [4]. Western blotting analysis confirmed that ectopic expression of transgenes did not affect the expression of the stemness marker, nestin (Fig. 4A), at passage number 5 in expansion phase (cells at this passage number were cryopreserved), control and differentiated cells, and corresponding passage 8 (P8).
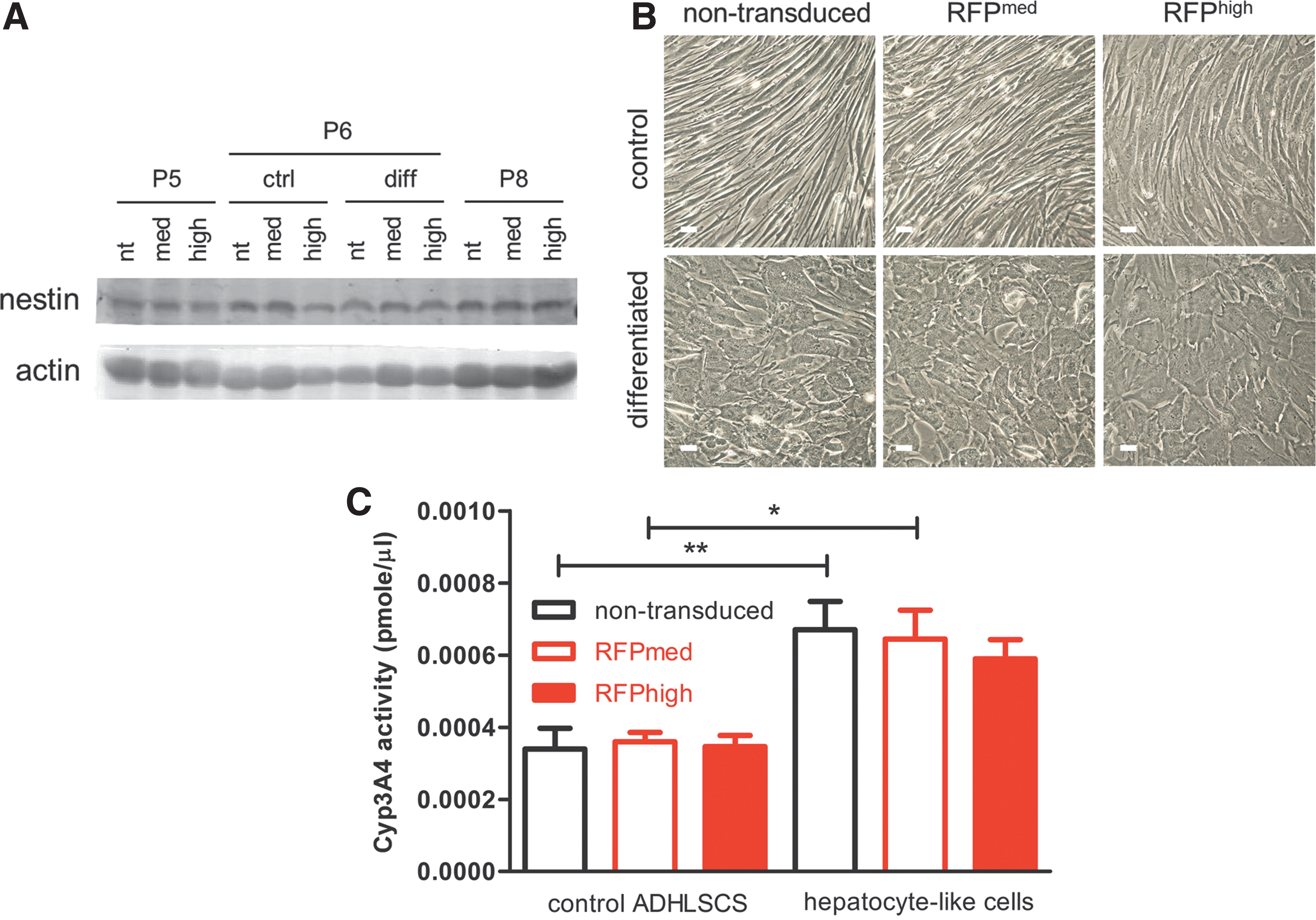
Lentiviral transduction did not affect stemness and hepatogenic differentiation of ADHLSCs. ADHLSCs at passage number 6 were grown on collagen-coated plates and were treated with control media or hepatogenic differentiation media, as described in the “Materials and Methods” section. After 3 weeks,
To confirm whether the differentiation capacity of ADHLSCs was affected by carrying transgenes, cell morphology and the activity of hepatocyte-specific enzyme were determined. As reported earlier [4], ADHLSCs became hepatocyte-like cells, as featured by polygonal shape with a granular cytoplasm, after differentiation protocol, and the same results were obtained with mRFPmed and mRFPhigh cells (Fig. 4B). In both nontransduced and RFPmed cells, Cyp3A4 activity of differentiated hepatocyte-like cells was significantly higher than that of control ADHLSCs (Fig. 4C). Moreover, no significant difference in Cyp3A4 activity between differentiated transduced and nontransduced cells was observed.
Altogether, lentiviral transduction neither affected stemness, in terms of nestin expression, nor hepatogenic differentiation, in terms of morphological change and Cyp3A4 activity.
Differentiation of ADHLSCs had no significant effect on mRFP expression and rluc activity
Stem cell differentiation is associated with epigenetic modification, which may downregulate the expression of a transgene [34], leading to gene instability or even permanent silencing [35] and may pose a concern for the fidelity of the triple fusion protein as a cell tracker. To anticipate the in vivo outcome of mRFP+ cells after transplantation, the stability of transgene expression was monitored at passage number 5 (P5) (used for cryopreservation) and 3 weeks after in vitro hepatogenic differentiation (P6-diff); cells kept on collagen-coated dish (P6-ctrl) and at expansion phase at the corresponding passage (P8) served as controls.
Not surprisingly, most sorted mRFPmed and mRFPhigh cells showed expression of mRFP at P4 (Fig. 5A-a, b). Indeed, the protein level of mRFP was higher in mRFPhigh than mRFPmed population at P5 (Fig. 5C). The number of mRFP-positive cells in both populations at P8 was comparable, as examined by fluorescence microscopy (Fig. 5A-g, h) and FACS (Fig. 5B). Besides, cells kept on collagen-coated plates maintained their %mRFP (Fig. 5A, B). These results suggested that the expression of the triple fusion reporter may become unstable along with in vitro passaging.
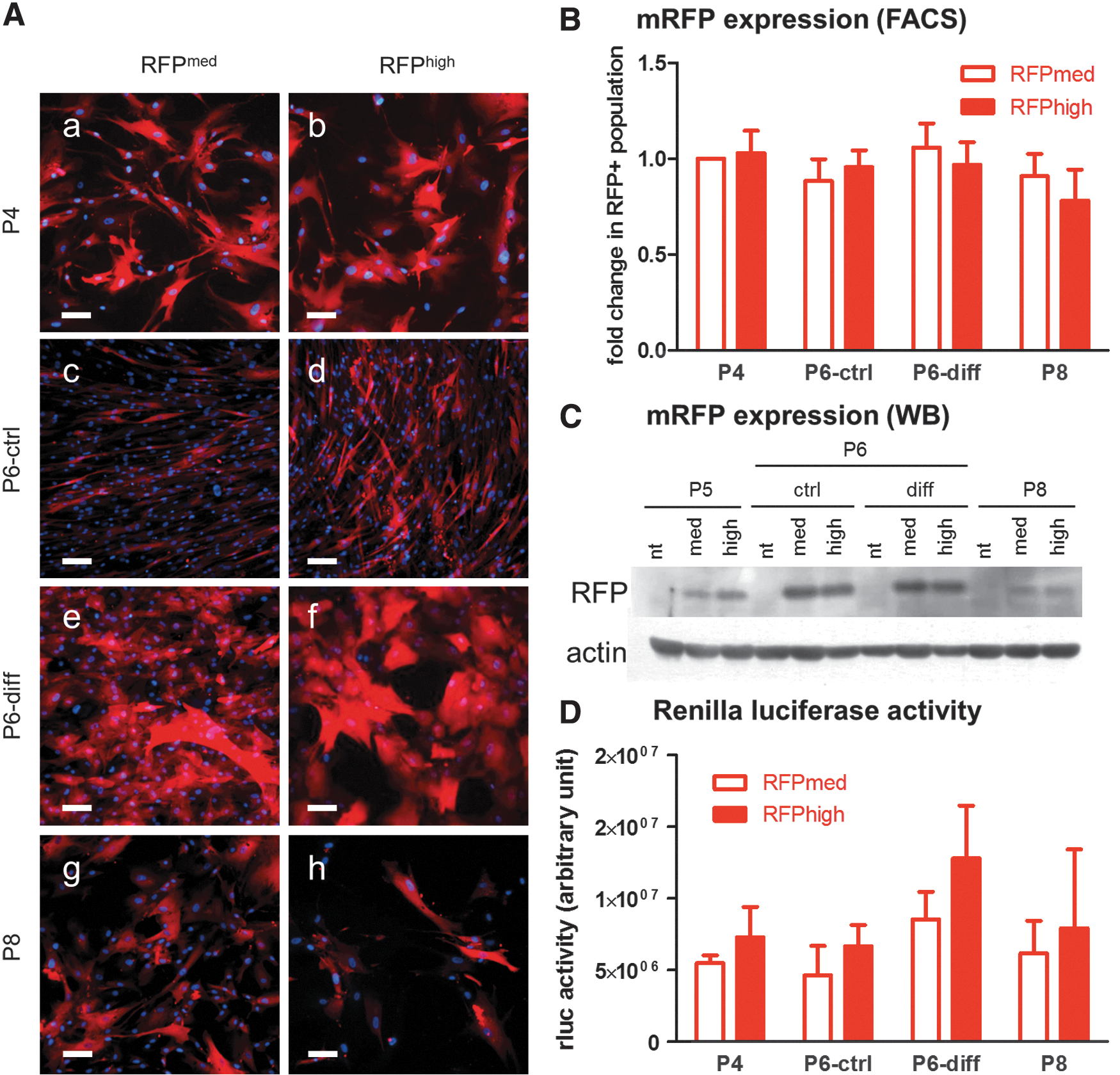
Duration of transgene was slightly altered along with expansion and differentiation protocol. Expression or activity of transgenes in ADHLSCs at P4 (the passage number for cryopreservation;
Furthermore, hepatogenic differentiation of ADHLSCs displayed comparable %mRFP (Fig. 5A, B) and protein expression (Fig. 5C) with cells at P6. Surprisingly, the protein expression level of mRFP was enhanced in cells grown on collagen-coated dishes (both ctrl and diff) for unknown reason. Furthermore, the activity of another reporter, renilla luciferase (Fig. 5D), was maintained in response to passaging or hepatogenic differentiation. These results implied that no significant silencing of transgene expression that might affect in vivo cell detection occurred during cell proliferation or differentiation.
Acute cell retention following different routes of administration
To monitor cell transplantation following IS and IH routes, ADHLSCs were preradiolabeled with 18F-FHBG, a specific substrate for HSV-1 ttk, before transplantation, to allow tracking of transduced cells with a cleaner signal. The resultant labeling efficiency was 17.2%. Mice were then imaged with PET and CT.
Immediately after the direct injection into the LLL or the spleen (Fig. 6), the majority of donor cells were detectable at the injection site (IS mouse 1 (depicted): 62% of whole mouse activity around the injection site, 1% in the bladder; IS mouse 2: 54% around injection site, 19% in the bladder; IH mouse 1 (depicted): 44% around the injection site, 26% in the bladder; IH mouse2: 66% around injection site, 25% in the bladder). Besides, radioactivity was observed in the urinary bladder, which may correspond to the efflux of nonphosphorylated 18F-FHBG, as PET images were acquired 1 h after radiolabeling. Also, the rate of radionuclide loss from radiolabeled cells into the supernatant was 9.6%.
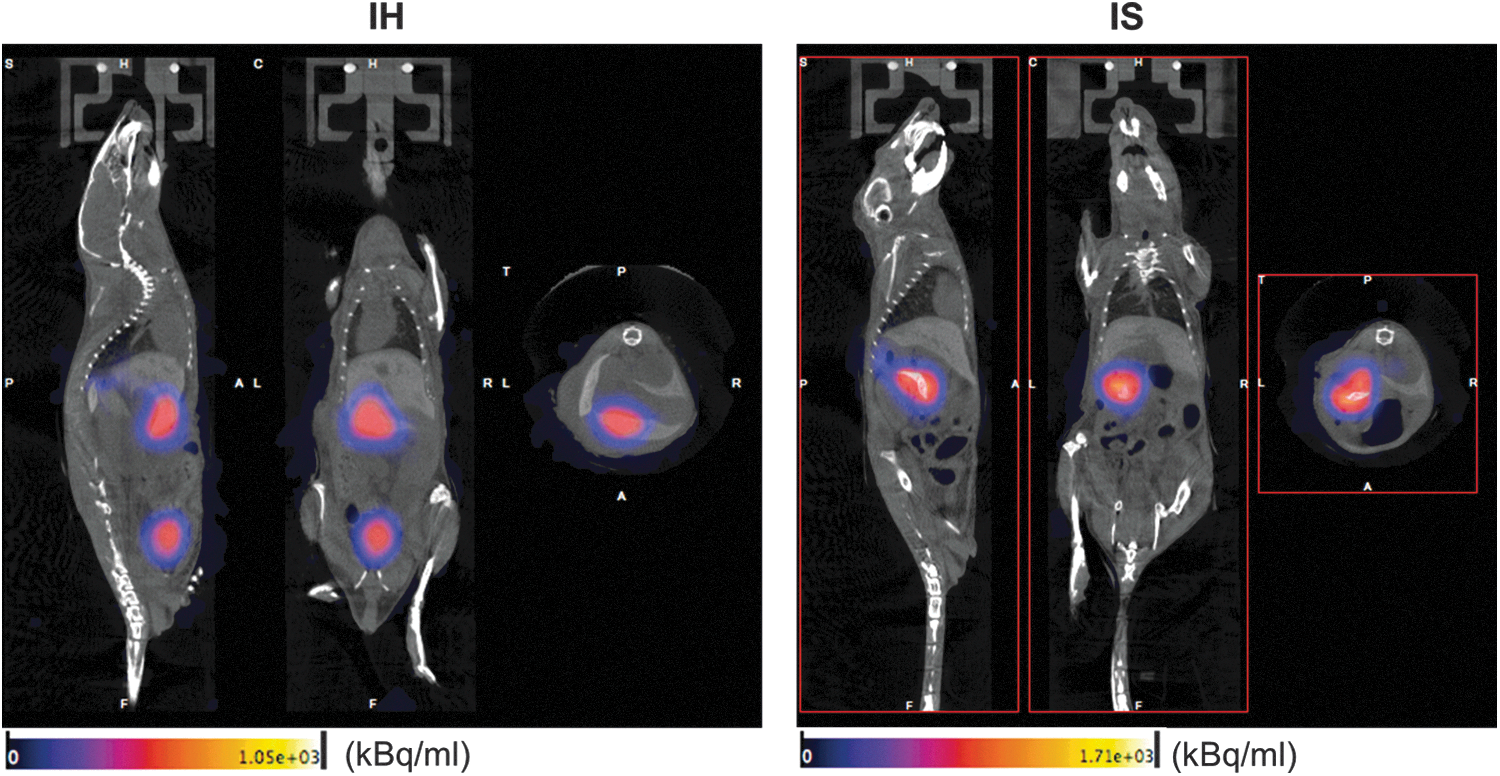
Acute retention of ADHLSCs after transplantation. Female SCID/beige mice were injected with 18F-FHBG-labeled ADHLSCs into either the left lateral lobe (LLL) of the liver (IH; n = 2) or the spleen (IS; n = 2). PET and CT imaging was acquired directly after surgery. CT, computed tomography; IH, intrahepatic injection; IS, intrasplenic injection; PET, positron emission tomography; SCID, severe combined immunodeficiency.
On day 4 and 7, the radiotracer was readministered by IV injection to investigate short-term donor cell engraftment. Unfortunately, only abdominal background was detectable in all mice (Supplementary Fig. S2), and these results were further confirmed by detection of the ex vivo radioactivity of isolated organs (data not shown). These results indicate that the donor cells may be distributed dispersedly through the liver or may be phagocytosed, so that the number of donor cells may not reach the detection limit of PET imaging during the follow-ups.
Longitudinal in vivo tracking of ADHLSCs by BLI and FLI
Taking advantage of a higher sensitivity than PET imaging [21], noninvasive BLI was repetitively performed to monitor biodistribution of donor cells. For practical reasons, cell transplantation was performed in two separate groups at a 2-week interval, so that the week 3 and 4 follow-up of the mice from group 2 could be performed at the same time as the week 1 and 2 follow-up of the mice from group 1 (Fig. 7). Of note, BLI is limited by depth of signal in the tissue and the dimension of the view significantly affects the detection of BLI signal. No signal from the ventral view of mice injected with ADHLSCs directly into LLL was observed on day 3 postsurgery, whereas the signal was detectable when the mice were placed left side up (n = 4). This suggested that the LLL-injected ADHLSCs mainly stayed at the sites of injection and merely disseminated into other lobes.
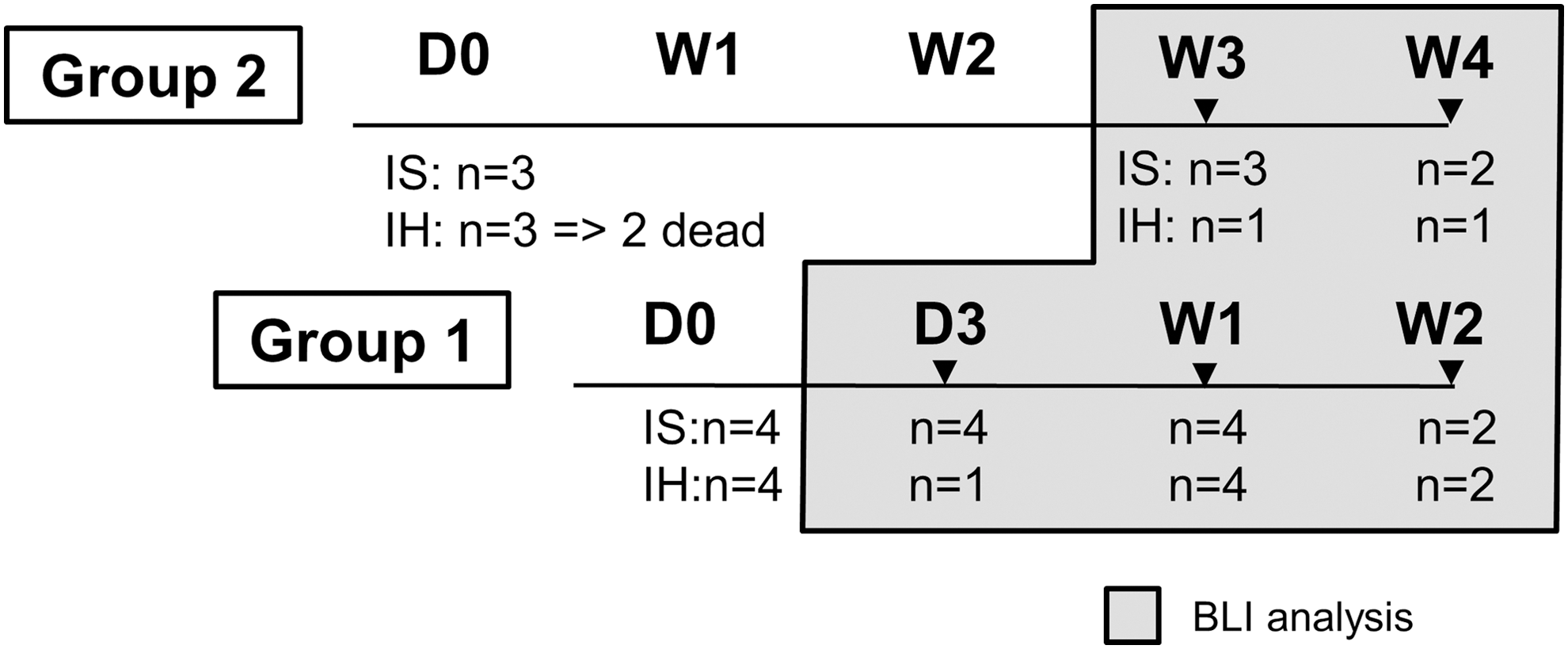
Experimental design for the in vivo BLI assay. BLI was performed to monitor biodistribution of donor cells following IS and IH injections of mRPFhigh ADHLSCs. For practical reasons, cell transplantation was performed in two separate groups at a 2-week interval, so that the week 3 and 4 follow-up of the mice from group 2 could be performed at the same time as the week 1 and 2 follow-up of the mice from group 1. Three to four mice were included per injection group. For IH route, a 1.5–2 cm transverse bilateral subcostal incision was performed to expose the LLL of the liver. The cell suspension was injected at two sites (50 μL each) of the LLL. For IS injection, a 1 cm left subcostal incision was performed, followed by handling of the surrounding adipose tissues with cotton swabs to expose the entire spleen. A volume of 100 μL of cell suspension was injected directly into the lower tip of the spleen. Before imaging, mice were shaved to decrease the light scattering of animal fur [31]. Each animal received ViviRen diluted with PBS/medium (pH 7) containing 0.1% BSA at 1 μg/g body weight via the tail vein. Mice were anesthetized with 2% of isoflurane and imaged with Photon Imager RT in CMMI. The luminescent signal was captured 2.5 min after administration for 5 min. On day 3, mice from the IH group were first imaged on their back, however, the ventral view appeared to be incorrect and mice had to be reimaged left side up. Due to difficulties in reinjecting the substrate, only one mouse was imaged at D3. In the second group, 2 of the IH mice died and only one could be imaged at W3 and W4. Mice were harvested at each time point for ex vivo BLI, fluorescence imaging, and histological analysis. BLI, bioluminescence imaging; CMMI, Center for Microscopy and Molecular Imaging.
On the contrary, IS mice showed strong BLI signal at the upper abdominal region from day 3 (Fig. 8A), implying that ADHLSCs were successfully delivered to the liver by this route. Furthermore, a hot spot remained at the left side of the body, which may correspond to the injection site, that is, the spleen. The left side and ventral view of the IH and IS mice were taken for further time points, respectively, to visualize the majority of the ADHLSCs.

Longitudinal monitoring of ADHSLCs following IH and IS transplantation. Female SCID/beige mice were injected into either the LLL of the liver or the spleen with 1 million of ADHLSCs expressing the triple fusion reporter.
From week 1 to 4, the IH group constantly showed confined signal, which may correspond to the injection site of the liver, with little migration to the right upper abdominal area at week 2. In contrast, the IS group displayed a more dispersed distribution at the level of the upper abdomen, very possibly in the liver (Fig. 8A). This BLI signal of IS mice was significantly higher than that of IH mice on day 3 (Fig. 8B). Besides, BLI signal in IS-injected mice was significantly increased from day 3 to week 1 (Fig. 8B). In addition, the BLI intensities generated by the donor cells were more pronounced in the IS-injected mice all throughout the observation periods despite a little decline in intensity (Fig. 8B).
Ex vivo fluorescence imaging showed intense signal in the liver in IH at 4 weeks posttransplantation (Fig. 8C). These results confirmed the localized BLI signal in the liver in IH group. However, the signal in IS group may be too dispersed to be detected.
RFP-assisted histological analysis
At different time points, fluorescence microscopy of mouse tissues was performed to validate the BLI results. By taking advantage of native mRFP emitted from transduced ADHLSCs, the biodistribution was readily detectable in the tissue samples (Fig. 9A). Colocalization of native mRFP with green fluorescent signal generated using antibodies targeting RFP confirmed the specificity of native red fluorescence as a cell marker (Fig. 9B).
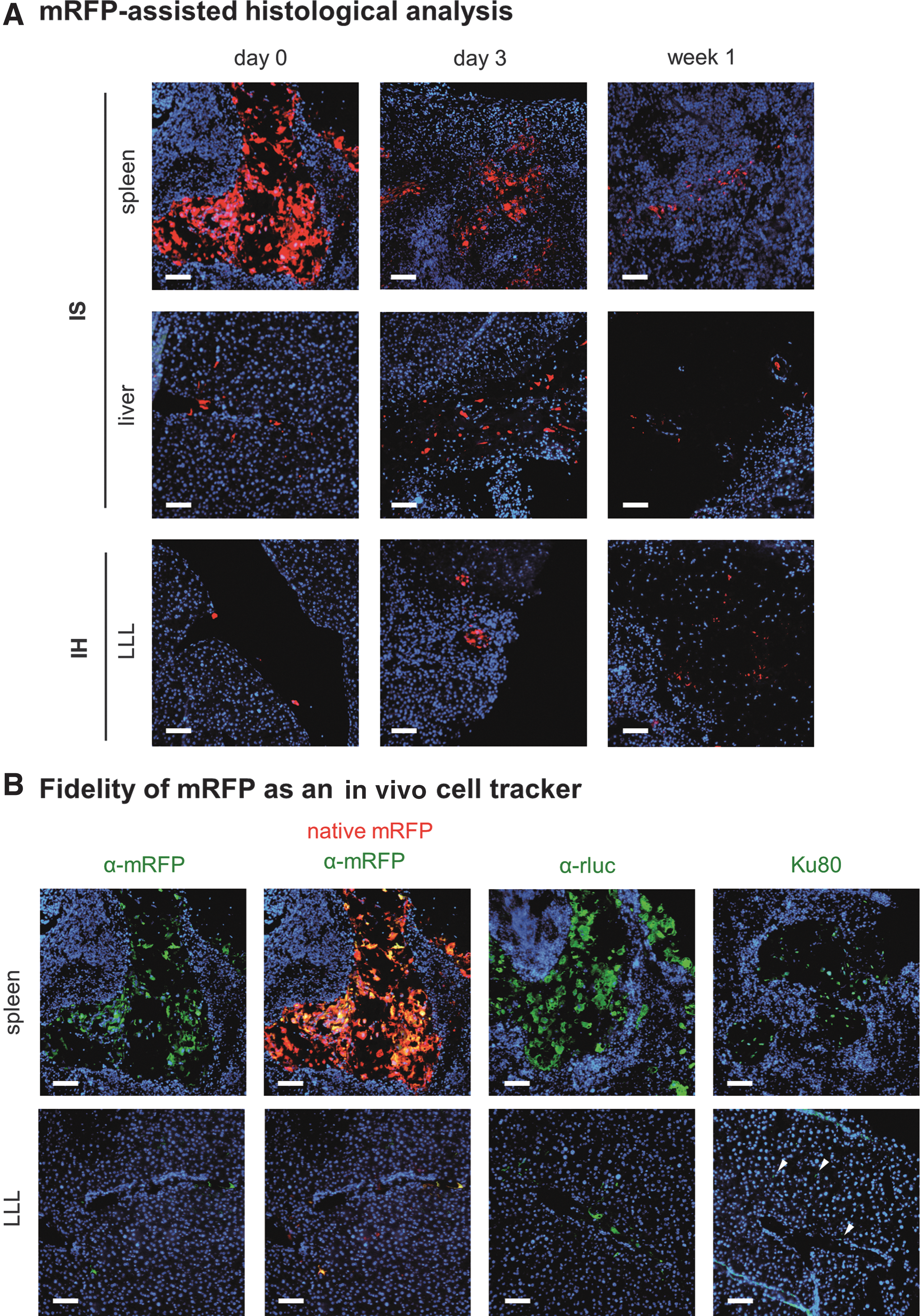
Ex vivo histological confirmation of in vivo BLI data. Female SCID/beige mice were injected into either the LLL of the liver (IH) or the spleen (IS) with 1 million of ADHLSCs expressing the triple fusion reporter. Organs were isolated at indicated time points.
Right after IS injection, the mRFP signals first accumulated in the spleen, and some signal was detected at the periportal area, confirming the successful delivery of ADHLSCs to the liver by this route (Fig. 9A). The mRFP signal gradually decreased in the spleen in the IS mice from day 0 to week 1 (Fig. 9A), suggesting that the majority of ADHLSCs may be cleared by the blood flow to reach the liver. Indeed, the mRFP signal was also detectable in the liver lobes at this time point (Fig. 9A).
In the IH group, the ADHLSCs were detectable in the liver from day 0 to week 1 (Fig. 9A). However, the mRFP signal was merely detectable in the tissue samples from week 2 to 4 (data not shown). Of note, the intensity of native mRFP was comparable throughout the observation periods after different routes of injection, implicating that the expression of transgene was stable in vivo. Besides, from day 3 to week 4, the disorganization of the hepatic cords was observed in the liver (data not shown).
Antibodies against rluc and primate ATP-dependent DNA helicase, Ku80, also validated the expression of transgene and human protein in the spleen and liver using immunofluorescence (Fig. 9B) and immunohistochemical (Supplementary Fig. S3) staining in the cryosections and paraffin-embedded sections, respectively. These results suggested that this triple fusion reporter was a reliable in vivo and ex vivo cell tracker. Of note, the native red fluorescence disappeared after heat-mediated antigen retrieval with citrate buffer when staining for rluc and Ku80. As a result, colocalization of native RFP and either rluc or Ku80 was not performed.
Discussion
The therapeutic potential of ADHLSCs depends on their ability to engraft into the host liver without spreading to untargeted organs. This study demonstrated the feasibility of using multimodality PET, contrast-enhanced CT, and BLI to detect ADHLSCs noninvasively in an immunocompromised mouse model without liver injury. The major findings of our study were the following: (1) ADHLSCs were transducible and could stably express transgene in vitro and in vivo; (2) BLI allowed long-term tracking in vivo for up to 4 weeks posttransplantation; and (3) when compared with the IH route, transplantation via IS seemed to allow a better donor cell distribution throughout the liver.
In this study, the passage number of ADHLSCs for transplantation was kept low, as high passaging may affect homing due to up- or downregulation of surface receptors during cell culture [36], and also lead to a potential downregulation of reporter genes. A second-generation VSV-G pseudotyped packaging plasmid encompassing a triple fusion reporter was used to track ADHLSCs in vivo. Its inability to produce replication-competent recombinants makes it safe to use. This pseudotyped virus enters the permissive cells by interaction with the LDL cell receptor and its family members [37], which are ubiquitously expressed in most cell types, leading to a broad tropism.
In accordance with previous findings [20], the activity of rluc, HSV1 ttk positively correlated with mRFP expression, allowing the sorting of mRFP+ ADHLSCs to potentially enrich the signal emitted by the other two reporter proteins. The mRFP signal in transduced ADHLSCs was maintained up to three months after transduction in vitro (data not shown), further implying the stability of transgenes. Besides, in accordance with the article by Love [38] et al., no effect of lentiviral transduction on the phenotype, proliferation, apoptosis, and hepatogenic differentiation of ADHLSCs was observed in vitro. Although the effect of transduction on these specific parameters may differ in vitro and in vivo, only transduced cells were used for in vivo experiments, as our primary goal was to study the biodistribution of the cells.
The reported enzymatic activity of each domain in this triple reporter is 54% for rluc, “high” for mrfp, and 149% for ttk, when compared to their full-length counterparts [20]. This construct was chosen in our study to maximize the detection with PET imaging, as it allows for the visualization of organs and therefore a more precise localization of the cells when it is used with a contrast reagent. Radiolabeled ADHLSCs were readily detectable by PET at the sites of injection. However, the results showed a leakage of radiotracer, as determined by an in vitro experiment and the nonspecific excretion of 18F-FHBG into the urinary bladder. The half-life for 18F-FHBG is rather short (109.8 min), thus allowing the detection of radiolabeled cells for only few hours.
Readministration of the radiotracer by IV injection on day 4 and 7 posttransplantation did not highlight any activity either in the liver or spleen, implicating that the number of cells may be below the technique's detection limit of a cell cluster consisting at least 104 cells [23,39] (other researchers even claimed that it requires 1 million HSV1-sr39tk-expressing cells in a 0.1 mL volume to be imaged by PET [40]). The application of 18F-FHBG as an investigational new drug (IND #61,880) has been approved by the U.S. Food and Drug Administration, which enables its use as an imaging tool in the clinic [41,42]. Therefore, it would be of great interest to perform infusion of 18F-FHBG-labeled ADHLSCs in a clinical setting, as the ADHLSC transplantation program delivered 6 × 107 cells per kg body weight [7], which would have a better chance to be detected by PET imaging.
In our hands, the in vivo biodistribution of ADHLSCs was readily detectable by BLI, and it allowed the imaging up to 4 weeks posttransplantation. An ∼10-fold loss of photon intensity can be expected for each centimeter of tissue depth [43]. Therefore, the intensity of the signal is negatively correlated with the distance between the detector and the BLI signal. Different sides of detection were taken depending on the route of injection, as the majority of the donor cells locate at the ventral and left side of the mice in IS- and IH-transplanted mice, respectively. However, despite these precautions, we cannot ensure that the localization and depth of the cells were exactly the same for all mice in the group and that hindrance of the signal by tissue was equivalent, which makes the interpretation of the data challenging.
BLI conveys a 2D information only; therefore, ex vivo BLI would need to be performed to gain information regarding the precise localization of the signal inside the body. However, due to technical issues, it could not be performed in this study.
Surprisingly, ex vivo fluorescence imaging showed intense mRFP signals in livers of IH mice, possibly at the site of injection. On the contrary, no mRFP signal was detectable in the IS-injected ones, implying that the dispersed distribution of the cells may be too low to be detected. In addition, the apparent discrepancy between the BLI results (stronger signal from the IS injections) and the ex vivo fluorescence results (stronger signal from the IH injections) may be due to a higher tissue depth of the cells, and therefore, a higher interference with the BLI signal in the IH group. Of note, the fluorescence signal is not affected by cell death, as the native mRFP remained detectable after paraformaldehyde fixation.
The choice of the delivery route of ADHLSCs is important in the determination of cell engraftment, which further affects therapeutic efficacy. The blood from the spleen carries the ADHLSCs through the splenic vein and converges into the hepatic portal vein, and subsequently to the liver. Indeed, abundant ADHSLCs were readily detectable in the spleen of the IS-injected mice (Fig. 9) immediately after transplantation and successfully migrated to the liver via the portal vein, as the ADHLSCs were also found adjacent to the veins in the liver of the same mice (Fig. 9).
In addition, the 18F-FDG-radiolabled ADHLSCs showed a migration to the right lobes of the liver at 80–90 min after IS transplantation (N = 3, Supplementary Fig. S4). These results were in accordance with previous evidence showing a dispersed distribution of MSCs in liver-injured mice after IS transplantation [8].
From day 0 to week 1, the cell retention in the spleen gradually decreased (Fig. 9), with a concomitant increase of the BLI signal in the upper abdominal liver area (Fig. 8A); this signal stayed up to 4 weeks posttransplantation in this immunocompromised mouse model without liver injury. Of note, the cells appeared to be dispersedly distributed throughout the liver, which may have prevented us from reaching the detection limit in the ex vivo BLI experiments and could explain the negative results, in addition to the lack of tissue oxygen for the chemical reaction.
As for IH transplantation, ADHLSCs remained mostly confined at the site of injection (Fig. 6). Little migration to the upper right abdominal area was found from 1 week posttransplantation. This is probably due to the trapping of the cells in the terminations of the hepatic veins with no connection to the portal system, which leads to cell retention in the parenchyma. Although the results will have to be confirmed with a larger number of mice, we found that cells could be detected in vivo by BLI up to 4 weeks posttransplantation (Fig. 8A).
However, most donor cells remained localized and allowed ex vivo mRFP detection up to 4 weeks posttransplantation (Fig. 8C). Although it has been demonstrated that MSCs are attracted to the sites of injury due to the secretion of chemoattractants [7,44], a study using bone marrow-derived mesenchymal stem cells (BMMSC) treatment of mice with acute liver injury [11] showed a donor cell distribution consistent with our results. These results implied that the localized distribution of the donor cells was not affected by the presence of liver injury.
In our study, the difference in donor cell distribution following IS and IH injection could be seen directly after transplantation and was sustained throughout the observation periods. However, liver diseases commonly exhibit diffused lesions. Conceivably, the IS transplantation may result in a better therapeutic effect due to efficient transport to the liver and dispersed distribution. On the contrary, IH transplantation not only damages the liver parenchyma and would worsen the ailments of a liver-diseased individual but it also shows fewer efficacies.
The transduced ADHLSCs were detectable at early time points in the tissue cryosections using mRFP. Although screening of native mRFP avoids the deparaffinization, rehydration, potential antigen retrieval steps, and antibody staining of paraffin-embedded samples, this procedure remains extremely time-consuming and the results largely depend on the sampling.
The ex vivo fluorescence imaging of the liver from week 1 to 4 showed confined and no signal in the IH and IS group (Fig. 8C), respectively. Although a strong signal was detectable in the IH group, the mRFP+ cells in liver tissue slices remained difficult to find. In contrast, the distribution of ADHLSCs may be too dispersed in the liver (Fig. 7A), and the chance of acquiring mRFP+ cells at 5-μm-thick scale was low.
Unlike Ku80 and mRFP staining, the fluorescence emitted by native mRFP and rluc staining stayed bright at least 3 months after mounting (data not shown). Observation of native mRFP and rluc staining in tissue samples was preferable to Ku80 staining, as the ADHLSCs possess a high cytoplasm to nucleus ratio and would be more readily detectable.
One could wonder why no cells were detected in the animals beyond the day of injection by PET and week 1 by immunohistochemistry (IHC), when such a strong signal was detected with BLI. However, it has to be noted that the limit of detection for BLI is between 3 and 103 cells depending on how deep the cells are, while the limit of detection of PET is 104. It is likely, based on the results of the IHC staining, that the number of viable cells was sufficient to produce a detectable signal by BLI but not sufficient to be detected by PET. Although no sham mouse was injected with ViviRen only to evaluate the background BLI signal produced in vivo, the lack of signal found in the IH mice when they were imaged on their back tends to show that the signal detected in the IS group is not due to background noise.
In concert with previous findings [11], the delivery of the MSCs to the liver caused the disorganization of the hepatic cords, as implicated by the nuclear staining with DAPI, eventually leading to the necrosis of the liver observed in our mice from day 3 onward (Fig. 9A). This phenomenon was observed in both IS and IH mice transplanted with transduced and nontransduced ADHLSCs (data not shown), exempting the roles of delivery route and the expression of ectopic genes in MSC-induced necrosis. Although SCID/beige mice do not have B, T lymphocytes and they are defective in natural killer cells [45], this phenomenon may be explained by the attraction of macrophages [46,47] or activation of liver-resident Kupffer cells [48] by MSCs to mediate the immune rejection.
In conclusion, we have demonstrated that ADHLSCs were susceptible to lentiviral transduction and allowed the expression of the triple fusion reporter, without significant alteration of phenotype, viability, proliferation, and hepatogenic differentiation capacity. The expression of the triple fusion reporter was stable and enabled noninvasive imaging of ADHLSC engraftment in an immunocompromised mouse model without liver injury. Besides, we showed that ADHLSCs could migrate from the spleen and successfully engrafted in the liver parenchyma for up to 4 weeks posttransplantation. Furthermore, the current model showed that cell transplantation by IS route may be preferable for hepatic targeting. However, the viability and activity of ADHLSCs after cell transplantation are not clear yet.
Therefore, the biological significance in a liver-diseased model will require further investigation. The understanding of the in vivo biodistribution and effects of ADHSLCs will be important for decision-making regarding transplantation parameters, such as the injection route for liver targeting, and consideration of using inhibitors against Kupffer cell activity to improve transplantation [48].
Footnotes
Acknowledgments
This investigation was supported by a grant from the Région Wallonne (Regenestem project, convention no. 1017264) and a grant from the F.R.S-FNRS (convention no. 3.4549.11), Belgium. Catherine Lombard and Joachim Ravau were paid by a grant from the Région Wallonne (Scaffostem project, no. 1318241). Pierre-Edouard Dollet received research funding from Promethera Biosciences. We acknowledge the technical supports provided by Dr. Anne Bol (Department of Molecular Imaging, Radiotherapy and Oncology, UCL) and Valérie Rosseels (Laboratory of Pediatric Hepatology and Cell Therapy, UCL), discussion with Dr. Véronique Roelants (Department of Nuclear Medicine, Cliniques Universitaires Saint-Luc), Dr. Christophe Deroose (Department of Nuclear Medicine, Katholieke Universiteit Leuven), and Dr. Zhong Lee (Department of Nuclear Medicine/Radiology, University Hospitals Case Medical Center, Case Western Reserve University). Besides, we are grateful to Nicolas Passon, Marie-Aline Laute, Dominique Egrise, Gilles Doumont, Sébastien Boutry, and Mathieu Roch (CMMI) for the assistance in the BLI and PET experiments. We thank Prof. Robert N. Muller, Scientific Director of the CMMI for his interest. The CMMI is supported by the European Regional Development Fund (ERDF), the Walloon Region, the Fondation ULB, the Fonds Erasme, and “Association Vinçotte Nuclear” (AVN).
Author Disclosure Statement
E.M.S. and M.N. are founders and scientific advisors for Promethera Biosciences and have founding shares and/or stock options. P.-E.D. received research funding from Promethera Biosciences. The other authors have no conflict of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.